

Abstract
A method for de novo assembly of data from the Oxford Nanopore MinION instrument is presented which is able to reconstruct the sequence of an entire bacterial chromosome in a single contig. Initially, overlaps between nanopore reads are detected. Reads are then subjected to one or more rounds of error correction by a multiple alignment process employing partial order graphs. After correction, reads are assembled using the Celera assembler. We show that this method is able to assemble nanopore reads from Escherichia coli K-12 MG1655 into a single contig of length 4.6Mb permitting a full reconstruction of gene order. The resulting assembly has 98.4% nucleotide identity compared to the finished reference genome.
The Oxford Nanopore MinION is a portable, single molecule genome sequencing instrument no larger than a typical smartphone. As this instrument directly senses native, individual DNA fragments without the need for amplification, it is able to sequence extremely long fragments (>10 kilobases) of DNA without a reduction in sequence quality 1.
The availability of very long reads is important when assembling genomes as they span repetitive elements and anchor repeat copies within uniquely occurring parts of the genome. Many bacterial genomes can be assembled into single contigs if reads greater than eight kilobases are available, as these reads span the conserved ribosomal RNA operon which is typically the longest repeat sequence in a bacterial genome 2.
The accuracy of the sequence reads is another potentially limiting step for genome assembly; at launch, data from the Pacific Biosciences sequencing instrument was hard to use for de novo assembly due to the lack of bioinformatics tools designed for long reads with a high error rate. Several algorithmic improvements have led to this platform becoming the gold standard for genome assembly; Koren et al. demonstrated that hybrid techniques could generate contiguous assemblies3. This method uses accurate short-read data, or Pacific Biosciences circular consensus reads, to correct error-prone long reads sufficiently for assembly 4. Non-hybrid de novo assemblies of PacBio data in absence of short-read data remained an open problem until Chin et al. developed the HGAP (Hierarchical Genome-Assembly Process) assembler 5. HGAP typically uses a subset of the longest reads in the sequencing data set to use as the input for the assembly process. This subset of long reads is corrected using the entire data set, and this corrected set of reads is assembled with the Celera Assembler. The final assembly is ‘polished’ using a signal-level consensus algorithm to high accuracy. This assembly strategy has revolutionized genome assembly of small genomes and is now being applied to large genomes 6,7.
Nanopore sequencing data has clear similarities to Pacific Biosciences data, with reads sufficiently long to be of great use in de novo assembly 7, 8. Recently, a hybrid approach to assembly which used Illumina short reads to correct nanopore reads prior to assembly was shown to give good results a bacterial and a yeast genome 9. However, non-hybrid assemblies have not yet been described. Recent versions of nanopore chemistry (R7.3) coupled with the latest base caller (Metrichor ≥ 1.9) permits read-level accuracies of between 78-85% 1,8. While this is slightly lower than the latest version of Pacific Biosciences chemistry 6, we hypothesised that a similar assembly approach where nanopore data reads are self-corrected may result in highly contiguous assemblies.
Overlaps can be detected more easily with higher accuracy data. Therefore for this study we exclusively used high-quality “passing filter” two-direction (2D) reads. DNA strands which have been read in both directions by the MinION and combined during base-calling are higher quality than the individual template and complement strands 8. All 2D reads from four separate MinION runs using R7.3 chemistry were combined. Each run was made using a freshly prepared sequencing library using library protocol SQK-MAP-003 (first run) or SQK-MAP-004 (three further runs). In total, 22,270 2D reads were used comprising 133.6Mb of read data, representing ˜29x theoretical coverage of the 4.6 megabase E. coli K-12 MG1655 reference genome (Supplementary Table 1).
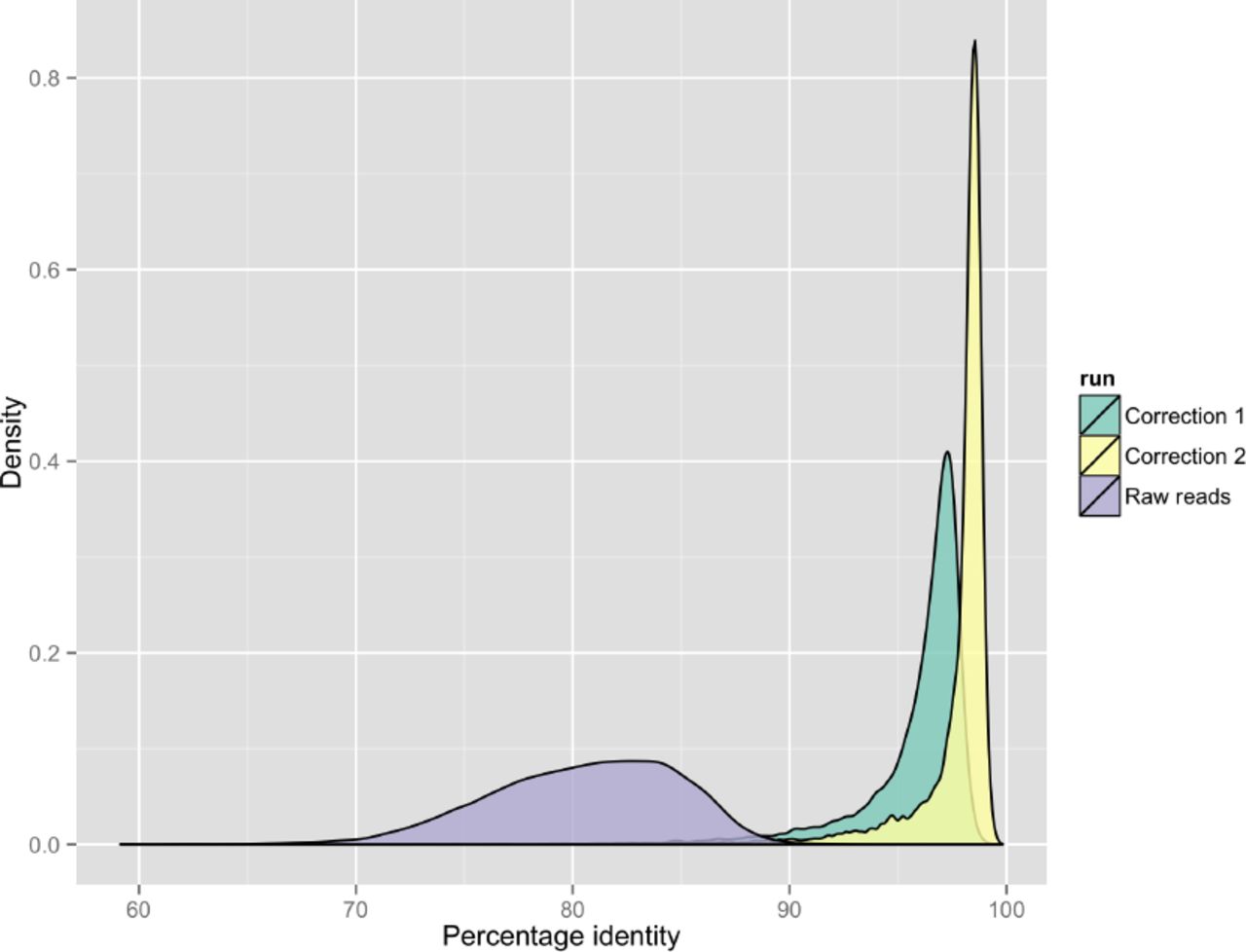
The FASTA sequences for reads were extracted using poretools 10. Potential overlaps between the reads were detected using the DALIGNER software11. Each read and the reads it overlaps were used as input to the partial order aligner (POA) software 12. POA uses a directed acyclic graph to compute a multiple alignment, which we use to determine a consensus sequence. The use of partial order graphs permits a more sensitive reconstruction of consensus sequences in the presence of large numbers of insertions or deletions. This step is analogous to the pre-correction step of the HGAP pipeline, which also uses partial order graphs (implemented in the pbdagcon software) to compute a consensus sequence. We run this correction process multiple times, using the corrected reads as the new input. The results of this correction process increased the mean percentage identity from 80.5% to 95.9% after the first iteration and 97.7% on the second iteration (Supplementary Figure 1).
The reads resulting from two rounds of correction were used as input to version 8.2 of the Celera Assembler 13. The Celera Assembler was run with settings specified in Supplementary Figure 4. The critical parameter for tuning is the overlap minimum identity, which should be double the raw read error rate, in this case set to 6% (ovlMinIdentity=0.06). This resulted in a highly contiguous assembly with four contigs, the largest being of length 4.6Mb and covering the entire E. coli chromosome (Figure 1). The other three contigs were small (6-15kb) and their sequences were present in the long contig and so we did not include them in further analysis.

Single contig assembly of E. coli K-12 MG1655. Panel A shows a dot-plot comparing the assembly presented here with the reference genome. Panel B shows read coverage as a histogram when nanopore reads are aligned against the assembled genome. Panel C shows read coverage and GC composition across the length of the assembled genome. Panel D shows an illustrative view of the read coverage for a randomly chosen section of the assembly genome and the underlying reads used to construct the assembly as viewed in the genome assembly tool Tablet 15.
The final assembly has 3,949 mismatches (85 per 100kb) and 47,395 indels of ≥ 1 base (1021 errors per 100kb), as determined by the QUAST validation tool when compared to the E. coli K-12 MG1655 reference genome (NC_000913.3) 14.
QUAST reported two misassemblies with respect to the reference sequence. On visual inspection of an alignment generated by the Mauve software we determined one large (> 500 base) region of difference between our assembly and the reference. This represented an insertion of a transposase-encoding region into our assembly. This sequence (approximately 750 bases long) is present nine times in the reference genome. Insertion events involving transposons are common in bacterial genomes and can affect wild-type strains commonly used in laboratory experiments, but are difficult to detect using draft assemblies from short-read sequencing technologies.
We explored the sequence context of errors in our assembly by comparing 5-mer counts in our assembled sequence and the reference genome. While most 5-mers are equally well-represented in our assembly and the reference, we found a significant under-representation of poly-nucleotide tracts in our assembly (Figure 4). As the MinION relies on a change in electric current to detect base-to-base transitions, which may not occur or may not be detectable as locally repetitive sequence transits the pore, this error mode is expected. We anticipate that the error profile of our assembly may be improved through an additional assembly ‘polishing’ stage employing signal-level information from the raw reads.

Kernel density plot showing the accuracy of reads from the four individual MinION runs used to generate the de novo assembly. The mean accuracy varies from 78.2% (run 3) to 82.2% (run 1).

Kernel density plot demonstrating the raw nanopore read accuracy and effect of two rounds of error correction on accuracy. The mauve area represents uncorrected sequencing reads, where the green area shows the improvement in accuracy after the first round of correction and the yellow shows improvement from the second round of correction. Further rounds of correction did not improve the accuracy further.

The left panel shows the correlation between 5-mer counts in the reference (x-axis) and the assembly (y-axis). Most 5-mers are equally well-represented in our assembly and the reference (Pearson’s r = 0.97) however some 5-mers are significantly underrepresented in our assembly. To explore these further, we identified 5-mers where the reference genome had ≥50% more occurrences than the assembly (red points in the left panel, grouped bars in the right panel). This indicated that the most underrepresented 5-mers in our assembly are those that consist of a single base (TTTTT, AAAAA, CCCCC, GGGGG) or contain a 4-base stretch of a single base (all others). In the right panel the bars are ordered by reference occurrence count.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Spec file for Celera Assembler.
We conclude that long read data from the Oxford Nanopore MinION can be used to assemble complete bacterial genomes to give an accurate view of gene order and synteny, without the need for complementary sequencing technologies. Further improvements in modeling sequence errors from this platform will be required to improve the base accuracy of the assembly. We have not evaluated the impact of greater sequencing coverage on accuracy, but this may result in further improvements. The software pipeline used to generate these assemblies is freely available online at https://github.com/jts/nanocorrect.
Competing Interests
NJL is a member of the MinION Access Programme (MAP) and has received free-of-charge reagents for nanopore sequencing presented in this study. JQ has received travel and accommodation expenses to speak at an Oxford Nanopore-organised symposium. NJL and JQ have ongoing research collaborations with Oxford Nanopore but do not receive financial compensation for this.
Oxford Nanopore MinION datasets used in this study.
Results from QUAST when comparing draft circular contig against the E. coil K12 MG1655 reference genome (accession NC_000913.3).
Acknowledgements
Data analysis was performed on the Medical Research Council Cloud Infrastructure for Microbial Bioinformatics (CLIMB) cyberinfrastructure. NJL is funded by a Medical Research Council Special Training Fellowship in Biomedical Informatics. JQ is funded by the NIHR Surgical Reconstruction and Microbiology Research Centre. JTS is supported by the Ontario Institute for Cancer Research through funding provided by the Government of Ontario. We are grateful for the staff of Oxford Nanopore for technical help and advice during the MinION Access Programme. We are grateful to the EU COST action ES1103 for funding the authors to attend a hackathon that kickstarted the work presented here.
References




