

Abstract
The rhabdoviruses are a diverse family of RNA viruses that includes important pathogens of humans, animals and plants. We have discovered the sequences of 32 new rhabdoviruses through a combination of our own RNA sequencing of insects and searching public sequence databases. Combining this with previously known sequences we reconstructed the phylogeny of 195 rhabdoviruses producing the most in depth analysis of the family to date. In most cases we know nothing about the biology of the viruses beyond the host they were isolated from, but our dataset provides a powerful way to phylogenetically predict which are vector-borne pathogens and which are specific to vertebrates or arthropods. This allowed us to identify 76 new likely vector-borne vertebrate pathogens among viruses isolated from vertebrates or biting insects. By reconstructing ancestral states, we found that switches between major groups of hosts have occurred rarely during rhabdovirus evolution, with single transitions giving rise to clades of plant pathogens, vertebrate-specific pathogens, and arthropod-borne pathogens of vertebrates. There are also two large clades of viruses that infect insects, including the sigma viruses, which are vertically transmitted. There are also few transitions between aquatic and terrestrial ecosystems. Our data suggest that throughout their evolution rhabdoviruses have occasionally made a long distance host jump, before spreading through related hosts in the same environment.
Introduction
RNA viruses are an abundant and diverse group of pathogens. In the past, viruses were typically isolated from hosts displaying symptoms of infection, before being characterized morphologically and then sequenced following PCR [1, 2]. PCR-based sequencing of novel RNA viruses is problematic as there is no single conserved region of the genome of viruses from a single family, let alone all RNA viruses. High throughput next generation sequencing technology has revolutionized virus discovery, allowing rapid detection and sequencing of divergent virus sequences simply by sequencing total RNA from infected individuals [1, 2]
One particularly diverse family of RNA viruses is the Rhabdoviridae. Rhabdoviruses are negative-sense single-stranded RNA viruses in the order Mononegavirales [3]. They infect an extremely broad range of hosts and have been discovered in plants, fish, mammals, reptiles and a broad range of insects and other arthropods [4]. The family includes important pathogens of humans and livestock. Perhaps the most well known is Rabies virus, which can infect a diverse array of mammals and causes a fatal infection; it kills 59,000 humans per year with an estimated economic cost of US$8.6 billion [5]. Other rhabdoviruses such as Vesicular Stomatitis Virus and Bovine Ephemeral Fever Virus are important pathogens of domesticated animals, whilst others are pathogens of crops [3].
Arthropods play a key role in transmission of many rhabdoviruses. Many viruses found in vertebrates have also been detected in arthropods, including sandflies, mosquitoes, ticks and midges [6]. The rhabdoviruses that infect plants are also often transmitted by arthropods [7]. Even the rhabdoviruses that infect fish have the potential to be vectored by ectoparasitic copepod sea-lice [8]. Rhabdoviruses replicate upon infection of insects (insects are not just mechanical vectors), which may explain why they are insects are common rhabdovirus vectors [7].
Other rhabdoviruses are insect-specific. In particular, the sigma viruses are a clade of vertically transmitted viruses that infect dipterans and are well-studied in Drosophila [9–11]. Recently, a number of rhabdoviruses have been found to be associated with a wide array of insect and other arthropod species, suggesting they may be common arthropod pathogens [12, 13]. Furthermore, a number of arthropod genomes contain integrated Endogenous Viral Elements (EVEs) with similarity to free-living rhabdoviruses, suggesting that these species have been infected with rhabdoviruses [14–17].
Here we aimed to uncover the diversity of the rhabdoviruses, and examine how they have switched between different host taxa during their evolutionary history. Insects infected with rhabdoviruses commonly become paralysed on exposure to CO2 [18–20]. We exploited this fact to screen field collections of flies from several continents for novel rhabdoviruses that were then sequenced using RNA-sequencing (RNA-seq). Additionally we searched for rhabdovirus-like sequences in publicly available RNA-seq data. We identified 34 novel rhabdovirus-like sequences from a wide array of invertebrates and plants, and combined them with recently discovered viruses to produce the most comprehensive phylogeny of the rhabdoviruses to date. For many of the viruses we do not know their true host range, so we used the phylogeny to identify a large number of new likely vector-borne pathogens and to reconstruct the evolutionary history of this diverse group of viruses.
Methods
Discovery of new rhabdoviruses by RNA sequencing
Diptera species (flies, mostly Drosophilidae) were collected in the field from Spain, USA, Kenya, France, Ghana and the UK (Data S1: http://dx.doi.org/10.6084/m9.figshare.1425432). Infection with Rhabdoviruses can cause Drosophila and other insects to become paralysed after exposure to CO2 [18–20], so we enriched our sample for infected individuals by exposing them to CO2 at 12°C for 15mins, only retaining individuals that showed symptoms of paralysis 30mins later. We extracted RNA from 79 individual insects (details in Data S1 http://dx.doi.org/10.6084/m9.figshare.1425432) using Trizol reagent (Invitrogen) and combined the extracts into two pools. RNA was then rRNA depleted with the Ribo-Zero Gold kit (epicenter, USA) and used to construct Truseq total RNA libraries (Illumina). Libraries were constructed and sequenced by BGI (Hong Kong) on an Illumina Hi-Seq 2500 (one lane, 100bp paired end reads, generating ∼175 million reads). Sequences were quality trimmed with Trimmomatic (v3); Illumina adapters were clipped, bases were removed from the beginning and end of reads if quality dropped below a threshold, sequences were trimmed if the average quality within a window fell below a threshold and reads less than 20 base pairs in length were removed. We de novo assembled the RNA-seq reads with Trinity (release 2013-02-25) using default settings and jaccard clip option for high gene density. The assembly was then blasted (tblastn) with rhabdovirus coding sequences as the query to identify rhabdovirus-like sequences. Contigs with hits were then reciprocally blasted against Genbank cDNA and RefSeq databases and only retained if they hit a virus-like sequence. Raw read data were deposited in the NCBI Sequence Read Archive (SRP057824). Putative viral sequences have been submitted to Genbank (accessions in Tables S1 and S2).
As the RNA-seq was performed on pooled samples, we assigned rhabdovirus sequences to individual insects by PCR. cDNA was produced using Promega GoScript Reverse Transcriptase and random-hexamer primers, and PCR performed using primers designed using the rhabdovirus sequences. Infected host species were identified by sequencing the mitochondrial gene COI. We were unable to identify the host species of the virus from a Drosophila affinis sub-group species (sequences appear similar to both Drosophila affinis and the closely related Drosophila athabasca), despite using other mitochondrial and nuclear genes to try and identify the species with certainty. We confirmed all sequences were only present in RNA using PCR, and so are likely free living viruses rather than being integrated into the insect genome (i.e. endogenous virus elements or EVEs [16]).
We identified sigma virus sequences in RNA-seq data from Drosophila montana [21]. We amplified the virus from an infected fly line by RT-PCR and carried out additional Sanger sequencing with primers designed using the RNA-seq assembly. Additional sequences from an RNA-seq analysis of pools of wild caught Drosophila: DImmSV from Drosophila immigrans (collection and sequencing described [22]), DTriSV from a pool of Drosophila tristis and SDefSV from Scaptodrosophila deflexa (both Darren Obbard, unpublished data), accessions in tables S1 and S2.
Discovery of rhabdoviruses in public sequence databases
Rhabdovirus L gene sequences were used to search (tblastn) against expressed sequence tag (EST) and transcriptome shotgun assembly (TSA) databases (NCBI). All hits were reciprocally blasted against Genbank cDNA and RefSeq databases and only retained if they hit a virus-like sequence. We used two approaches to examine whether sequences were present as RNA but not DNA. First, where assemblies of whole-genome shotgun sequences were available, sequences were blasted to check whether they were integrated into the host genome. Second, for the virus sequences in the butterfly Pararge aegeria and the medfly Ceratitis capitata we were able to obtain infected samples to confirm the sequences are only present in RNA by performing PCR on both genomic DNA and cDNA (samples kindly provided by Casper Breuker/Melanie Gibbs, and Philip Leftwich respectively)
Phylogenetic analysis
All available rhabdovirus-like sequences were downloaded from Genbank (accessions in Data S2: http://dx.doi.org/10.6084/m9.figshare.1425419). Amino acid sequences for the L gene (encoding the RNA Dependent RNA Polymerase or RDRP) were used to infer the phylogeny (L gene sequences: http://dx.doi.org/10.6084/m9.figshare.1425067), as they contain conserved domains that can be aligned across this diverse group of viruses. Sequences were aligned with MAFFT [23] under default settings and then poorly aligned and divergent sites were removed with either TrimAl (v1.3 strict settings, implemented on Phylemon v2.0 server, alignment: http://dx.doi.org/10.6084/m9.figshare.1425069) [24] or Gblocks (v0.91b selecting smaller final blocks, allowing gap positions and less strict flanking positions to produce a less stringent selection, alignment: http://dx.doi.org/10.6084/m9.figshare.1425068) [25]. These resulted in alignments of 1492 And 829 amino acids respectively.
Phylogenetic trees were inferred using Maximum Likelihood in PhyML (v3.0) [26] using the LG substitution model [27], with a gamma distribution of rate variation with four categories and using a sub-tree pruning and regrafting topology searching algorithm. Branch support was estimated using Approximate Likelihood-Ratio Tests (aLRT) that is reported to outperform bootstrap methods [28]. Figures were created using FIGTREE (v. 1.4) [29].
Reconstruction of host associations
Viruses were categorised as having one of four types of host association: arthropod-specific, vertebrate-specific, arthropod-vectored plant, or arthropod-vectored vertebrate. However, the host association of some viruses are uncertain when they have been isolated from vertebrates, biting-arthropods or plant-sap-feeding arthropods. Due to limited sampling it was not clear whether viruses isolated from vertebrates were vertebrate specific or arthropod-vectored vertebrate viruses; or whether viruses isolated from biting-arthropods were arthropod specific viruses or arthropod-vectored vertebrate viruses; or if viruses isolated from plant-sap-feeding arthropods were arthropod-specific or arthropod-vectored plant viruses. We omitted three viruses that were isolated from hosts outside of these four categories from our analyses.
We simultaneously estimated both the current and ancestral host associations, and the phylogeny of the viruses, using a Bayesian analysis, implemented in BEAST v1.8 [30, 31]. Since accurate branch lengths are essential for this analysis, we used a subset of the sites and strains used in the Maximum Likelihood analysis. We retained 189 taxa (all rhabdoviruses excluding the divergent fish-infecting novirhabdovirus clade and the virus from Hydra, as well as the viruses from Lolium perenne and Conwentzia psociformis, which had a large number of missing sites). Sequences were trimmed to a conserved region of 414 amino acids where data was recorded for most of these viruses (the Gblocks alignment trimmed further by eye: http://dx.doi.org/10.6084/m9.figshare.1425431). We used the host-association categories described above, which included ambiguous states. To model amino acid evolution we used an LG substitution model with gamma distributed rate variation across sites [27] and an uncorrelated lognormal relaxed clock model of rate variation among lineages [32]. To model the evolution of the host associations we used an asymmetric transition rate matrix (allowing transitions to and from a host association to take place at different rates) and a strict clock model. We used a constant population size coalescent prior for the relative node ages and the BEAUti v1.8 default priors for all other parameters [30] (BEAUti xml http://dx.doi.org/10.6084/m9.figshare.1431922). Convergence was assessed using Tracer v1.6 [33], and a burn-in (of 30%) removed prior to the construction of a consensus tree, which included a description of ancestral host associations. High effective sample sizes were achieved for all parameters (∾200).
The maximum clade credibility tree estimated (http://dx.doi.org/10.6084/m9.figshare.1425436) for the host association reconstruction was very similar to the independently estimated maximum likelihood phylogeny (http://dx.doi.org/10.6084/m9.figshare.1425083), which made no assumptions about the appropriateness or otherwise of applying a clock model. The minor topological differences may be expected in reconstructions that differ in their assumptions about evolutionary rates. In Figure 2 we have transferred the ancestral state reconstruction from the BEAST tree to the maximum likelihood tree.
Results
Novel rhabdoviruses from RNAEseq
To search for new rhabdoviruses we collected a variety of different species of flies, screened them for CO2 sensitivity and sequenced total RNA of these flies by RNA-seq. We identified rhabdovirus-like sequences from a deEnovo assembly by BLAST, and used PCR to identify which samples these sequences came from.
This approach resulted in eleven rhabdovirus-like sequences from nine (possibly ten) species of fly. Seven of these viruses were previously unknown and four had been reported previously from shorter sequences (Tables S1 and S2). Rhabdoviruses known from other species of Drosophila typically have genomes of ∼12.5Kb [11, 34], and six of our sequences were approximately this size, suggesting they are near-complete genomes. None of the viruses discovered in our RNA-seq data appeared to be integrated into the host genome (see Methods for details).
To investigate the putative gene content of the viruses, we predicted genes based on open reading frames (ORFs). For the viruses with apparently complete genomes (Figure 1), we found that those from Drosophila ananassae, Drosophila affinis, Drosophila immigrans and Drosophila sturtvanti contained the five core genes found across all rhabdoviruses, with an additional gene between the P and M genes. This is the location of the X gene found in sigma viruses, and in three of the four viruses it showed sequence homology to the X gene of other sigma viruses. The virus from Drosophila busckii did not contain an additional ORF between the P and M genes, but instead contained an ORF between the G and L gene. Using the gene content and the phylogeny described below, we have classified our newly discovered viruses as either sigma viruses or other rhabdoviruses and named them after the host species they were isolated from (Figure 1) [35]. We also found one other novel mononegavirales-like sequence from Drosophila unispina that groups with a recently discovered clade of arthropod associated viruses (Nyamivirus clade [12], see Table S5 and the full phylogeny: http://dx.doi.org/10.6084/m9.figshare.1425083), confirming our approach can detect a wide range of divergent viruses.
[Figure 1 here]
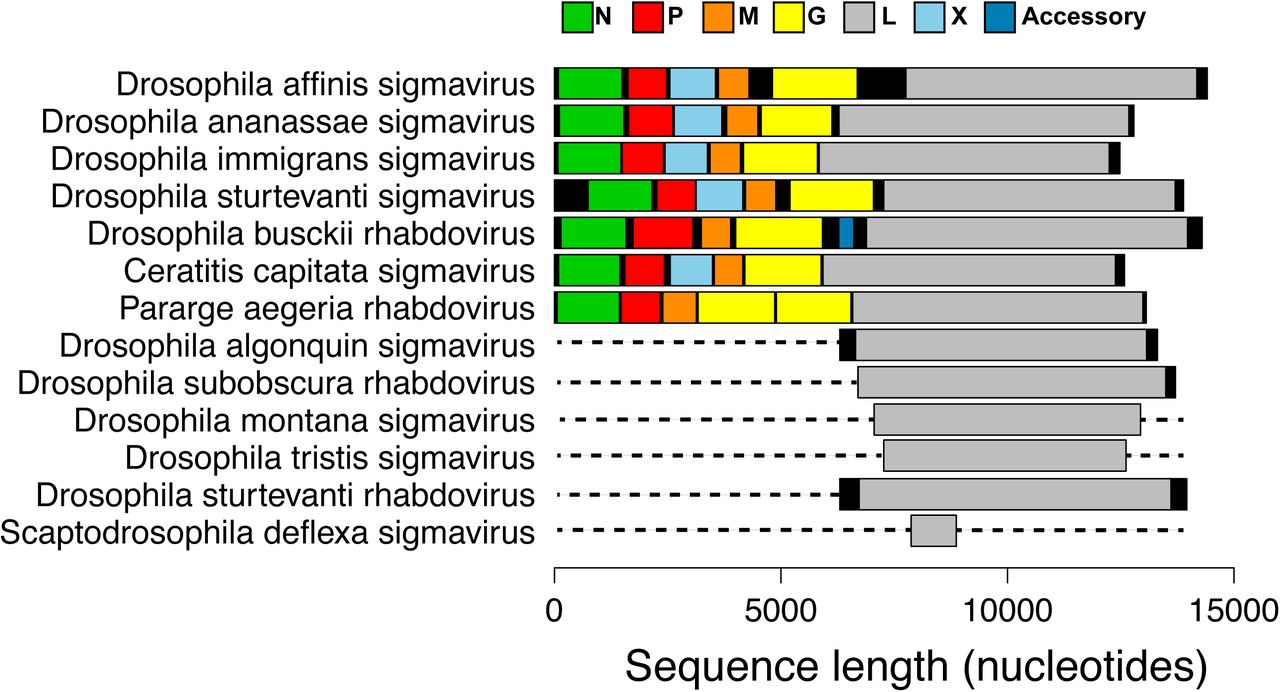
Genome organization of newly discovered viruses. Predicted ORFs are shown in colour, non-coding regions are shown in black. ORFs were designated as the first start codon following the transcription termination sequence (7 U’s) of the previous ORF to the first stop codon. Dotted lines represent parts of the genome not sequenced. These viruses were either from our own RNA-seq data, or were first found in in public databases and key features verified by PCR and Sanger sequencing. Rhabdovirus genomes are typically ∼12-13kb long and contain five core genes 3’N-P-M-G-L-5’ [3]. However, a number of groups of rhabdoviruses contain additional accessory genes [46]. Online version in colour.
New Rhabdoviruses from public databases
We identified a further 26 novel rhabdovirus-like sequences by searching public databases of assembled RNA-seq data with BLAST. These included 19 viruses from arthropods (Fleas, Crustacea, Lepidoptera, Diptera), one from a Cnidarian (Hydra) and 5 from plants (Table S3). Of these viruses, 19 had sufficient amounts of coding sequence (>1000bp) to include in the phylogenetic analysis (Table S3), whilst the remainder were too short (Table S4).
Four viruses from databases had near-complete genomes. These were from the moth Triodia sylvina, the house fly Musca domestica (99% nucleotide identity to Wuhan house fly virus 2 [12]), the butterfly Pararge aegeria and the medfly Ceratitis capitata, all of which contain the five core rhabdovirus genes. The sequence from C. capitata had an additional gene predicted between the P and M genes with sequence similarity to the X gene in sigma viruses. There were several unusual sequences. Firstly, in the virus from P. aegeria there appear to be two full-length glycoprotein genes between the M and L gene (we confirmed the stop codon between the two genes was not an error by Sanger sequencing). Secondly, the Agave tequilana transcriptome contained an L gene ORF on a contig that was the length of a typical rhabdovirus genome but did not appear to contain typical gene content, suggesting it has very atypical genome organization, has been misassembled or is integrated into its host plant genome [36]. Finally, the virus from Hydra magnipapillata contained six predicted genes, but the L gene (RDRP) was unusually long. Some of the viruses we detected may well be EVEs inserted into the host genome and subsequently expressed [17]. For example, this is likely the case for the sequence from the silkworm Bombyx mori that we also found in the silkworm genome, and the L gene sequence from Spodoptera exigua that contains stop codons. Assuming viruses integrated into the host genome once infected those hosts, this does not affect our conclusions below about the host range of these viruses [14–16]. We also found nine other novel mononegavirale-like sequences that group with recently discovered clades of insect viruses [12] (see Table S5 and http://dx.doi.org/10.6084/m9.figshare.1425083).
Rhabdovirus Phylogeny
To reconstruct the evolution of the Rhabdoviridae we have produced the most complete phylogeny of the group to date (Figure 2). We used an alignment of the relatively conserved L gene (RNA Dependant RNA Polymerase) from our newly discovered viruses with sequences of known rhabdoviruses to give an alignment of 195 rhabdoviruses (and 26 other mononegavirales as an outgroup). We reconstructed the phylogeny using different sequence alignments and methodologies, and these all gave qualitatively similar results with the same major clades being reconstructed (Gblocks: http://dx.doi.org/10.6084/m9.figshare.1425083, TrimAl: http://dx.doi.org/10.6084/m9.figshare.1425082 and BEAST: http://dx.doi.org/10.6084/m9.figshare.1425436). The branching order between the clades in the dimarhabdovirus supergroup was generally poorly supported and differed between the methods. Eight sequences that we discovered were not included in this analysis as they were considered too short, but their closest BLAST hits are listed in Table S4.
We recovered all of the major clades described previously (Figure 2), and found that the majority of known rhabdoviruses belong to the dimarhabdovirus clade (Figure 2b). The RNA-seq viruses from Drosophila fall into either the sigma virus clade (Figure 2b) or the arthropod clade sister to the cyto- and nucleo-rhabdoviruses (Figure 2a). The viruses from sequence databases are diverse, coming from almost all of the major clades with the exception of the lyssaviruses.
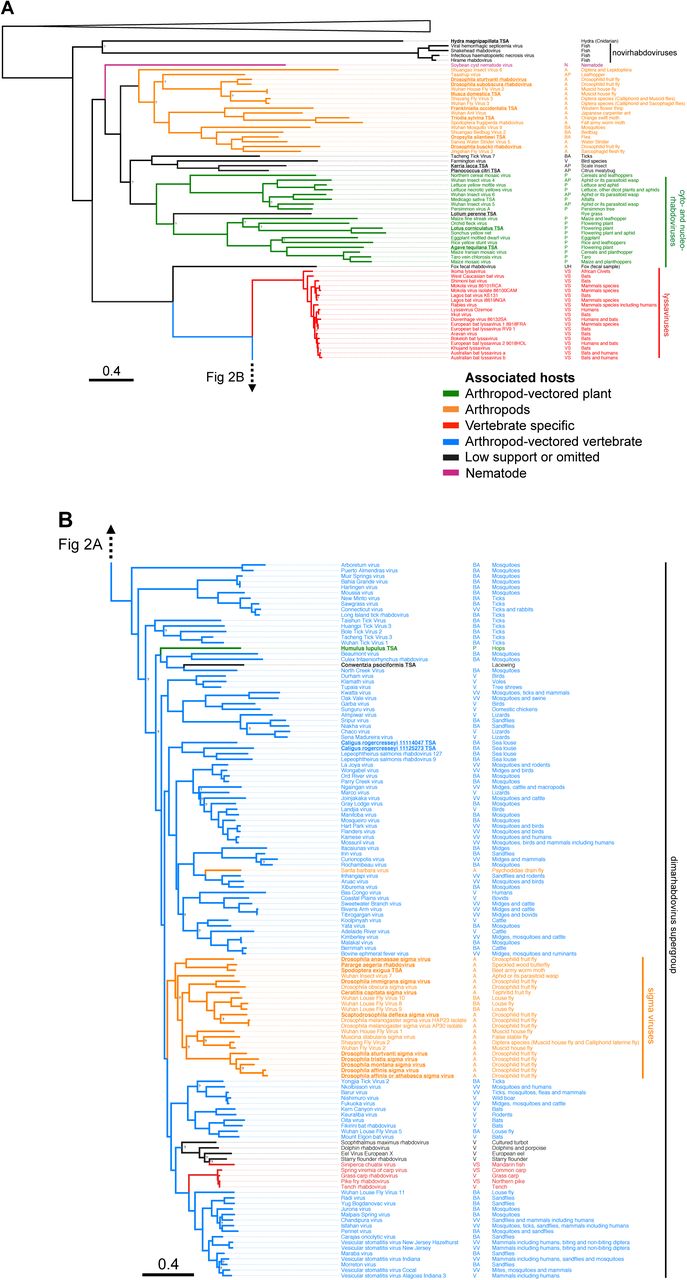

Maximum likelihood phylogeny of the Rhabdoviridae. A shows the basal fish-infecting novirhabdoviruses, an unassigned group of arthropod associated viruses, the plant infecting cyto- and nucleo-rhabdoviruses, as well as the vertebrate specific lyssaviruses. B shows the dimarhabdovirus supergroup, which is predominantly composed of arthropod-vectored vertebrate viruses, along with the arthropod specific sigma virus clade. Branches are coloured based on the Bayesian host association reconstruction analysis. Black represents taxa omitted from host-state reconstruction or associations with <0.95 support. The tree was inferred from L gene sequences using the Gblocks alignment. The columns of text are the virus name, the host category used for reconstructions, and known hosts (from left to right). Codes for the host categories are: vs= vertebrate-specific, vv= arthropod-vectored vertebrate, a= arthropod specific, ba = biting-arthropod (ambiguous state), v = vertebrate (ambiguous state) and ap=plant-sap-feeding-arthropod (ambiguous state). Names in bold and underlined are viruses discovered in this study. The tree is rooted with the Chuvirus clade (root collapsed) as identified in [12]. Nodes labelled with question marks represent nodes with aLRT (approximate likelihood ratio test) values less than 0.75. Scale bar shows number of amino-acid substitutions per site. Online version in colour.
Changes in host species
With a few exceptions, rhabdoviruses are either arthropod-vectored pathogens of plants or vertebrates, or are vertebrate- or arthropod-specific. In many cases the only information about a virus is the host from which it was isolated. Therefore, it is not clear whether viruses isolated from vertebrates are vertebrate-specific or arthropod-vectored, or whether viruses isolated from biting arthropods (e.g. mosquitoes, sandflies, ticks, midges and sea lice) are arthropod specific or also infect vertebrates. Likewise, it is not clear whether viruses isolated from sap-sucking insects (all Hemiptera: aphids, leafhoppers, scale insect and mealybugs) are arthropod-specific or arthropod-vectored plant viruses. However, in the absence of these data, we used the phylogenetic relationships of the viruses to predict both the ancestral and present host associations (http://dx.doi.org/10.6084/m9.figshare.1425436).
This approach identified a large number of viruses that are likely to be new arthropod-vectored vertebrate pathogens (Figure 2b). 87 of 92 viruses with ambiguous host associations were assigned a host association with strong posterior support (>0.95). Of the 52 viruses found in biting arthropods, 45 were predicted to be arthropod-vectored vertebrate viruses, and 6 to be arthropod-specific. Of the 33 viruses found in vertebrates, 31 were predicted to be arthropod-vectored vertebrate viruses, with none being vertebrate-specific. Of the 7 viruses found in plant-sap-feeding arthropods (Figure 2a), 3 were predicted to be plant-associated and 2 arthropod-associated.
We were also able to infer the ancestral host association of 182 of 188 of the internal nodes on the phylogenetic tree with strong support (>0.95). In addition to the switches of host-type that occur deep in the tree, there are a small number of changes on the terminal branches of the phylogeny. These could either represent errors in the host assignment (e.g. cross-species contamination), or recent host shifts.
A striking pattern that emerged from our reconstructions of host associations is that switches between major groups of hosts have occurred rarely during the evolution of the rhabdoviruses, excluding a few rare transitions on terminal branches (Figure 2). There has been one clear switch to become insect-vectored plant pathogens (cyto- and nucleo-rhabdoviruses). A single virus isolated from the hop plant Humulus lupulus sits in the dimarhabdovirus clade, but this may be because the plant was contaminated with insect matter, as the same RNA-seq dataset contains COI sequences with high similarity to thrips. A single transition to being vertebrate–specific has occurred in the lyssaviruses clade [3]. There has also been a single transition to vertebrate viruses that are vectored by arthropods in the dimarhabodovirus clade.
There are two main clades of arthropod-specific viruses. The first clade is a sister group to the large plant virus clade. This novel group of largely insect-associated viruses are associated with a broad range of insects, including flies, butterflies, moths, ants, thrips, bedbugs, fleas, mosquitoes, water striders and leafhoppers. The mode of transmission and biology of these viruses is yet to be examined. The second clade of insect associated viruses is the sigma virus clade [10, 11, 18, 34]. These are derived from vector-borne dimarhabdoviruses that have lost their vertebrate host and become vertically transmitted pathogens of insects [10]. They are common in Drosophilidae, and our results suggest that they may be widespread throughout the Diptera, with occurrences in the Tephritid fruit fly Ceratitis capitata, the stable fly Muscina stabulans, several divergent viruses in the housefly Musca domestica and louse flies removed from the skin of bats. All of the sigma viruses characterised to date have been vertically transmitted [10], but some of the recently described virus may be transmitted horizontally – it has been speculated that the viruses from louse flies may infect bats [37] and Shayang Fly Virus 2 has been reported in two fly species [12] (although contamination could also explain this result). For the first time we have found sigma-like viruses outside of the Diptera, with two Lepidoptera associated viruses and a virus from an aphid/parasitoid wasp. Drosophila sigma virus genomes are characterised by an additional X gene between the P and M genes [34]. Interestingly the two louse fly viruses with complete genomes, Wuhan insect virus 7 from an aphid/parasitoid and Pararge aegeria rhabdovirus do not have an X gene. Overall, our results suggest sigma-like viruses may be common in a wide array of insect species.
The rhabdoviruses cluster on the phylogeny not only by the hosts they infect, but also by whether they are found in terrestrial or aquatic environments. There has been one shift from terrestrial to aquatic hosts during the evolution of the basal novirhabdoviruses, which have a wide host range in fish. There have been other terrestrial to aquatic shifts in the dimarhabdoviruses: in the clade of fish and cetacean viruses and the clade of viruses isolated from sea-lice. The sea-lice viruses may be crustacean-specific as the two viruses from Lepeophtheirus salmonis do not seem to infect the fish they parasitise and are present in all developmental stages of the lice suggesting they may be transmitted vertically [38].
Discussion
Viruses are ubiquitous in nature and recent developments in high-throughput sequencing technology have led to the discovery and sequencing of a large number of novel viruses in arthropods [12, 13]. Here we have identified 43 novel virus-like sequences, from our own RNA-seq data and public sequence repositories. Of these, 32 were rhabdoviruses, and 26 of these were isolated from arthropods. Using these sequences we have produced the most extensive phylogeny of the Rhabdoviridae to date, including a total of 195 virus sequences.
In most cases we know nothing about the biology of the viruses beyond the host they were isolated from, but our analysis provides a powerful way to predict which are vector-borne pathogens and which are specific to vertebrates or arthropods. We have identified a large number of new likely vector-borne pathogens – of 85 rhabdoviruses isolated from vertebrates or biting insects we predict that 76 are arthropod-borne viruses of vertebrates (arboviruses). Along with the known arboviruses, this suggests the majority of known rhabdoviruses are arboviruses, and all of these fall in a single clade known as the dimarhabdoviruses. In addition to the arboviruses, we also identified two clades of likely insect-specific viruses associated with a wide range of species, suggesting rhabdoviruses may be common arthropod pathogens.
We found that shifts between distantly related hosts are rare in the rhabdoviruses, which is perhaps unsurprising as both rhabdoviruses of vertebrates (rabies virus in bats) and invertebrates (sigma viruses in Drosophildae) show a declining ability to infect hosts more distantly related to their natural host [39–41]. It is thought that sigma viruses may sometimes jump into distantly related but highly susceptible species [40, 42, 43], but our results suggest that this rarely happens between major groups such as vertebrates and arthropods. It is nonetheless surprising that arthropod-specific viruses have arisen rarely, as one might naively assume that there would be fewer constraints on vector-borne viruses losing one of their hosts. Within the major clades, closely related viruses often infect closely related hosts (Figure 2). For example, within the dimarhabdoviruses viruses isolated from mosquitoes, ticks, Drosophila, Muscid flies, Lepidoptera and sea-lice all tend to cluster together (Figure 2B). However, it is also clear that the virus phylogeny does not mirror the host phylogeny, suggesting that following major transitions between distantly related host taxa, viruses preferentially shift between more closely related species.
There has been a near four-fold increase in the number of rhabdovirus sequences in the last five years. In part this may be due to the falling cost of sequencing transcriptomes [44], and initiatives to sequence large numbers of insect and other arthropods [45]. The use of high-throughput sequencing technologies should reduce the likelihood of sampling biases associated with PCR based discovery where people look for similar viruses in related hosts. This suggests that the pattern of viruses forming clades based on the host taxa they infect is likely to be robust. However, these efforts are disproportionately targeted at arthropods, and it is possible that there may be a great undiscovered diversity of viruses in other organisms.
Rhabdoviruses infect a diverse assortment of host species, including a large number of arthropod species. Our limited search has unearthed a large number of novel rhabdovirus genomes, suggesting we are only just beginning to uncover the diversity of these viruses.
Data accessibility
All data has been made available in public repositories:
NCBI Sequence Read Archive Data: SRP057824
Data S1, sample information: http://dx.doi.org/10.6084/m9.figshare.1425432
Data S2, virus ID, Genbank accessions and host information: http://dx.doi.org/10.6084/m9.figshare.1425419
L gene sequences fasta: http://dx.doi.org/10.6084/m9.figshare.1425067
TrimAl alignment fasta: http://dx.doi.org/10.6084/m9.figshare.1425069
Gblocks alignment fasta: http://dx.doi.org/10.6084/m9.figshare.1425068
Phylogenetic tree Gblocks alignment: http://dx.doi.org/10.6084/m9.figshare.1425083
Phylogenetic tree TrimAl alignment: http://dx.doi.org/10.6084/m9.figshare.1425082
BEAST alignment fasta: http://dx.doi.org/10.6084/m9.figshare.1425431
BEAUti xml file: http://dx.doi.org/10.6084/m9.figshare.1431922
Bayesian analysis tree: http://dx.doi.org/10.6084/m9.figshare.1425436
Supplementary materials: Tables S1-5 List of newly discovered viruses.
Competing interests
We have no competing interests.
Contributions
BL and FMJ conceived and designed the study. BL and JD carried out molecular work. BL, WJP, DJP and DJO carried out bioinformatic analysis. BL, GGRM and JJW carried out phylogenetic analysis. BL and FMJ wrote the manuscript with comments from all other authors. All authors gave final approval for publication.
Funding
BL and FMJ are supported by a NERC grant (NE/L004232/1), a European Research Council grant (281668, DrosophilaInfection), a Junior Research Fellowship from Christ’s College, Cambridge (BL). GGRM is supported by an MRC studentship. The metagenomic sequencing of viruses from D. immigrans, D. tristis and S. deflexa was supported by a Wellcome Trust fellowship (WT085064) to DJO.
Acknowledgments
Many thanks to Mike Ritchie for providing the DMonSV infected fly line; Casper Breuker and Melanie Gibbs for PAegRV samples and Philip Leftwich for CCapSV samples. Thanks to everyone who provided fly collections.





{kind=link}
{kind=link}
{kind=link}