

Abstract
Approximately 2-4% of the human genome is in non-Africans comprised of DNA intro-gressed from Neanderthals. Recent studies have shown that there is a paucity of introgressed DNA around functional regions, presumably caused by selection after introgression. This observation has been suggested to be a possible consequence of the accumulation of a large amount of Dobzhansky-Muller incompatibilities, i.e. epistatic effects between human and Neanderthal specific mutations, since the divergence of humans and Neanderthals approx. 400-600 kya. However, using previously published estimates of inbreeding in Neanderthals, and of the distribution of fitness effects from human protein coding genes, we show that the average Neanderthal would have had at least 40% lower fitness than the average human due to higher levels of inbreeding and an increased mutational load, regardless of the dominance coefficients of new mutations. Using simulations, we show that under the assumption of additive dominance effects, early Neanderthal/human hybrids would have experienced strong negative selection, though not so strong that it would prevent Neanderthal DNA from entering the human population. In fact, the increased mutational load in Neanderthals predicts the observed reduction in Neanderthal introgressed segments around protein coding genes, without any need to invoke epistasis. The simulations also predict that there is a residual Neanderthal derived mutational load in non-African humans, leading to an average fitness reduction of at least 0.5%. Although there has been much previous debate about the effects of the out-of-Africa bottleneck on mutational loads in non-Africans, the significant deleterious effects of Neanderthal introgression have hitherto been left out of this discussion, but might be just as important for understanding fitness differences among human populations. We also show that if deleterious mutations are recessive, the Neanderthal admixture fraction would gradually increase over time due to selection for Neanderthal haplotypes that mask human deleterious mutations in the heterozygous state. This effect of dominance heterosis might partially explain why adaptive introgression appears to be widespread in nature.
1 Introduction
In recent years, prodigious technological advances have enabled extraction of DNA from the remains of our extinct Neanderthal relatives [1]. This ancient DNA has revealed that Neanderthals had lower genetic diversity than any living human population [2, 3]. By analyzing patterns of divergence between distinct Neanderthal haplotypes, Prüfer et al. inferred that Neanderthals experienced a long population bottleneck, maintaining an effective population size around Ne = 1,000 during the 400,000 years before their extinction [2]. This Neanderthal bottleneck lasted approximately ten times longer than the out-of-Africa bottleneck that reduced diversity in modern humans who migrated out of Africa [4, 5, 6].
A classical consequence of population bottlenecks is that they interfere with natural selection by increasing evolutionary stochasticity [7, 8]. When effective population size is small and genetic drift is therefore strong, weakly deleterious alleles have a tendency to persist in the population as if they were neutral. Neanderthal exome sequencing has confirmed this prediction, providing direct evidence that purifying selection was weaker in Neanderthals than in humans [3, 9]. Compared to humans, Neanderthals have a relatively high ratio of nonsynonymous (NS) to synonymous (S) variation within proteins, indicating that they probably accumulated deleterious NS variation at a faster rate than humans do.
It is an open question whether archaic hominins’ deleterious mutation load contributed to their decline and extinction. However, there is clear evidence that Neanderthals escaped total genetic extinction by interbreeding with the anatomically modern humans who left Africa between 50 and 100 thousand years ago [1]. In Europeans and Asians, haplotypes of Neanderthal origin have been inferred to comprise 2–4% of each individual’s genome. When pooled together, these Neanderthal haplotypes collectively span about 30% of the human reference sequence [10, 11].
The introgression of Neanderthal alleles related to hair, skin pigmentation, and immunity appear to have provided non-Africans with adaptive benefits, perhaps because Neanderthals had preadapted to life in Europe for thousands of years before humans left Africa [10, 11, 12, 13]. However, these positively selected genes represent a tiny fraction of Neanderthal introgression’s genetic legacy. A larger number of Neanderthal alleles appear to have deleterious fitness effects, with putative links to various diseases as measured by genome-wide association studies [10].
The distribution of deleterious mutations in humans has been the subject of much recent research. A controversial question is whether the out-of-Africa bottleneck created differences in genetic load between modern human populations [14, 15]. Some previous studies concluded that this bottleneck saddled non-Africans with potentially damaging genetic variants that could affect disease incidence across the globe today [16, 17], while other studies have concluded that there is little difference in genetic load between Africans and non-Africans [18, 9]. Although previous studies have devoted considerable attention to simulating the accumulation of deleterious mutations during the out-of-Africa bottleneck, none to our knowledge have incorporated the fitness effects of introgression from Neanderthals into non-Africans.
In this paper, we quantify the deleterious effects on humans of introgression with Neanderthals with a high mutational load. We present simulations showing that archaic introgression may have had fitness effects comparable to the out-of-Africa bottleneck, saddling non-Africans with weakly deleterious alleles that accumulated as nearly neutral variants in Neanderthals.
2 Results
To estimate the fitness effects of Neanderthal introgression on a genome-wide scale, we used forward-time simulations incorporating linkage, exome architecture, and population size changes to model the flux of deleterious mutations across hominin species boundaries. We describe three main consequences of this flux, which are not mutually exclusive and whose relative magnitudes depend on evolutionary parameters such as the distribution of dominance coefficients and fitness effects of new mutations. One consequence is strong selection against early human/Neanderthal hybrids, implying that the initial contribution of Neanderthals to the human gene pool may have been much higher than the contribution that persists today. A second consequence is depletion of Neanderthal ancestry from conserved regions of the genome, a pattern that has been previously inferred from genetic data [10, 11] and interpreted as evidence for partial reproductive incompatibilities between humans and Neanderthals. A third consequence is the persistence of deleterious alleles in present-day humans, creating a difference in mutation load between non-Africans (who experienced Neanderthal admixture) and Africans who did not.
2.1 The Reduced Fitness of Neanderthals
Our first step toward quantifying these three consequences of introgression was to estimate preadmixture mutation loads in humans and Neanderthals. We accomplished this using simulations where all humans and Neanderthals experience deleterious mutations drawn from the same distribution of fitness effects (DFE), so that any differences in mutation load are driven by differences in demographic history. Because the fitness effects of noncoding mutations are difficult to measure, we restricted our attention to deleterious mutations that alter protein coding sequences (nonsyn-onymous or NS mutations). There have been several estimates of the distribution of sleection coefficients in human protein coding genes [19, 20, 21, 22]. We here use the estimates of Eyre-Walker, et al. who found that the DFE of human NS mutations is gamma-distributed with shape parameter 0.23 and mean selection coefficient −0.043 [23]. Although it is probably unrealistic to neglect the fitness effects of synonymous and non-exonic mutations, it is also conservative in that additional deleterious mutations would only increase the human/Neanderthal load difference beyond the levels estimated here.
Using the UCSC map of exons from the hg19 reference genome, we assume that each exon accumulates NS mutations at a rate of 7.0 × 10−9 per site per generation, with fitness effects sampled from the distribution estimated by Eyre-Walker et al. No deleterious mutations occur between exons, but recombination does occur at a rate of 1.0 × 10−8 crossovers per site per generation. We implemented this genetic architecture within the simulation program SLiM [24] by using the recombination map feature built into the simulator. Specifically, for each pair of adjacent exons separated by a gap of b base pairs, we represent this gap as a single base pair with recombination rate b × 10−8 per generation. Similarly, each boundary between two chromosomes is encoded as a single base pair with a recombination rate of 0.5 crossovers per generation. We chose to focus on the dynamics of the 22 autosomes, neglecting the more complex evolutionary dynamics of the X and Y chromosomes.
We allowed the mutation spectrum of this exome to equilibrate in the ancestral human/ Neanderthal population by simulating an ancestral population of size 10,000 for 44,000 generations. After this mutation accumulation period, the ancestral population splits into a human population of size 10,000 plus a Neanderthal population of size 1,000. The Humans and Neanderthals then evolve in isolation from each other for 16,000 more generations (a divergence time of 400,000–470,000 years assuming a generation time between 25 and 29 years). To a first approximation, this is the history inferred by Prüfer, et al. from the Altai Neanderthal genome using the Pairwise Sequentially Markov Coalescent [2].
We made two different sets of simulations that differed in their assumptions regarding dominance coefficients of de novo mutations: one with fully additive effects and one with fully recessive effects. We expect that the true distribution of dominance effects falls somewhere in between these two extreme models. Throughout, we assume log-additive interactions among loci. In other words, the fitness of each simulated individual can be obtained by adding up the selection coefficients at all sites to obtain a sum S and calculating the fitness to be exp(–S). The fitness of individual A relative to individual B is the ratio of their two fitnesses.
In the simulation with additive fitness effects, the median Neanderthal was found to have fitness 0.63 compared to the median human (Figure 1A). Assuming recessive fitness effects, the excess load accumulated by Neanderthals was even greater, with a median Neanderthal fitness of 0.39 compared to the median human (Figure 1B). In each case, the fitness differential was caused by accumulation of weakly deleterious mutations with selection coefficients ranging from 5 × 10−5 (nearly neutral in the larger human population) to 2 × 10−3 (nearly neutral in the smaller Neanderthal population). This agrees with asymptotic predictions that mutations with 2Ns > 1 are not affected by a bottleneck with minimum population size N [25]. To illustrate, we divided selection coefficient space into several disjoint intervals and measured how each interval contributed to the fitness reduction in Neanderthals. For each interval of selection coefficients  , and each individual Gv, we calculated the mutation load summed across derived alleles with selection coefficients between
, and each individual Gv, we calculated the mutation load summed across derived alleles with selection coefficients between  and
and  to obtain a load value
to obtain a load value  . Given that the median human has load
. Given that the median human has load  , the fitness reduction due to Si-mutations in individual Gv is
, the fitness reduction due to Si-mutations in individual Gv is  . Figures 1C and D show the distribution of this fitness reduction, which is more variable between individuals for strong-effect mutations than for weak-effect mutations.
. Figures 1C and D show the distribution of this fitness reduction, which is more variable between individuals for strong-effect mutations than for weak-effect mutations.
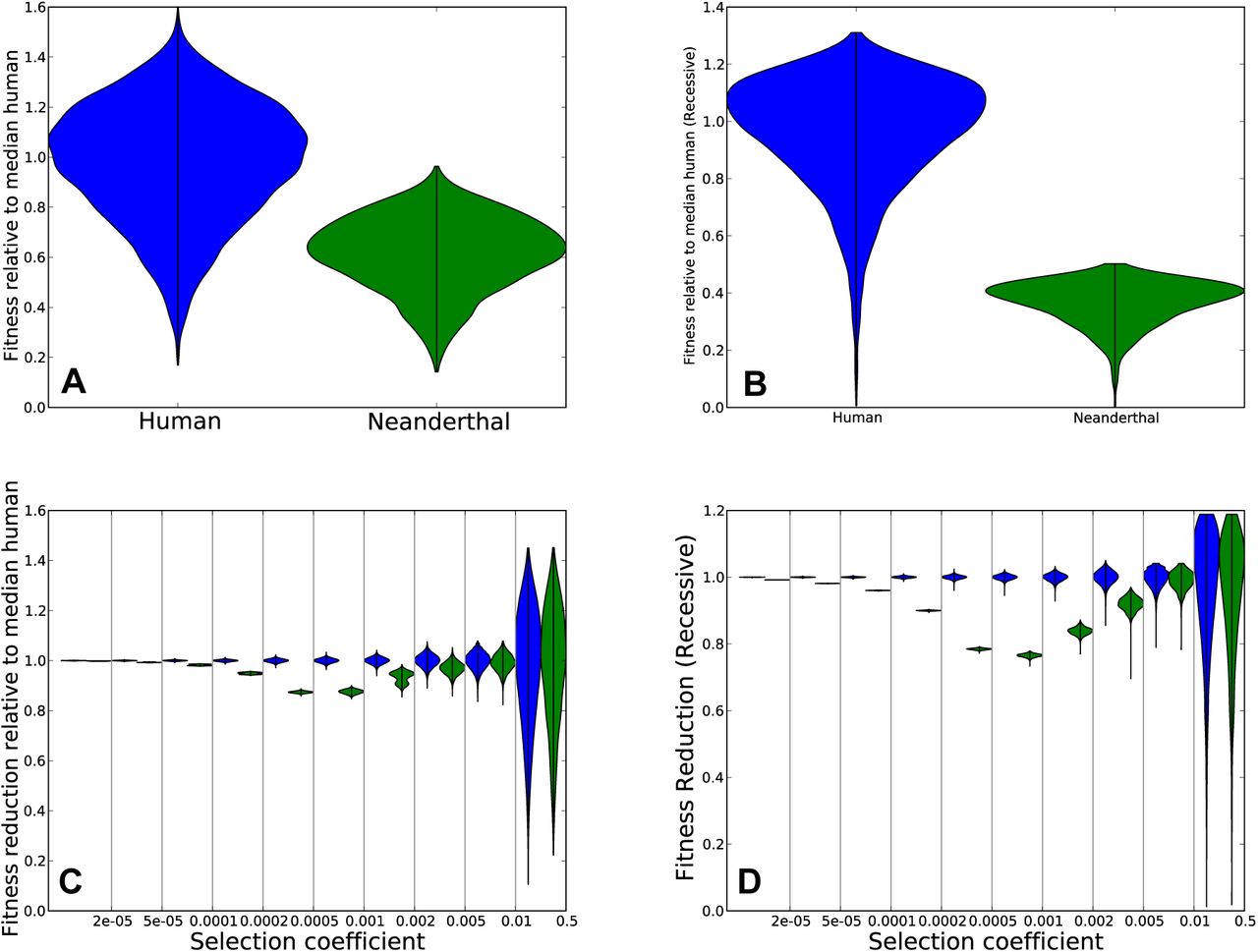
Panel A shows the distribution of fitness in Neanderthals versus non-admixed humans, assuming that their effective population sizes differed ten-fold since their divergence 16,000 generations ago, and assuming additive mutation effects. After simulating the two populations using SLiM, we calculated each individual’s fitness relative to the median human. The violin plots of the two distributions show significant variance within each population but lower fitness in Neanderthals. Panel B shows the same but for a model of recessive mutations. Panels C and D show the same data as in A and B, respectively, but stratified into different bins of selection coefficients. The fitness reduction due to mutations with s between 2 × 10−4 and 5 × 10−4, is much greater in Neanderthals than in humans. In contrast, the fitness reduction due to very weak effect mutations s < 2 × 10−5 is similar between the two populations, as is the fitness reduction due to strongly deleterious mutations with s > 0.005. This is expected as mutations outside this range are either effectively neutral in both populations or strongly deleterious in both populations.
Under an additive model, a recessive model, or anything in between, the severe reduction in fitness of Neanderthals would have doomed them to quick extinction if they had been competing for the same niche with humans under conditions of reproductive isolation.
2.2 Recessive Mutations Lead to Positive Selection for Neanderthal DNA
We model Neanderthal gene flow as a discrete event associated with an admixture fraction f, sampling Nf Neanderthals and N(1 – f) humans from the gene pools summarized in Figure 1, and then allowing this admixed population to mate randomly for 2,000 additional generations. A Neanderthal gene flow date of 2,000 generations before the present is compatible with Fu, et al.’s claim that the admixture occurred 52,000–58,000 years ago [26], assuming a human generation time between 26 and 29 years. To simulate the out-of-Africa bottleneck, which affected humans around the time of admixture, we used a model based on the history inferred by Gravel, et al. from the site frequency spectrum of the 1000 Genomes data [5]. At the time of admixture (2,000 generations ago), the non-African population size drops from N = 10, 000 to N = 1, 861. 900 generations later, the size is further reduced to N = 1,032 and begins exponentially growing at rate 0.38% per generation. We discretized this exponential growth such that the population size increases in a stepwise fashion every 100 generations (Figure 2). Because forward-time simulations involving large numbers of individuals are very time and memory intensive, we also capped the population size at N = 20, 000 (the size that is achieved 300 generations before the present). Although recent, explosive population growth may have increased the abundance of young deleterious mutations in the human population [17, 27, 28], it probably had minimal effects on the trajectory of older alleles introduced by Neanderthal admixture.

This cartoon shows the demographic history used to simulate Neanderthal and human genomes. A single ancestral population of size 10,000 is simulated for 44,000 generations to let the distribution of deleterious mutations reach equilibrium. At this point, a Neanderthal population of size 1,000 splits off. After 16,000 generations of isolation, Neanderthals and humans admix, followed by the out of Africa bottleneck and piecewise-constant exponential growth.
In the recessive-effects case, we found that the Neanderthal admixture fraction increased over time at a logarithmic rate (Figure 3A). To quantify this change in admixture fraction, we added neutral marker mutations (one every 105 base pairs) to the initial admixed population that were fixed in Neanderthals and absent from humans (Figure 3B). The average allele frequency of these markers started out equal to the admixture fraction f, but was observed to increase monotonically. An initial admixture fraction of 1% was found to be consistent with a present-day admixture fraction around 3%, with most of the increase occurring over the first 500 generations. The selection favoring Neanderthal alleles is an example of dominance heterosis [29, 30, 31, 32].

A. This plot depicts Neanderthal admixture over 2,000 generations in a simulation where all mutations are recessive. The initial admixture fraction is only 1%, but rises to 3% genomewide due to selection for Neanderthal haplotypes that protect against human recessive mutations. B. This cartoon illustrates how the Neanderthal admixture fraction is estimated as a function of time from the simulation output. Fixed neutral marker mutations (one every 105 base pairs) are introduced into the Neanderthal population before admixture, making it possible to measure changes in their average frequency over time in response to selection on linked deleterious mutations.
Before admixture, most deleterious alleles are private to either humans or Neanderthals, leading introgressed Neanderthal alleles to be hidden from purifying selection when they are introduced at low frequency. Because Neanderthal haplotypes rarely have deleterious alleles at the same sites that human haplotypes do, they are protective against deleterious human variation, despite the fact that they have a much higher recessive burden than human haplotypes. Several studies have pinpointed archaic genes that appear to be under positive selection in humans [10, 12, 33, 34] because they confer resistance to pathogens or are otherwise strongly favored. Examples of recent adaptive introgression also abound in both animals and plants [35, 36, 37, 38]. Although many introgressed alleles confer clear adaptive benefits, we note that some positive selection on foreign DNA may have less to do with adaptation than with heterosis.
2.3 Additive Fitness Effects Lead to Strong Selection Against Early Hybrids
If most deleterious mutations have additive fitness effects instead of being recessive, different predictions emerge. The reduced fitness of Neanderthals is not hidden, but imposes selection against hybrids in the human population. Such selection against negative deleterious mutations could potentially be offset by positive selection or by associative overdominance due to linked recessive mutations. In the absence of these effects, however, we found that an initial admixture fraction of 10% Neanderthals was necessary to observe a realistic value of 2.5% Neanderthal ancestry after 2,000 generations. Most of the selection against Neanderthal ancestry occurred within the first 20 generations after admixture, at which point the average frequency of the Neanderthal markers had already declined below 3% (Figure 4). During the first 20 generations the variance in admixture fraction between individuals is relatively high, permitting efficient selection against the individuals who have more Neanderthal ancestry than others. However, once all individuals have nearly the same admixture fraction but have retained Neanderthal DNA at different genomic locations, Hill-Robertson interference slows down the purging of foreign deleterious alleles [39, 40, 41]. This suggests that introgression of Neanderthal DNA into humans would have been possible without positive selection, despite the high mutational load, but would require a large initial admixture fraction, perhaps close to 10%.

This plot depicts the mean and variance of the Neanderthal ancestry fraction in an admixed population where fitness effects are additive, starting with 10% Neanderthals in generation 1. During the first 10–20 generations after admixture, both the mean and variance in Neanderthal ancestry decrease quickly due to selection against individuals whose Neanderthal ancestry fraction is higher than the population average. After 20 generations, however, all individuals have nearly the same amount of Neanderthal ancestry and selection against its deleterious component becomes less efficient.
2.4 Persistence of Deleterious Neanderthal Alleles in Modern Humans
Figures 1 and 4 illustrate two predictions about Neanderthal introgression: first, that it probably introduced many weakly deleterious alleles, and second, that a large fraction of deleterious alleles with additive effects were probably eliminated within a few generations. However, it is not clear from these figures how many deleterious Neanderthal alleles are expected to persist in the present day human gene pool. To address this question, we simulated a control human population experiencing additive mutations that has undergone the out of Africa bottleneck without also experiencing Neanderthal introgression.
At a series of time points between 0 and 2000 generations post-admixture, we recorded each individual’s total load of weakly deleterious mutations (s < 0.0005) as well as the total load of strongly deleterious mutations (s > 0.0005). The three quartiles of the fitness reduction due to weakly deleterious mutations are plotted in Figure 5A, while the three quartiles of the strongly deleterious fitness reduction are plotted in Figure 5B. Neither the out of Africa bottleneck nor Neanderthal admixture has much effect on the strong load. However, both the bottleneck and admixture exert separate effects on the weak load, each decreasing fitness on the order of 1%.
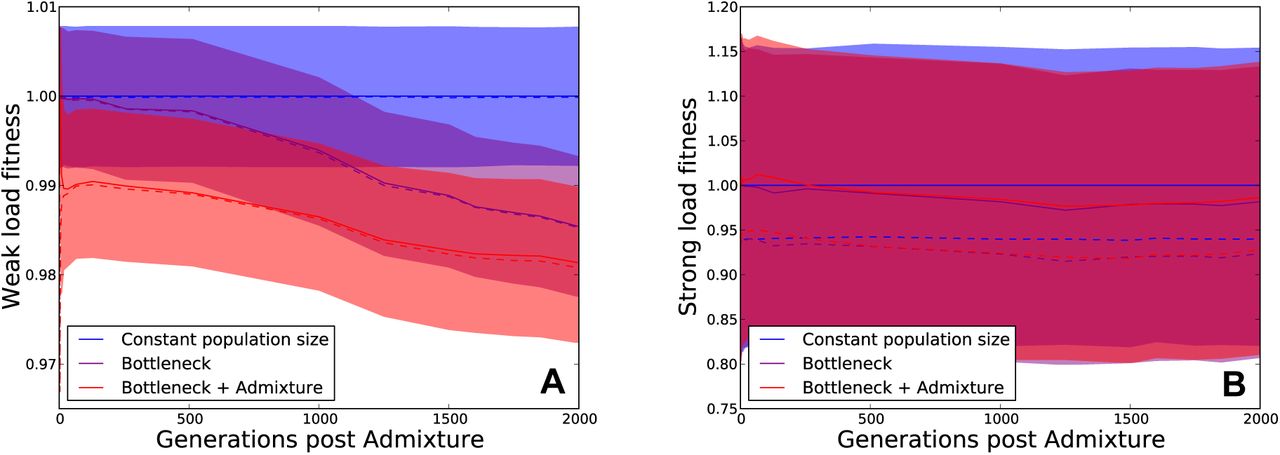
These plots show the mutation load in three simulated human populations, one with constant population size, one that experiences only the out of Africa bottleneck and a third that experiences the bottleneck along with Neanderthal admixture (see Figure 2), assuming additive fitness effects. We partition each individual’s mutation load into two components: the weak load due to mutations with selection coefficient less than 0.0005, and the strong load due to mutations with selection coefficient greater than 0.0005. At time t, each individual’s weak-load fitness and strong-load fitness are normalized relative to the median individual in the constant-size population. The dotted lines show the median in each respective population, while solid lines shiw the mean and the shaded area encompasses the 25th through 75th percentiles. Panel A shows that the admixed population suffers the greatest fitness reduction due to weak mutations, even 2,000 generations after admixture. Panel B shows that neither the bottleneck nor admixture affects the strong load.
The excess weak load attributable to Neanderthal admixture is much smaller than the variance of strong mutation load that we observe within populations, which is probably why the excess Neanderthal load decreases in magnitude so slowly over time. However, the two load components have very different genetic architectures–the strong load consists of rare variants with large fitness effects, whereas the weak load is enriched for common variants with weak effects. Although surviving Neanderthal alleles are unlikely to affect the risks of Mendelian diseases with severe effects, they may have disproportionately large effects on polygenic traits that influence fitness.
2.5 Depletion of Neanderthal Ancestry near Genes can be Explained without Reproductive Incompatibilities
Two empirical studies of Neanderthal-human sequence similarity found that Neanderthal ancestry appears to be depleted from conserved regions of the genome [10, 11]. In particular, Sankararaman, et al. found that the Neanderthal ancestry fraction appears to be negatively correlated with the B statistic, a measure of the strength of background selection as a function of genomic position. In the quintile of the genome that experiences the strongest background selection, they observed a median Neanderthal ancestry fraction around 0.5%, while in the quintile that experiences the weakest background selection, they calculated a median admixture fraction around 2%. This has been interpreted as evidence for epistatic reproductive incompatibilities between humans and Neanderthals [10, 11].
In light of the strong selection against Neanderthal DNA we have predicted on the basis of demography, we posit that reproductive incompatibilities are not required to explain much of the Neanderthal ancestry depletion observed near conserved regions of the genome. Conserved regions are regions where mutations have a high probability of being deleterious and thus being eliminated by natural selection; these are the regions where excess weakly deleterious mutations are most likely to accumulate in Neanderthals. This suggests that selection will act to reduce Neanderthal ancestry in conserved regions even if each allele has the same fitness in both populations.
To model depletion of Neanderthal ancestry in the neighborhood of conserved DNA, we simulated a set of “Neanderthal” genomes that were fixed for 1,000 evenly spaced weakly deleterious mutations (one mutation per 3 megabases), introgressing these genomes into a “human” population containing no deleterious variants. For simplicity, all deleterious mutations were given the same selection coefficient s = 5 × 10−4, such that the fitness of a Neanderthal relative to a human is exp(−600 · 5 × 10−4) ≈ 0.74, similar to the fitness difference obtained from our mutation accumulation simulations.
Assuming that Neanderthals are fixed for a deleterious variant that is absent from humans before introgression, it is straightforward to calculate the expected admixture fraction as a function of time at a neutral locus linked to the deleterious one. Given a neutral allele a located L base pairs away from a deleterious allele of selection coefficient s, the frequency fa of allele a is expected to decrease every generation until the deleterious allele recombines onto a neutral genetic background. Letting r denote the recombination rate per site per generation and fa (T) denote the frequency of allele a at time T, then

If the deleterious allele is not fixed in Neanderthals before introgression, but instead has Neanderthal frequency fN and human frequency fH, the expected admixture fraction after T generations is instead

This can be viewed as a case of associative overdominance as described by Ohta, where linked deleterious alleles reduce the expected frequency of a neutral allele down to a threshold frequency that is determined by the recombination distance between the two loci [42].
The simulation scenario is more complex than this two-locus admixture scenario because all deleterious and neutral loci are linked for the first few generations until long Neanderthal haplotypes are broken up by recombination. However, essentially zero 3-megabase haplotypes are expected to survive through 2,000 generations of recombination, meaning that most neutral mutations will spend the majority of the time linked to only one deleterious variant. Figure 6 compares the 2-locus expectation to the simulated admixture fraction as a function of distance to the nearest deleterious variant. In each scenario, the 20% of the genome that lies closest to a deleterious Neanderthal variant ends up with a mean admixture fraction of only 1%, compared to 3% in the 20% of the genome that lies farthest from a deleterious Neanderthal variant. This is reminiscent of the Sankararaman, et al.’s observation that the median admixture fraction in the genome with the lowest B statistic quintile is only a quarter of the median admixture fraction inferred within the highest B statistic quintile.

Each colored curve plots the average fraction of Neanderthal ancestry after N generations of admixture (N = 20, 800, or 2,000) as a function of distance from an introgressed deleterious mutation, and assuming an additive model of dominance effects. After 2,000 generations, the approximate elapsed time since Neanderthals interbred with humans, the average Neanderthal admixture fraction is only 1% in the 20% of the genome that lies closest to a deleterious variant, compared to 3% averaged over the 20% of the genome that is farthest from the nearest such deleterious variant. This figure approximately recapitulates the results of Sankararaman, et al. who estimated a negative correlation between admixture fraction and the conservation-indicative B statistic. Black dotted lines show that the decay rate of admixture fraction with distance is correctly predicted by Equation (1).
3 Discussion
Our simulations show that an increased additive mutational load due to low population size is sufficient to explain the paucity of Neanderthal admixture observed around protein coding genes in modern humans. However, our results do not preclude the existence of Dobzhansky-Muller incompatibilities between Neanderthals and humans. Other lines of evidence hinting at such incompatibilities lie beyond the scope of this study. One such line of evidence is the existence of Neanderthal ancestry “deserts” where the admixture fraction appears near zero over stretches of several megabases. Another is the depletion of Neanderthal ancestry near testis-expressed genes [10, 11] and recent chromosomal rearrangements [43]. However, these patterns could be explained by a relatively small number of negative epistatic interactions between human and Neanderthal alleles, as only 10–20 deserts of Neanderthal ancestry have been identified.
Depletion of Neanderthal DNA from the X chromosome has also been cited as evidence for reproductive incompatibilities, perhaps in the form of male sterility [10]. However, we note that the X chromosome may have experienced more selection due to its hemizygous inheritance in males that exposes recessive deleterious mutations. We have shown that selection against the first few generations of hybrids is determined by the load of additive (or hemizygous) mutations, and that the strength of this initial selection determines how much Neanderthal DNA remains long-term. This implies that the admixture fraction on the X chromosome should be lower than on the autosomes if some deleterious mutations are recessive, even in the absence of recessive incompatibility loci that are thought to accumulate on the X according to Haldane’s rule [44, 45, 46].
A model assuming that the general pattern of selection is caused by epistatic effects would involve hundreds or thousands of subtle incompatibilities in order to explain the genome-wide negative correlation of Neanderthal ancestry with background selection. Given the relatively recent divergence between humans and Neanderthals and the abundant evidence for their admixture, it seems unlikely that this divergence could have been given rise to hundreds of incompatible variants distributed throughout the genome. In contrast, our results show that it is highly plausible for the buildup of weakly deleterious alleles to reduce the fitness of hybrid offspring, causing background selection to negatively correlate with admixture fraction.
The distribution of dominance effects in humans is not well characterized. But it is likely that introgressed Neanderthal DNA has been subject to a selective tug-of-war, with selection favoring Neanderthal DNA in regions where humans carry recessive deleterious mutations and selection disfavoring Neanderthal alleles that have additive or dominant effects. In a sense, this is the opposite of the tug-of-war that may occur when a beneficial allele is linked to recessive deleterious alleles that impede the haplotype from sweeping to high frequencies [47, 48].
If most mutations have additive dominance effects and multiplicative effects across loci, initial admixture would have to have been as high as 10% to explain the amount of admixture observed today. In contrast, if most mutations are recessive, an initial admixture fraction closer to 1% appears most plausible. We did not model selection for new beneficial mutations here, and it is possible that such selection might also have helped facilitate introgression, particularly in the first generations where selection against hybrids would otherwise have been strong. As more paleolithic human DNA is sequenced, it may become possible to measure how admixture has changed over time and extract information from this time series about the distribution of dominance coefficients. This information could also help resolve confusion about the fitness effects of the out of Africa bottleneck, which is predicted to have differently affected the burdens of additive versus recessive variants [15, 25].
We do not claim to have precisely estimated the deleterious Neanderthal load that remains in non-Africans today, as this would require better estimates of the DFE across different genes and more exploration of the effects of assumptions regarding recent demographic history. However, our results suggest that Neanderthal admixture should be incorporated into models exploring mutational load in humans to more accurately predict the mutation load difference between Africans and non-Africans. Association methods have already revealed correlations between Neanderthal alleles and several human diseases [10]. Our results on mutations with addititve dominance effects suggest that introgression reduced non-African fitness about as much as the out-of-Africa bottleneck did.
Introgression of recessive mutations is predicted to affect fitness in a more complex way. Some adaptive benefits will result from Neanderthal and human haplotypes masking one another’s deleterious alleles, but Hill-Robertson interference may also hurt fitness as overdominant selection at recessive sites drags linked dominant Neanderthal alleles to higher frequency. In addition, Neanderthal haplotypes are predicted to have worse recessive burdens than human ones if they become homozygous due to selection or inbreeding.
Our results have implications for conservation biology as well as for human evolution, as they apply to any case of secondary contact between species with different effective population sizes. When an outbred population experiences gene flow from a more inbred population, we predict an increase in genetic entropy where deleterious alleles spill rapidly into the outbred population and then take a long time to be purged away by selection. This process could magnify the effects of outbreeding depression caused by genetic incompatibilities [49, 50, 51] and acts inversely to the genetic rescue process, in which individuals from an outbred population are artificially transplanted into a threatened population that has been suffering from inbreeding depression [52, 53, 54]. These results suggest that care should be taken to prevent two-way gene flow when genetic rescue is being attempted to prevent lasting damage to the fitness of the outbred population.
4 Acknowledgements
We thank Joshua Schraiber for manuscript comments and members of the Nielsen and Slatkin labs for helpful discussions. K.H. received support from a Ruth L. Kirschstein National Research Service Award from the National Institutes of Health (award number F32GM116381). K.H. and R.N. also received support from NIH Grant IR01GM109454-01 to R.N., Yun S. Song, and Steven N. Evans. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- [1].↵
- [2].↵
- [3].↵
- [4].↵
- [5].↵
- [6].↵
- [7].↵
- [8].↵
- [9].↵
- [10].↵
- [11].↵
- [12].↵
- [13].↵
- [14].↵
- [15].↵
- [16].↵
- [17].↵
- [18].↵
- [19].↵
- [20].↵
- [21].↵
- [22].↵
- [23].↵
- [24].↵
- [25].↵
- [26].↵
- [27].↵
- [28].↵
- [29].↵
- [30].↵
- [31].↵
- [32].↵
- [33].↵
- [34].↵
- [35].↵
- [36].↵
- [37].↵
- [38].↵
- [39].↵
- [40].↵
- [41].↵
- [42].↵
- [43].↵
- [44].↵
- [45].↵
- [46].↵
- [47].↵
- [48].↵
- [49].↵
- [50].↵
- [51].↵
- [52].↵
- [53].↵
- [54].↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}