

Introduction
Recent efforts aggregating the genomes and exomes of tens of thousands of individuals have provided unprecedented insights into the landscape of rare human genetic variation1,2 and generated critical resources for clinical and population genetics. The recently announced U.S. Precision Medicine Initiative raises the prospect of growing these databases to encompass hundreds of thousands of human genomes. In the context of these ambitious efforts, it is important to quantify the power of large sequencing projects to discover rare functional genetic variants3. In particular, we need to understand, as we sequence ever larger cohorts of individuals, how many new variants we can expect to identify and their expected allele frequencies. Accurate estimates of these quantities will enable better study design and quantitative evaluation of the potential and limitations of these datasets for precision medicine.
Results
Predicting the number of new variants we expect to identify in larger cohorts requires accurate estimates of allele frequencies of all the genetic variation in the human population, including the rare variants that have not been observed in the current sequencing cohorts4-6. The population frequencies of the unobserved rare variants determine the discovery rate of new variants as the cohort sizes increase. We developed a new method, UnseenEst, to estimate the frequency distribution of all variants using the observed site frequency spectrum (SFS) of the current cohort. The method is based on linear program estimators of the SFS7, and our mathematical analysis shows that it enables accurate extrapolation of the SFS from current data to cohort sizes more than an order of magnitude larger (Supplementary Information).
Protein-coding variants represent the most readily interpretable and medically relevant slice of human genetic variation, and have been assessed in large sample sizes through the widespread application of exome sequencing approaches2. We leveraged data from the Exome Aggregation Consortium (ExAC)8 to estimate the discovery rates of different classes of protein coding variants in larger cohorts. We validated UnseenEst by training it on random 10% of the alleles in ExAC and then used the estimated frequency distribution to predict the number of distinct variants that we can identify in the entire ExAC cohort. For every variant type (Supp. Figure 1) and every population (Supp. Figure 2), UnseenEst accurately predicted the number of unique variants that were identified in the entire ExAC cohort as well as the empirical SFS of ExAC (Supp. Table 1).
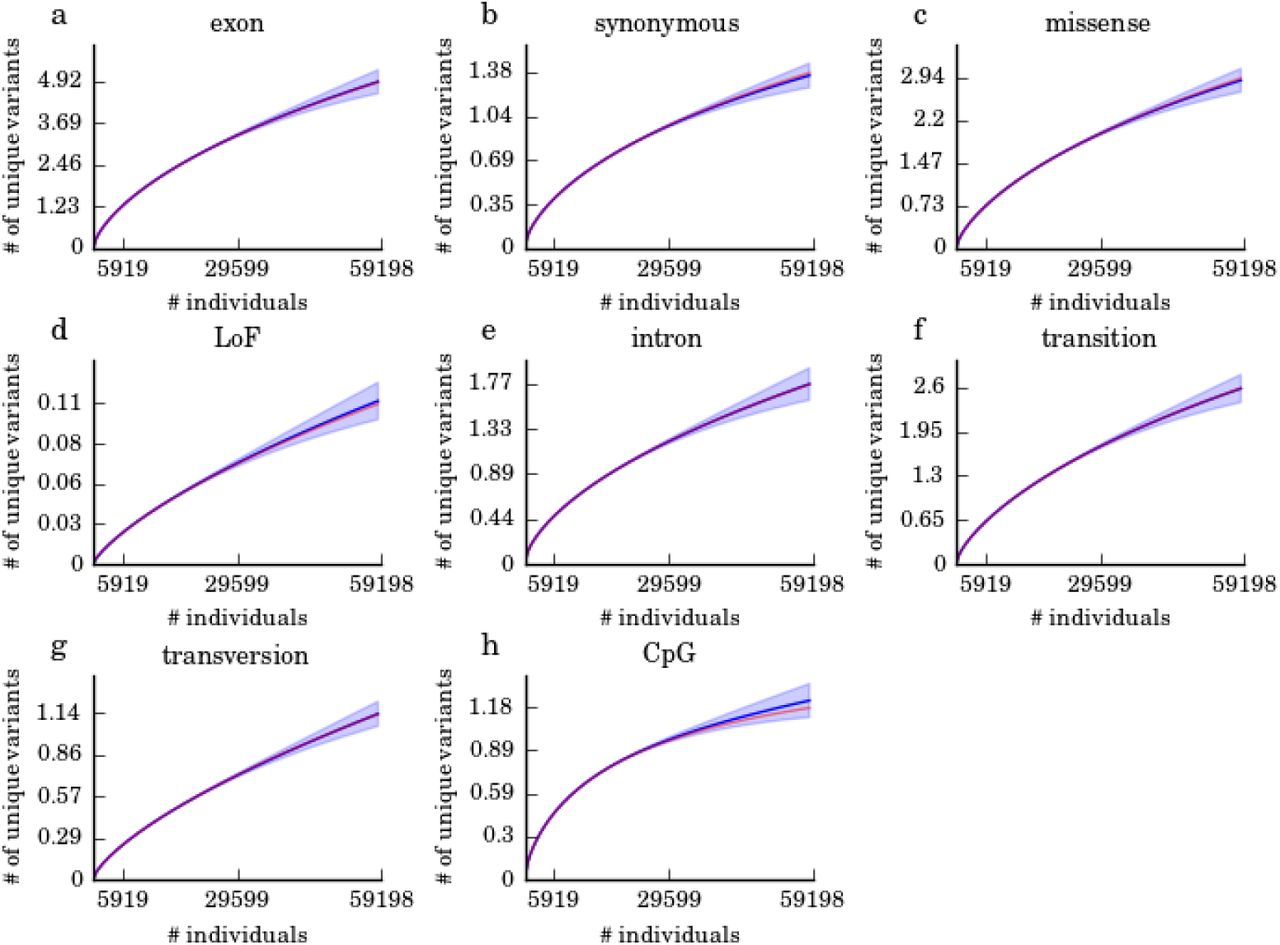
Each panel corresponds to one variant type. For each variant type, we applied UnseenEst on 10% of the ExAC alleles (5919 individuals) to predict the number of unique variants that we would expect to observe in a cohort of size less than or equal to ExAC (59198 individuals). The blue curves are the average predictions over the different 10% sub-samples and the blue shaded regions correspond to one standard deviation from the average. The red curves are the actual number of unique variants observed in ExAC. For all variant types, the predicted number of unique variants is in good agreement with the observed number of unique variants.

For each of the ExAC populations, we trained UnseenEst on random 10% of the alleles and applied it to predict the total number of unique variants in the entire population. The x-axis of each panel indicate the number of individuals of that population; the first mark (e.g. 5919 in (a)) indicate the size of the training set and the last mark (e.g. 59198 in (a)) is the total cohort size of that population in ExAC. The blue curves are the average predictions over the different 10% sub-samples and the blue shaded regions correspond to one standard deviation from the average. The red curves are the actual number of unique variants observed in ExAC. For all variant types, the predicted number of unique variants is in good agreement with the observed number of unique variants.
Blue rows are the number of ExAC variants with empirical allele counts in bins of 0-10, 10-100, 100-1000, and greater than 1000. Red rows are the predicted allele counts based on UnseenEst trained on 10% of the samples. The standard deviations are shown in the parentheses.

We trained UnseenEst on the U.S. Census-matched ExAC cohort (“current”) and predicted the number of unique variants we expect to find in up to 500K individuals. The number of unique variants in the cohort were estimated for synonymous, missense and lose-of-function (LoF) variants in (a), and for CpGs, transitions and transversions in (b). The shaded regions correspond to one standard deviation around the estimates. (c) A gene is classified as LoF on a given allele if that allele contains at least one variant that introduces a stop codon, disrupts a splice donor/receptor site, or disrupts the reading frame. Genes are partitioned into bins based on their LoF allele frequencies: less than 10−5, 10−5 to 10−4, 10−4 to 10−3, and greater than 10−3. The y-axis indicates the number of genes with LoF allele frequency belonging to each bin. Error bars correspond to one standard deviation. (d) Estimated number of genes with at least 10 and 20 LoF alleles.
From the full ExAC dataset, we generated a cohort of 33778 healthy individuals that matched the ancestral population breakdown of the 2010 U.S. Census (Supp. Table 2). We trained UnseenEst on this U.S. Census-matched cohort and predicted the frequency distributions of variants in the entire population (Supp. Figure 3). In particular, we estimated the number of distinct variants we expect to identify in cohorts of up to 500K individuals. These results provide a quantitative framework to evaluate the power and limitations of precision medicine initiatives in discovering rare coding variants.

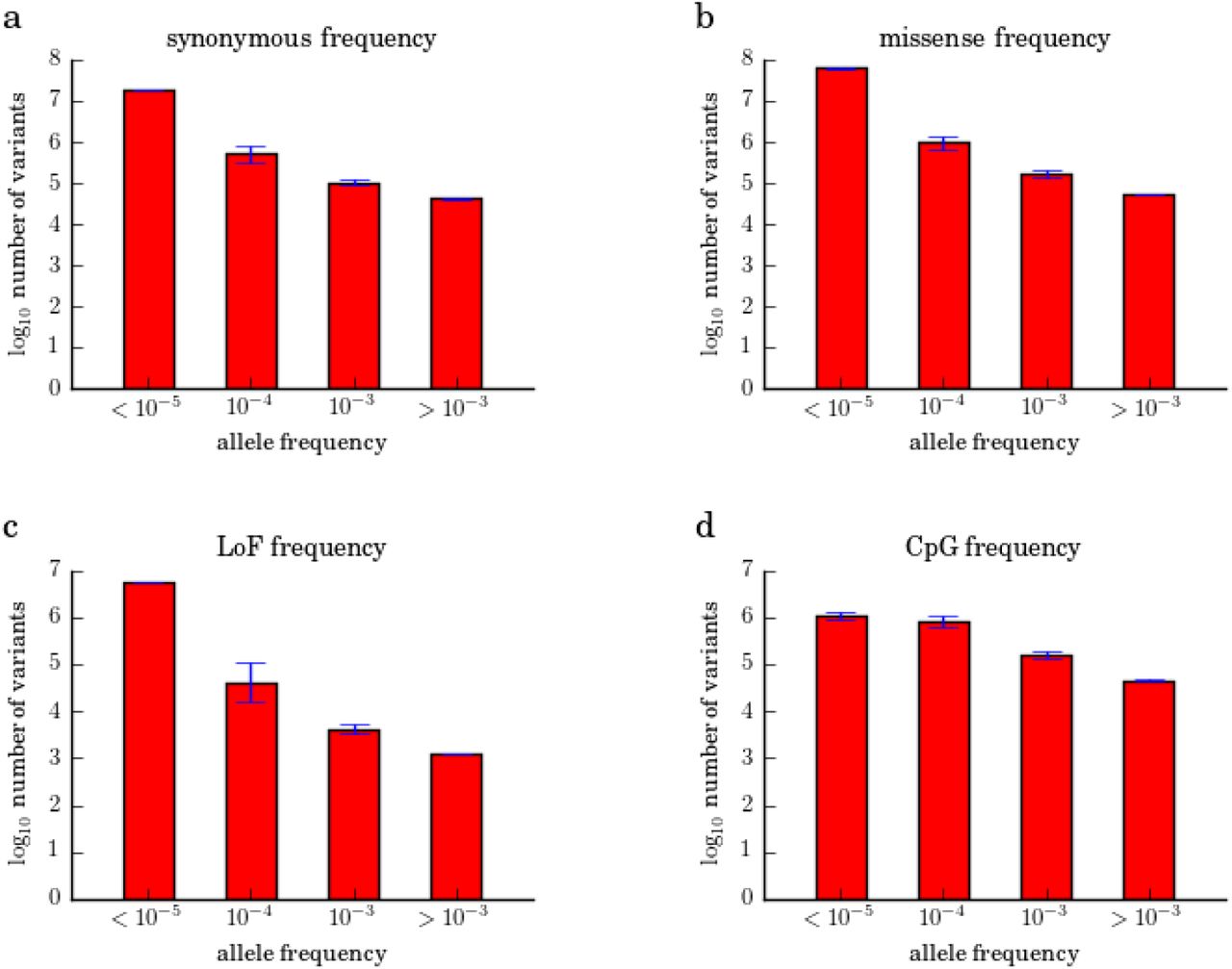
UnseenEst was trained on the U.S. Census matched ExAC cohort and the synonymous (a), missense (b), LoF (c) and CpG (d) allele frequencies were estimated for the US population. The variants are grouped into bins based on allele frequency: less than 10−5, 10−5 to 10−4, 10−4 to 10−3, and greater than 10−3. The y-axes indicate the log10 number of variants in each bin. The error bars correspond to one standard deviation.
The top row shows the number of individuals of each ancestry in the 2010 U.S. Census. The middle row shows the ancestry composition of the ExAC cohort. The bottom row shows the number of individuals of each ancestry in the ExAC cohort that was adjusted to match the 2010 U.S. Census.
We categorized the variants by their predicted functional consequence—synonymous, missense, and loss-of-function (LoF), which is defined as point substitutions that introduce stop codons or disrupt splice donor/acceptor sites (Figure 1a). The discovery rate of LoF variants is the lowest, reflecting the fact that LoFs are likely to be deleterious and hence tend to occur comparatively rarely in the healthy population. With 500K individuals, we expect to identify 400K distinct LoF variants or 7.5% of all possible LoF point mutations in the human exome. In the same cohort, we expect to identify 3.4 million synonymous and 7.5 million missense variants, corresponding to 18% and 12% of possible synonymous and missense variants respectively. These estimates indicate that the discovery rates of rare LoF, missense and synonymous variants are far from saturation, even with 500K individuals. We note that slightly higher numbers of distinct synonymous and missense variants (Supp. Figure 4) would be discovered if the 500K individuals were instead sampled from the same ancestral composition as the current ExAC cohort, which contains higher fractions of South and East Asian individuals than the U.S., indicating that the overall discovery rate of rare variants can be boosted by optimizing the population composition of the sequencing cohort.

The x-axis indicates the number of individuals in the cohort and the y-axis indicates the fraction of possible variants that we expect to observe at in a cohort of that size. We trained UnseenEst on the full ExAC dataset and made the predictions for synonymous (grey), missense (orange) and loss-of-function (brown) variants.
We additionally classified the variants by their biochemical properties (Figure 1b). With the 34K individuals of the current cohort, we can already identify close to 50% of all possible variants at CpG sites (the most highly mutable substitution class), and the discovery rate for this class of variant quickly saturates as cohorts grow larger. Transversions, in contrast, are discovered much more slowly—attaining 7.6% of all possible transversions with 500K individuals—which is consistent with their much lower mutation rate. We further applied UnseenEst to quantify the number of distinct missense variants we expect to discover in specific gene families of interest, for example genes near GWAS hits and known drug target genes (Supp. Figure 5). Missense mutations in drug target genes are particularly suppressed, suggesting that these genes are more likely to be essential to humans.

We trained the model on the cohort that matches U.S. demographics and predicted the fraction of possible missense variants in each gene family that we can expect to observe in cohorts of size up to 500K individuals. (a) Recessive genes (red) and dominant genes (blue). (b) All genes (red) and genes with cerebral specific expression (blue). (c) Genes associated with GWAS loci (red) and drug target genes (blue).
LoF variants likely disrupt the normal function of genes and by studying individuals carrying such variants, we can quantify the phenotypic consequence of disrupting particular genes. Therefore, a catalogue of the number of human alleles harboring candidate LoF variants for each gene is an important resource for drug development and disease diagnosis. We applied UnseenEst to estimate the LoF frequency of genes in the U.S. population (Figure 1c, Supp. Figure 6). About 2900 genes have LoF allele frequency lower than 10−5, consistent with strong intolerance to inactivation, whereas 1700 genes are expected to harbor LoF variants in at least 0.1% of the population. With 250K individuals, we expect to identify 14K genes that harbor LoFs in at least 10 individuals, substantially expanding the current catalog of 10K such genes in ExAC (Figure 1d, Supp. Figure 7). We estimate that the discovery rate of these genes with multiple LoF occurrences will saturate around 16K, providing an upper bound on the number of genes that can tolerate LoF variants on one allele.

We trained UnseenEst on random subsamples of 10% of the alleles in the U.S. Census matched cohort and applied it to estimate the number of genes with at least 10 LoF alleles in the entire cohort. The red curve is the actual number of genes with at least 10 LoF alleles and the blue curve is the average predictions over the different subsamples. The shaded blue region corresponds to one standard deviation of the predictions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
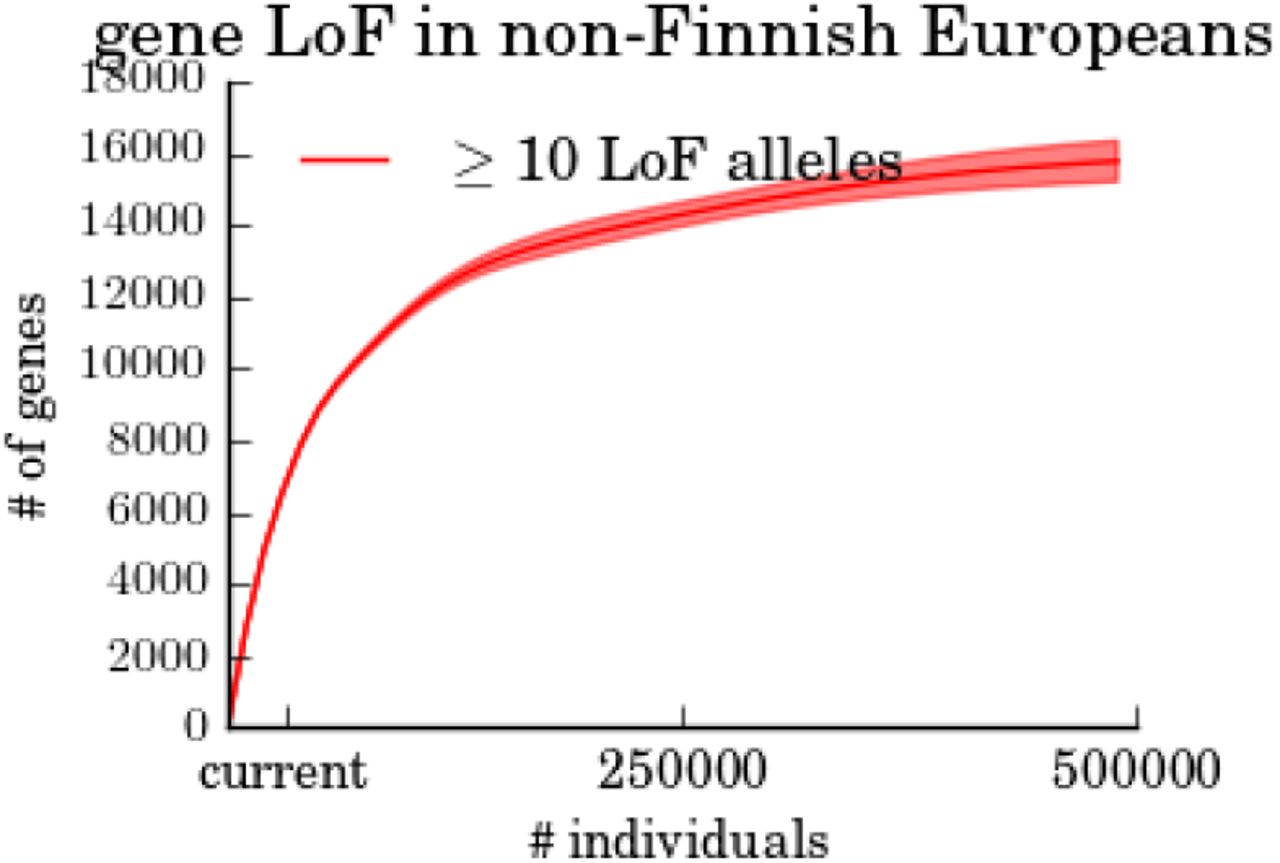
Estimated number of genes with at least 10 LoF alleles in non-Finnish Europeans as a function of the sample size. The number of genes with at least 10 LoF alleles saturates around 16K genes, in agreement with the saturation level of LoF genes in the U.S. Census-matched population (Figure 1d).
Discussion
We describe a framework for estimating the power of sequencing cohorts to discover protein-coding variants. We apply it to the largest available collection of sequenced individuals to estimate the discovery power of much larger cohorts such as the ones proposed by the Precision Medicine Initiative. While our predictions here assumed that the samples are representative of the U.S. demography, UnseenEst can be directly applied to estimate the discovery rate of cohorts with different ancestral composition. Our results show that sequencing a cohort of 500K randomly selected U.S. individuals would provide access to over 12% of all possible missense variants and 7.5% of all possible LoF variants, thereby permitting exploration of a substantial fraction of human biological diversity.
References
References




