

Abstract
Background Tardigrades are meiofaunal ecdysozoans that may be key to understanding the origins of Arthropoda. Many species of Tardigrada can survive extreme conditions through adoption of a cryptobiotic state. A recent high profile paper suggested that the genome of a model tardigrade, Hypsibius dujardini, has been shaped by unprecedented levels of horizontal gene transfer (HGT) encompassing 17% of protein coding genes, and speculated that this was likely formative in the evolution of stress resistance. We tested these findings using an independently sequenced and assembled genome of H. dujardini, derived from the same original culture isolate.
Results Whole-organism sampling of meiofaunal species will perforce include gut and surface microbiotal contamination, and our raw data contained bacterial and algal sequences. Careful filtering generated a cleaned H. dujardini genome assembly, validated and annotated with GSSs, ESTs and RNA-Seq data, with superior assembly metrics compared to the published, HGT-rich assembly. A small amount of additional microbial contamination likely remains in our 135 Mb assembly. Our assembly length fits well with multiple empirical measurements of H. dujardini genome size, and is 120 Mb shorter than the HGT-rich version. Among 23,021 protein coding gene predictions we found 216 genes (0.9%) with similarity to prokaryotes, 196 of which were expressed, suggestive of HGT. We also identified ~400 genes (<2%) that could be HGT from other non-metazoan eukaryotes. Cross-comparison of the assemblies, using raw read and RNA-Seq data, confirmed that the overwhelming majority of the putative HGT candidates in the previous genome were predicted from scaffolds at very low coverage and were not transcribed. Crucially much of the natural contamination in both projects was non-overlapping, confirming it as foreign to the shared target animal genome.
Conclusions We find no support for massive horizontal gene transfer into the genome of H. dujardini. Many of the bacterial sequences in the previously published genome were not present in our raw reads. In construction of our assembly we removed most, but still not all, contamination with approaches derived from metagenomics, which we show are very appropriate for meiofaunal species. We conclude that HGT into H. dujardini accounts for 1–2% of genes and that the proposal that 17% of tardigrade genes originate from HGT events is an artefact of undetected contamination.
Background
Tardigrades are a rather neglected phylum of endearing, microscopic animals, also known as waterbears or moss piglets [1]. They are members of the superphylum Ecdysozoa [2], and moult during both pre-adult and adult growth. They are part of the Panarthropoda, and current thinking places them as a sister phylum to Onychophora (velvet worms) and Arthropoda [3, 4]. They, like onychophorans, have lobopod limbs, with all species having four pairs. There are about 800 described species of tardigrade [1], though many more are likely to be as yet undescribed [5]. All are small (tardigrades are usually classified in the meiofauna) and are found in sediments and on vegetation from the Antarctic to the Arctic, from mountain ranges to the deep sea, and in salt and fresh water. Their dispersal in terrestrial habitats may be associated with the ability of many (but not all) species to enter environmentally resistant stasis, where the tardigrade can lose almost all body water, and thus resist extremes of temperature, pressure and desiccation [6–9], including deep space vacuum [10] and irradiation [11]. Research interests in tardigrades include their utility as environmental and biogeographic marker taxa, the insight their cryptobiotic mechanisms may yield for biotechnology, and exploration of their development compared to other Ecdysozoa, especially the well-studied Nematoda and Arthropoda.
Hypsibius dujardini (Doyère, 1840) is a limnetic tardigrade that is an emerging model for evolutionary developmental biology [4, 12–21]. It is easily cultured in the laboratory, is largely see-through (aiding analyses of development and anatomy; Figure 1), and has a rapid life cycle. H. dujardini is a parthenogen, and so is intractable for traditional genetic analysis, though reverse-genetic approaches are being developed [17]. We, and others, have been using H. dujardini as a genomic study system, revealing the pattern of ecdysozoan phylogeny [3, 4] and the evolution of small RNA pathways [22]. H. dujardini is not known to be cryptobiotic, but serves as a useful comparator for tardigrades that have evolved this fascinating physiology [9].

A A whole, dorsal view of a living H. dujardini adult, taken under differential interference contrast microscopy. The head, with two eyes, is to the top right. The green colouration in the centre is algal food in the gut. Within the body, numerous coleomocytes and strands corresponding to the unicellular muscles (see D) can be seen. Six of the eight legs are under the body
B, C Identification of the species in the Sciento culture was confirmed as H. dujardini by comparing the morphology of the doubleclaws on the legs (B) and of the pharyngeal armature (the stylets and placoids) to the standard systematic key [68] (C).
D H. dujardini has readily accessible internal anatomy. In this fluorescence micrograph, the animal has been stained with rhodamine-phalloidin to label the actin bundles, especially in the muscles. The arrangement of these muscles can be followed through the three dimensional animal, mapping central and distal attachment points. The bright component to the left is the triradial myoepithelial pharynx. (This image is one of a stacked confocal series).
E, F DIC and matching fluorescence confocal image of a H. dujardini stained with bis-benzimide (Hoechst 3342) and biodipy ceramide. The bis-benzimide (blue) labels nuclei, while the biodipy ceramide labels lipid membranes and particularly membranes of neural cells. This ventral view shows the paired ventral nerve cords that link the four segmental ganglia. Each leg has a focus of nuclei associated with gland cells. (This image is one of a stacked confocal series).
The scale bar in F is 40 micrometres.
A recent high profile study based on de novo genome sequencing came to the startling conclusion that 17% of genes in H. dujardini arose by horizontal gene transfer (HGT) from non-metazoan taxa [13]. This is twice as high as predictions for the previous most extensive animal HGT example, the asexual bdelloid rotifers Adineta vaga [23] and Adineta ricciae [24]. In bdelloid rotifers it has been suggested that HGT was instrumental in allowing the continued adaptation and survival of an asexual lineage over large evolutionary timescales [23–27]. HGT can potentially bring to a recipient genome an array of new biochemical capacities, and contrasts with gradualist evolution of endogenous genes to new function. Surveys of published genomes have revealed many cases of HGT [28], including several where HGT brought important new functions to the host. For example, tylenchomorph plant parasitic nematodes deploy a suite of plant cell wall degrading enzymes and other effectors acquired from bacterial and fungal sources [29–31]. The reported H. dujardini HGT gene set involved functions associated with stress resistance and it was suggested that anhydrobiosis itself might be part of the mechanism that permitted high levels of HGT [13].
However, claims of functional HGT must be carefully backed by several lines of evidence [32–35]. Animal genomes can accrete horizontally transferred DNA from a range of sources, especially symbionts that travel with the germline [34], but the majority of these transfers are non-functional, with the DNA fragments “dead-on-arrival” and subsequently evolving neutrally. The common nuclear insertions of mitochondrial genes are one example of dead- on-arrival HGT, but other examples from a range of bacteria, especially Wolbachia, are well established [34, 36, 37]. While it is possible that noncoding HGT fragments may affect host genome regulation, examples of functional noncoding HGT are largely lacking [34]. Biological incorporation of a bacterial gene into an animal genome requires a series of adaptations to the new transcriptional environment [32, 35]. An array of evidence is required to support claims of functional HGT, including linkage to other, known host-genome-resident genes, ecological or phylogenetic perdurance (presence in all, or many individuals of a species, and presence in related taxa), phylogenetic proof of foreignness, acquisition of spliceosomal introns, acclimatisation to host genome base composition and codon usage biases, and evidence of active transcription (for example in mRNA sequencing data) [32]. Phylogenetic evidence of “foreignness” does not in itself constitute strong evidence of HGT, as contaminant sequences will, obviously, map to the clade-of-origin of the genome from which they are derived.
Genomic sequencing of small target organisms that are not grown axenically differs from projects focused on larger species (where careful dissection can yield contamination-free, single-species samples), or from species where fully axenic samples (such as cell cultures) are available. For small organisms it is necessary to pool many individuals, and thus also pool their associated microbiota. This microbiota will include gut as well as adherent and infectious organisms. Adult H. dujardini have only ~103 cells, and thus only a very small mass of bacteria is required to yield equivalent representation of bacterial genomes in raw sequencing data. Contaminants can negatively affect assembly in a number of ways, mainly because they generate scaffolds that do not derive from the target genome, which subseuently compromise downstream analyses. Because the contaminants are unlikely to be at the same stoichiometry as the target genome, assemblers that try to optimise assembly by tracing paths through a De Bruijn graph based on expected coverage may perform suboptimally [38]. Contamination can also result in chimaeric contigs with contaminant and target genes in apparent physical linkage. Cleaned datasets result in better assemblies (as judged by numerical scores such as N50 length) [39, 40], but care must be taken not to accidentally eliminate real target genome data (for example HGT fragments).
Given the potential challenge to accepted notions of the integrity and phylogenetic independence of animal genomes, the suggestion that 17% of the genes of H. dujardini are the products of HGT [13] requires strong experimental support. Here we present detailed analyses of the evidence presented [13], including comparison to an independently generated assembly we had generated from the same original cultivar using approaches designed for low-complexity metagenomic and meiofaunal genome projects [39, 40]. We find no evidence for massive horizontal gene transfer into the genome of H. dujardini.
This is the second version of this preprint, and includes new analyses made possible by the release of additional raw data.
Results and Discussion
Assembly of the genome of H. dujardini
Using propidium iodide flow cytometry, we estimated the genome of H. dujardini to be ~110 Mb, similar to a previously published estimate [20]. Other tardigrade genomes have been estimated at 40 Mb to 800 Mb (http://www.genomesize.com/) [41].
Despite careful cleaning of animals before extraction, genomic DNA samples prepared for sequencing of H. dujardini were contaminated with other taxa, mainly bacteria and algal food. Our initial, non-optimised assembly of our (trimmed and adapter cleaned) raw short read data (nHd.1.0) spanned 185.8 Mb, significantly larger than expected. We used taxon-annotated GC-coverage plots (TAGC plots or blobplots) to screen the nHd.1.0 assembly for contaminants [39, 40]. We identified at least five distinct sets (“blobs”) of likely contaminant data, derived from a variety of Bacteria (Figure 2). These blobs had very different coverage and/or GC% to the main blob that had sequence similarity with tardigrade ESTs and GSSs and arthropod proteins. Contigs with bacterial, non-eukaryote identification, variant GC% and different coverage were selected, and reads mapping to these flagged for removal. We screened potential contaminant contigs for mitigating evidence and conservatively retained any that had eukaryote-like sequences, and any read pairs that had conflicting assignments. Several large contigs spanning ~25 Mbp were removed because they matched known Bacteroidetes sequences with high identity across their entire length. There was minimal contamination with C. reinhardtii, the food source, and this was removed using the C. reinhardtii reference genome [42]. Further rounds of assembly and blobplot analyses revealed a small number of additional contaminant contigs, generated from previously unassembled reads or taxonomically unannotated contigs. This is not unusual [40]. These were also removed. We further removed contigs and scaffolds below 500 bp. The resultant assembly, nHd.2.3, is likely to still contain contaminant data (see below) but was coherent with respect to coverage, GC% and taxonomic identity of best BLAST matches. Importantly, more RNA-Seq data mapped to the cleaned assembly than did to the raw assembly nHd.1.0 (92.8% versus 92.6%). Similarly the mapping of assembled transcripts [12] was similar between nHd.1.0 and nHd.2.3 (92.1% vs 91.2% and 93.8% vs 93.6% for the two transcriptome assemblies). We conclude that we have not over-cleaned the assembly.
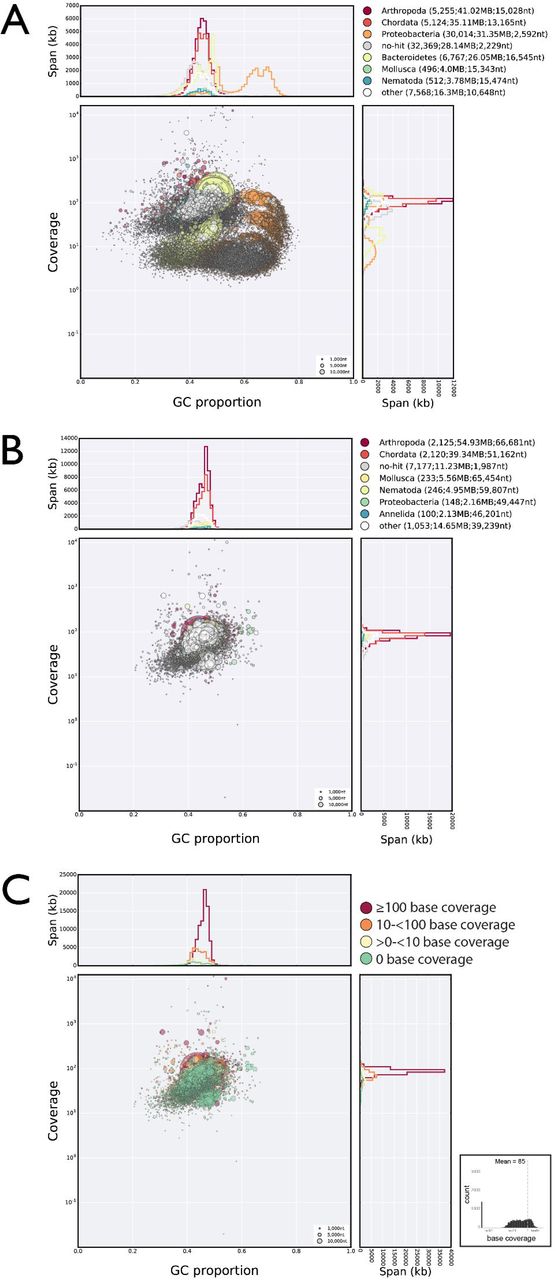
A Blobplot of the initial assembly of the trimmed short insert raw read data, identifying significant contamination with a variety of bacterial genomes. All short insert raw reads were mapped back to this assembly. The panel in the top right is a key to the colours used for each phylum, and the number of contigs or scaffolds assigned to that phylum, the span of these contigs or scaffolds, and the N50 length of the contigs or scaffolds.
B Blobplot of the nHd.2.3 assembly derived from the third cycle of read cleaning. All short insert raw reads were mapped back to this assembly. Remaining scaffolds that have sequence similarities to Bacteria rather than Metazoa have very similar coverage to the H. dujardini scaffolds.
C Blobplot of the nHd.2.3 assembly, with scaffold points plotted as in B but coloured by average base coverage from mapping of RNA-Seq data. The histogram at the bottom right shows the frequency of scaffolds at different average RNAseq base coverages.
High resolution versions of each panel are available in Supplemental Information.
The current interim assembly (version nHd.2.3) had a span of 135 Mb, with N50 length 50.5 kb (Table 1). The assembly had good representation of core conserved eukaryotic genes (CEGMA [43]; 97.2% partial and 88.7% complete, with a duplication rate of 1.3—1.5) and of our ESTs (96% mapped) and GSSs (97% mapped). The majority (>91%) of the RNA-Seq and assembled transcriptome data mapped to the genome. Unmapped RNA-Seq and transcriptome data are likely to derive from intron-spanning reads and fragments rather than erroneously removed genome sequence. We produced a high-confidence, Augustus-predicted set of 23,021 proteins. The number of genes may be inflated because of fragmentation of our assembly. For example, only 20,370 of the proteins were predicted to have a start methionine. Many of the 2,651 proteins lacking methionine were on short scaffolds, are themselves short, and may be either fragments or mispredictions.
Hypsibius dujardini assembly comparison
The assembly of the H. dujardini genome was not a simple task, and the nHd.2.3 assembly is likely to still contain contamination despite our best data filtering efforts. We identified 327 scaffolds spanning 5.0 Mb where the sum of best BLAST or diamond blastp matches across all the genes on the scaffold suggested attribution to bacterial source genomes (Supplemental file 1). Some of these scaffolds also encode eukaryote-like genes, and may represent HGT events. One hundred and ninety five scaffolds identified as bacterial (spanning 1.4 Mb) had only bacterial or no genes and were likely unfiltered contamination. No scaffolds with matches to bacterial 12S or 16S rRNAs were identified. We identified three scaffolds in the nHd.2.3 assembly that contained two instances each of the H. dujardini small subunit (SSU) and large subunit (LSU) rRNAs (one scaffold contained both). We also identified an 11 kb scaffold that had best matches to SSU and LSU from bodonid kinetoplastid protozoa. We identified two additional small scaffolds (6 kb and 1 kb) that encoded kinetoplastid genes (a retrotransposon and histone H2A, respectively). The presence of these scaffolds in the high-coverage portion of the assembly likely resulted from the multicopy nature of the encoded loci and the remainder of any bodonid genome is likely to have not been assembled because of low coverage. Below we describe the use of additional raw sequence data to identify remaining contaminants.
The genome was made openly available to browse and download (including raw data, filtered subsets thereof, and intermediate analysis files) on a dedicated BADGER genome exploration environment [44] server at http://www.tardigrades.org (Figure 3). We have currently simply flagged likely contaminant contigs (Supplemental file 2) in the BADGER genome explorer. Our assembly and annotation data were released publicly on this server in April 2014.

The Hypsibius dujardini genome has been loaded into a dedicated BADGER genome exploration environment at http://www.tardigrades.org. The explorer will be updated as new analyses are performed.
An alternative genome assembly of H. dujardini is proposed to contain 17% HGT
Boothby et al. [13] published their estimate of the genome of H. dujardini, based on a subculture of the same Sciento culture we sampled for nHd.2.3, in November 2015. We were surprised by three headline claims made for H. dujardini based on this assembly: that the genome was 252 Mb in span, that the tardigrade had 39,532 protein coding genes and that over 17% of these genes (6,663) had been derived from “massive” horizontal gene transfer into the H. dujardini genome from a range of prokaryotic and microbial eukaryotic sources. We reviewed these striking differences between the Boothby et al. genome (called the University of North Carolina, or UNC genome hereafter) and our Edinburgh genome, nHd.2.3, using both our raw data and the UNC raw and assembled data, made available after publication.
Surprisingly, the UNC assembly, despite the application of two independent long read technologies (Moleculo, based on single-molecule assembly of shorter Illumina reads, and Pacific Biosciences SMRT [PacBio] single molecule reads) and abundant short read data, had poorer metrics than nHd.2.3 (Table 1). Specifically, the scaffold N50 length was one third that of nHd.2.3, despite the UNC authors having discarded all scaffolds shorter than 2,000 bases. The lack of contiguity is unlikely to be due to heterozygosity, as H. dujardini is parthenogenetic and likely strongly bottlenecked. The span of the UNC assembly was 1.9 times that of nHd.2.3, and in direct conflict with the UNC authors’ own and our estimates of the H. dujardini genome size. Tellingly, the UNC span was greater than the span of our nHd.1.0, generated before filtering of bacterial contamination. The UNC protein prediction set was 1.7 times as large as ours, likely because of uncritical acceptance of all predictions from a range of gene finding algorithms. The UNC assembly had equivalent scoring for complete and partial representation of the core eukaryotic gene set assessed by CEGMA (Table 1), but was estimated to carry over 3 copies of each typically single-copy gene. This apparent multiplicity of representation can result from uncollapsed haploid genome estimates in very heterozygous species, and from erroneous inclusion of non-target genome data. Bacteria have proteins that are identified by CEGMA, and gain respectable CEGMA scores. The UNC genome span and the number of gene predictions were misreported in the previous paper [13] as 212 Mbp and 38,145 genes respectively (Boothby, pers. comm.).
The claim for massive HGT was based on a number of analyses [13]: the presence in the assembly of sequences that display features of bacterial and other origins by application of a BLAST bit-score based HGT index [45], the development of phylogenies for possible HGT genes to show their affinity with non-metazoan taxa, coverage, GC proportion, codon usage similarities, and PCR-based affirmation of some of the junctions between candidate HGT genes.
Two of these tests do not allow explicit support or rejection of HGT in the absence of proof of integration into a host genome. The HGT index [45] compares BLAST bit scores of a candidate HGT sequence derived from searches of probable recipient taxon and likely donor taxon databases where there is prior evidence of integration of the tested sequence in a eukaryotic genome. Screening of transcriptomes generated from poly(A)-selected mRNA, a process that will exclude bacterial and archaeal sequences a priori, is a credible use. Its application to genomic sequence is incorrect, as a bacterial contaminant gene will have a high HGT index simply because it is a bacterial gene. Similarly, phylogenetic analysis of a contaminant gene will affirm the phylogenetic position of the gene within the clade of its source species, not that it is of HGT origin.
Boothby et al. [13] assessed integration in two ways, both focussed on whether there was sequence evidence of linkage between putative HGT loci. Many of these tests did not explicitly assess HGT, as they examined relationships between pairs of bacterial genes. Rather obviously, bacterial genes will have bacterial neighbours in bacterial genomes. We classified each UNC protein prediction as viral, bacterial, archaeal, non-metazoan eukaryotic, metazoan, or unclassified in the case of conflicting signal. We screened the neighbourhood of each non-metazoan locus and identified 713 non-metazoan–metazoan and 294 non-eukaryote-eukaryote junctions. Long-read PacBio data are ideal for direct confirmation of linkage between HGT and resident genes. The UNC PacBio data were of relatively low quality (a mean length of 1.8 kb and a N50 length of 2.0 kb), and the PacBio assembly provided by the authors spanned only 120 Mb (with an N50 of 3.3 kb). PacBio data affirmed only 26 non-metazoan–metazoan linkages of 713 total, and 10 non-eukaryote-eukaryote junctions out of 294 possible junctions.
Boothby et al. [13] also assessed genomic integration of 107 candidates directly, using PCR amplification of predicted junction fragments. Their 107 candidates included 38 bacterial-bacterial, 8 archaeal-bacterial, and 61 non-metazoan–metazoan or non-eukaryotic-eukaryotic gene pairs (Table 2). Confirmation was achieved for most junctions, but PCR products were only analysed electrophoretically (several had faint or multiple products), and none were sequenced to confirm the expected amplicon sequence. We confirmed only 32 of the 107 putative HGT linkages in UNC PacBio data. Our assessment of the taxonomic origin of the loci in these pairs suggested some of classifications were in error, and we identified 49 bacterial–bacterial pairs. The existence of these 49 junctions in tardigrade culture DNA does not prove HGT, as contaminant prokaryotic genomes will carry such pairs. We found no expression of the 49 bacterial–bacterial pairs, further confirming that they are contaminants rather than examples of HGT.
Assessment of PCR-tested putative HGT loci in the UNC H. dujardini assembly
We classified the remaining 58 as 24 prokaryotic–eukaryotic, 27 non-metazoan eukaryotic–metazoan and 7 viral–eukaryotic junction pairs (Table 2). Of the 24 prokaryotic–eukaryotic junctions, two of the putative eukaryotic neighbours have marginal assignment to Eukaryota. The 27 non-metazoan eukaryotic–eukaryotic junctions include 6 where the assignment of the focal locus (non-metazoan eukaryotic) is unclear. The 7 viral–eukaryotic junctions include one where the assignment of the neighbouring gene is uncertain, and we note that 6 of the 7 viral candidates involved homologues of the same protein (carrying domain of unknown function DUF2828) from a series of Mimiviridae. Mimiviruses are well known for their acquisition of foreign genes, and thus these scaffolds may derive from mimivirus infection of one of the several species in the multi-xenic culture rather than tardigrade genome insertions. All 58 loci had read coverage in the Edinburgh raw data, and we observed the same genomic environment in nHd.2.3 in 51 gene pairs. We found evidence of expression from 49 of these loci (Table 2).
Within the set of 107 putative HGT genes approximately half were found to have predicted introns by Boothby et al. [13]. However, eukaryotic gene finders will “invent” introns in prokaryotic DNA, and thus the finding of eukaryote-like splice donor and acceptor sequences in prokaryotic DNA can simply be a process artefact resulting from the prediction algorithm. Codon usage assessment was limited to comparison to other Metazoa and Bacteria, rather than to bona fide tardigrade genes, and thus did not specifically address HGT.
In sum, the evidence for “massive” HGT into the UNC assembly is not compelling, and many tested loci were not confirmed by PacBio data, our Edinburgh read data or expression. Given these concerns, we compared our raw data and nHd.2.3 assembly to the UNC data, and independently predicted potential HGT in the nHd.2.3 assembly.
Raw read data do not support massive HGT in H. dujardini
We compared taxon-annotated GC-coverage plots for the UNC raw data (which we trimmed and adapter cleaned) and our trimmed and adapter cleaned, but otherwise unfiltered, raw data mapped to both assemblies (Figure 4). In all blobplots, a large blob at relatively high coverage and a GC proportion of ~45% corresponded to the H. dujardini genome. The best BLAST or diamond blastx matches of scaffolds in this blob were to existing H. dujardini sequences (ESTs and GSS from our laboratory), arthropods and other Metazoa, with a few matching bacteria (see below). The high-coverage scaffolds that matched existing H. dujardini sequences corresponded to the mitochondrion [4] and ribosomal RNAs, as would be expected.

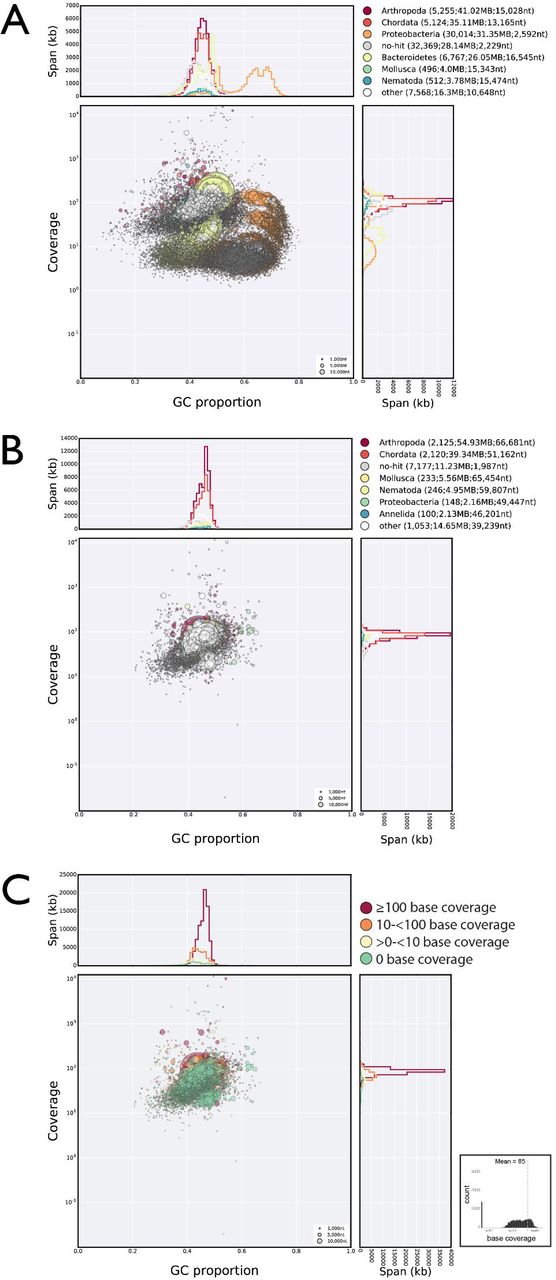
A Blobplot showing the UNC assembly contigs distributed by GC proportion and coverage derived from all the UNC raw genomic sequence data (data files TG-300, TG-500 and TG-800). Scaffold points are scaled by length, and coloured based on taxonomic assignment of the sum of the best BLAST and diamond matches for all the genes on the scaffold. Taxonomic assignments are summed by phylum. Scaffolds flagged as bacterial tend to have low coverage in the UNC raw data.
B Blobplot showing the UNC assembly contigs distributed by GC proportion and coverage derived from the UNC TG-300 raw genomic sequence data. Scaffold points are plotted as in A.
C Blobplot showing the UNC assembly contigs distributed by GC proportion and coverage derived from the UNC TG-500 raw genomic sequence data. Scaffold points are plotted as in A.
D Blobplot showing the UNC assembly contigs distributed by GC proportion and coverage derived from the UNC TG-800 raw genomic sequence data. Scaffold points are plotted as in A.
Comparison B, C and D showed that many of the bacterial scaffolds had different relative stoichiometries in the three UNC libraries.
E Blobplot showing the UNC assembly contigs distributed by GC proportion and coverage derived from the Edinburgh raw short insert genomic sequence data. The scaffolds flagged as bacterial also tend to have very low coverage in these data compared to the main set of H. dujardini scaffolds. Scaffold points are plotted as in A.
F Blobplot (as in B) with the scaffold points coloured by average RNA-Seq base coverage. The low-coverage scaffolds have no or very little RNA-Seq evidence of transcription. The histogram in the bottom right shows the frequency of scaffolds at different average RNAseq base coverages.
High resolution versions of each panel are available in Supplemental Information.
7,334 scaffolds, spanning 68.9 Mb, ~27% of the UNC assembly, had zero or very low (<10) coverage of reads mapped from either the UNC raw data (Figure 4 A [all data] and B-D [split by library]) or Edinburgh raw data (Figure 4 E) and therefore cannot be part of the target tardigrade genome (which was sequenced to much greater depth in both projects). Bacterial genomes in low-complexity metagenomic datasets often assemble with greater contiguity than does the target metazoan genome, even when sequenced at low coverage, because bacterial DNA usually has higher per-base complexity (i.e. a greater proportion is coding) [39, 40], and thus the longest scaffolds in the UNC assembly were bacterial (Figure 5 A). The largest span of UNC contaminants matched Bacteriodetes, and had uniformly low coverage. A second group from Proteobacteria had a wide dispersion of coverage, from ~10 fold higher than the H. dujardini nuclear mean to zero. Most proteobacterial scaffolds had distinct GC% and coverage compared to bona fide H. dujardini scaffolds. It was striking that many of the putatively bacterial scaffolds had close to zero coverage in both UNC and Edinburgh data (Figure 5 B). The wide spread of coverage of UNC proteobacterial scaffolds in the Edinburgh data may reflect the presence of related but not identical contaminants in the UNC and Edinburgh cultures. The variable coverages make it unlikely that these are common symbionts. We identified 15 scaffolds in the UNC assembly with robust matches to 12S and 16S genes from Armatimonadetes, Bacteroidetes, Chloroflexi, Planctomycetes, Proteobacteria and Verrucomicrobia (Table 3).

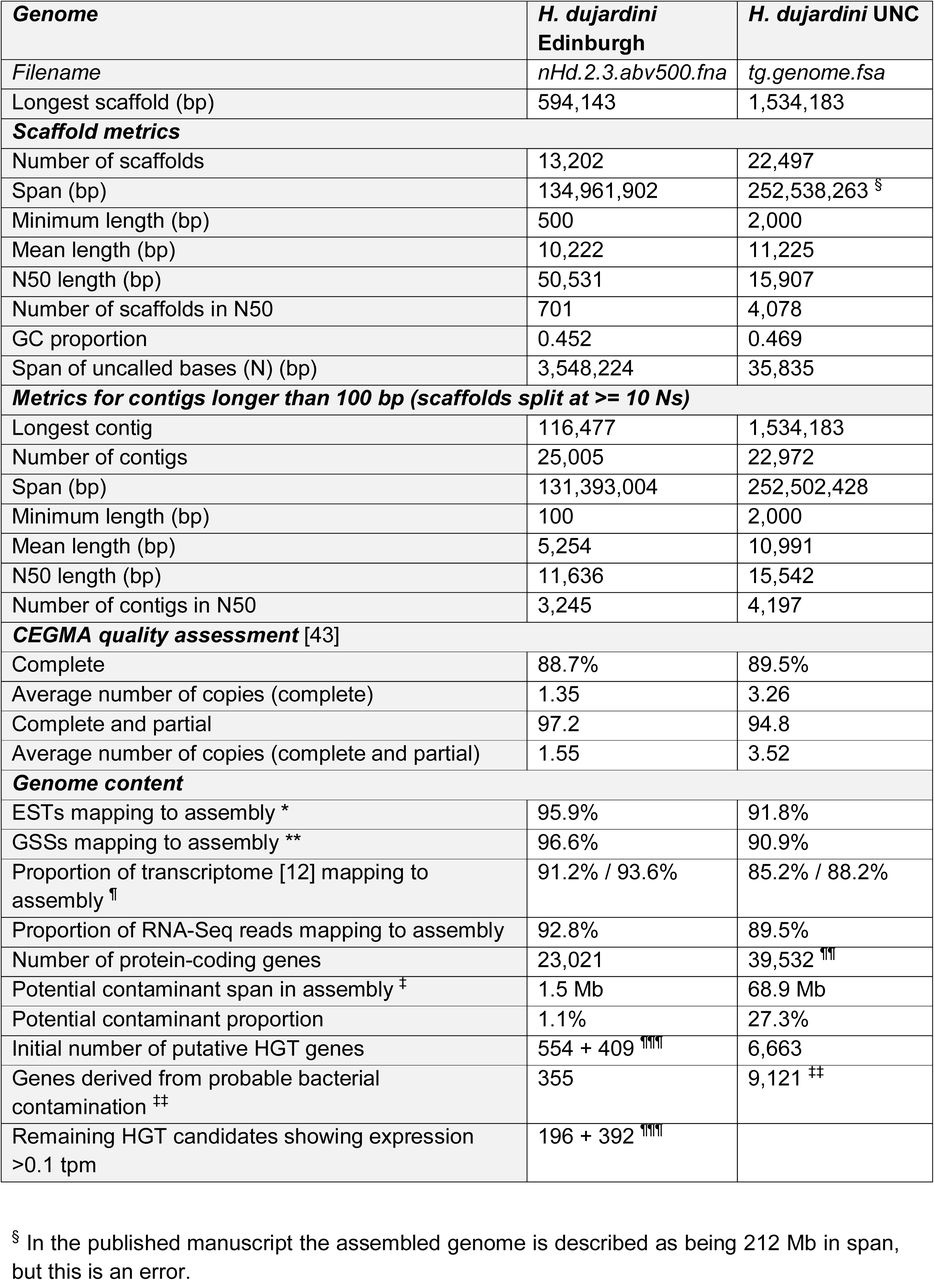
A Scaffolds from the UNC and nHd.2.3 assemblies were grouped by size class the proportion of each size class that was scored as being derived from Bacteria, Archaea, Viruses, Eukaryota and those with no-hits plotted as a stacked histogram. The nHd.2.3 assembly has no scaffolds >1 Mb. The longest scaffolds (>0.5 Mb) in the UNC assembly were bacterial.
B Each scaffold in the UNC assembly was plotted based on its coverage in the Edinburgh short insert data a (X-axis) and in the pooled UNC short insert dat (Y-axis). If these scaffolds were a true part of the H. dujardini genome, we expect them to have the same coverage in both raw dataset. Scaffolds placed off the diagonal (i.e. different relative coverage in the two datasets) and especially those with very low coverage in one or the other dataset are unlikely to be true parts of the H. dujardini genome. Scaffolds were coloured by inferred phylum of origin. Above the plot is a histogram of summed span of scaffolds at each coverage for the Edinburgh data, and to the right a histogram of spans for the UNC data. Many bacterial scaffolds were unique to the UNC raw data, and most others have limited representation in both datasets. Eukaryota scaffolds spanned 173 Mb and had an N50 of 17.3 kb. Bacteria scaffolds spanned 70.7 Mb and had an N50 of 13.3 kb. Archaea scaffolds spanned 0.1 Mb and had an N50 of 12.7 kb. Virus scaffolds spanned 0.01 Mb and had an N50 of 6.3 kb. No-hit scaffolds spanned 8.5 Mb and had an N50 of 7.2 kb.
C Each scaffold in the nHd.2.3 assembly was plotted based on its coverage in the Edinburgh short insert data (X-axis) and in the pooled UNC short insert data (Y-axis), as in B. Some bacterial scaffolds were unique to the Edinburgh raw data, and are remaining contaminants in nHd.2.3. Eukaryota scaffolds spanned 118 Mb and had an N50 of 58.3 kb. Bacteria scaffolds spanned 5.0 Mb and had an N50 of 37.7 kb. Archaea scaffolds spanned 0.02 Mb and had an N50 of 1.5 kb. Virus scaffolds spanned 0.03 Mb and had an N50 of 22.3 kb. Nohit scaffolds spanned 11.2 Mb and had an N50 of 2.0 kb.
D Expression of “soft” HGT candidates, “hard” HGT candidates and all other genes was estimated by mapping 351 M RNA-Seq reads to the nHd.3.2 assembly with kallisto. Expression was quantified as transcripts per million (tpm). For each tpm expression bracket, the proportion of genes in each category was plotted.
Scaffolds containing bacterial and eukaryotic ribosomal RNA sequences in the UNC H. dujardini assembly.
Seven UNC scaffolds had matches to H. dujardini SSU and LSU, with four containing both subunits. We identified two very similar ~20 kb scaffolds (scaffold2445_size21317 and scaffold2691_size20337) that both contained two, tandemly repeated copies of the ribosomal cistron of a bdelloid rotifer closely related to Adineta vaga (for which a genome sequence is available [23]). We screened the UNC genome for additional matches to the A. vaga genome, and found many, but given that A. vaga is robustly reported to contain a large proportion of bacterially-derived HGT genes [23, 25] we treated these matches with caution. However a total of 0.5 Mb of scaffolds had best sum matches to Rotifera rather than to any bacterial source (Supplemental file 1). Six mimiviral-like proteins were identified (as noted above).
We mapped H. dujardini RNA-Seq read data to the UNC assembly (Figure 4 F). As these RNA-Seq data were derived via poly(A) selection, mapping constitutes strong evidence of eukaryotic transcription. Only nine UNC scaffolds that had low or no read coverage in our raw genome data had any RNA-Seq reads mapped (Figure 4 F) at very low levels (between 0.19 and 31 transcripts per million). One scaffold (scaffold1161) had two genes for which expression was >0.1 transcript per million (tpm), but all the genes on this scaffold had best matches to Bacteria. Comparison to the RNA-Seq density plot for the nHd.2.3 assembly (Figure 2 C) showed that the pattern of high, low and no RNA-Seq mapping scaffolds observed in the area we attributed to the tardigrade genome in the UNC assembly blobplots was reflected in the nHd2.3 mapping. The RNA-Seq data thus give no support to gene expression from the low coverage, bacterial-like contigs in the UNC assembly.
The scaffolds identified as likely bacterial contaminants in the UNC assembly include 9,872 protein predictions, including 9,121 bacterial, 480 eukaryotic and 271 unassigned. In UNC scaffolds that were strongly supported by coverage ≥10 in both raw sequence datasets (Figure 5 B), Boothby et al. predicted 29,660 protein-coding genes, but only 566 of these had highest similarity to bacterial proteins.
Our analyses constituted sensitive tests of three of the expected characteristics of functional HGT for the UNC assembly: perdurance, physical linkage to sequences clearly identifiable as host, and expression. Many scaffolds had low coverage compared to bona fide tardigrade scaffolds (Figure 4 A) and different relative coverage in different libraries (Figure 4 B-D). They thus do not travel with the tardigrade scaffolds in a stoichiometric manner, and are unlikely to be part of the same genome. Absence from our raw data (Figure 4 E) showed that the putative HGT scaffolds were not found in all animals subcultured from the Sciento stock. The dominance of sequence with strong bacterial similarity and absence of all but marginal similarity to metazoan sequence also suggests that these contigs are not co-assemblies of bacterial and metazoan components. RNA-Seq mapping failed to support expression of genes on bacterial-like scaffolds (Figure 4 F). While some putative HGT loci were confirmed, these are a very small proportion of the proposed total.
Low levels of horizontal gene transfer in H. dujardini
We screened our current nHd.2.3 assembly for potential HGT. The RNA-Seq and transcriptome assembly data mapped more poorly to the UNC assembly than it did to our nHd.3.2 assembly (Table 1), and, as noted above, the mapping of transcriptome data to the cleaned assembly was equivalent to our pre-cleaning nHd.1.0 assembly. The nHd.2.3 assembly was thus not missing expressed coding genes compared to the UNC assembly, and is unlikely to have been over-aggressively filtered of HGT candidates. Indeed nHd.2.3 contained 43 of the 53 informative junctional fragments analysed by Boothby et al. [13] (Table 4).
Putative HGT genes in the nHd2.3 Hypsibius dujardini assembly
Blobplot analyses highlighted scaffolds in nHd.2.3 that had bacterial similarities, but coverages similar to the bona fide tardigrade scaffolds. Forty-eight nHd.2.3 scaffolds, spanning 0.23 Mb and including 41 protein-coding genes, had minimal coverage in UNC data (coverage of <10; Figure 5 C), suggesting contaminant status. The remaining 13,154 nHd.2.3 scaffolds spanned 134.7 Mb. We screened these for possible HGT in two ways: by examining scaffolds labelled in blobplots as having high coverage in both datasets, but a majority consensus likely bacterial origin based on BLAST and diamond matches, and by examining each of our gene predictions for higher similarity to non-metazoan rather than metazoan sequences. For potential HGT candidates we sought supporting evidence in RNA-Seq expression data.
Of the 23,021 protein coding genes predicted in nHd.2.3, about half (10,161 proteins) had unequivocal signatures of being metazoan. These included sequences with best matches in a range of phyla, including Arthropoda and Nematoda as was expected, but also in lophotrochozoan (Mollusca, Annelida), deuterostome (Chordata) and basal metazoan (Cnidaria) phyla. A priori these might be candidates for metazoan–metazoan HGT. However, as H. dujardini is the first tardigrade to be sequenced, this pattern of diverse similarity may just reflect a lack of close relatives in the public databases. For most of the remainder (11,655 proteins) we identified best matches in a wide range of non-metazoan eukaryotes, but there were often also metazoan matches with similar scores and, sometimes, bacterial matches. It is likely, again, that these proteins are tardigrade, in large measure, but fail to find a best match in Metazoa because of phylogenetic distance. This suggests that H. dujardini may be a “long branch” taxon, as observed in analyses of its mitochondrial genome [4]. Some non-metazoan eukaryote-like proteins may have arisen from non-elimination of remaining contamination. For instance, we identified bodonid ribosomal RNA loci, and also two proteins from short kinetoplastid-like scaffolds.
We found 564 bacterial-metazoan HGT candidates in nHd.2.3, from 328 scaffolds. In these scaffolds, 166 contained only genes predicted to be bacterial (totalling 350 gene predictions). While some of these scaffolds also contained genes that had equivocal assignment (eukaryotic vs non-eukaryotic), we regard these loci and scaffolds as likely remaining contaminants, and at best “soft” candidates for HGT. Gene expression from these soft HGT candidates was very low (Figure 5 D). There were 214 genes with similarity to bacteria and linkage to genes with eukaryotic similarities on 162 scaffolds with GC% and coverage similar to the tardigrade genome in both datasets (Table 4). Of these candidates, 196 had expression >0.1 tpm (Figure 5 D). These genes, 0.9% of the total predicted for H. dujardini nHd.2.3, are “hard” candidates for HGT. We also screened the nHd.2.3 proteome for loci with highest similarity to non-metazoan eukaryotes. We identified 409 genes (1.8% of all genes), and 392 of these had expression >0.1 tpm, and 333 >1 tpm. We caution that these loci may in fact be normal tardigrade genes for which simple similarity measures are insufficient to identify true HGT. Proof of HGT status requires data from additional tardigrades, and careful phylogenetic analysis, now underway.
Within the high-coverage blob of assembly scaffolds supported by both Edinburgh and UNC raw data, blobtools analyses identified 358 scaffolds with majority similarity to bacterial sequences (black points in Figure 5 C). Fifty-two of these scaffolds carried no predicted genes, and 77 contained protein predictions that were identified as eukaryote or unassigned. These were classified as bacterial based on marginal nucleotide similarities to bacterial sequences. The remaining 229 scaffolds were also flagged in the gene-based analysis as containing HGT candidates. Our assembly thus still contained contaminating sequences, mainly from bacteria but also including some from identified eukaryotes. Cross-comparison of the Edinburgh and UNC datasets, currently ongoing, will permit robust elimination of such “difficult” contamination, and true estimation of HGT proportions.
Conclusions
We have generated a good quality, first-draft genome for the model tardigrade H. dujardini. The assembly has good numerical and biological credibility scores. We have identified areas for improvement of our assembly, particularly with respect to removal of remaining contaminant-derived sequences. We approached the initial data as a low complexity metagenomic project, sampled from an ecosystem rather than a single species. This approach is going to be ever more important as genomics approaches are brought to bear on new systems less amenable to culture and isolation. The blobtools package [39, 40] and related toolkits such as Anvi’o [46] promise to ease the significant technical problem of separating microbial genomes from those of other target species. Our strategy for improving the assembly was boosted by the recent release of additional large-scale data for H. dujardini from the same strain [13].
Our analyses of gene content and the phylogenetic position of H. dujardini and by inference Tardigrada are at an early stage, but are already yielding useful insights. Early, open release of the data has been key. The H. dujardini EST data have already been exploited by others in deep phylogeny analyses that place Tardigrada in Panarthropoda [3]. The H. dujardini mitochondrial genome was isolated and fully sequenced based on the EST data, and phylogenetic analysis along with onychophoran and diverse arthropod mitochondrial genomes gave support for Panarthropoda [4]. A P2X receptor identified in the ESTs was shown to have an intriguing, unique mix of electrophysiological properties [16]. The presence of this ancient class of ligand gated ion channels in a tardigrade implies that it has been lost independently in nematodes and arthropods. H. dujardini, as a limnetic species, does not readily enter cryptobiosis, and thus analysis of its genome for functions associated with this phenomenon, often erroneously associated with all tardigrades, requires sequencing of species that do. H. dujardini has been used as a non-cryptobiotic comparator for exploration of cryptobiosis in other tardigrade species, in particular mining the EST data for proteins that could be associated with ice nucleation and other mechanisms [6–9]. Our ESTs were briefly summarised previously [20]. A study of the evolution of opsin loci in H. dujardini compared sequences derived from RNA-Seq to the nHd.2.3 assembly [12]. We are working to improve the H. dujardini assembly incorporating UNC data and improved contamination screening processes.
Our analyses of the H. dujardini genome conflict with the published UNC draft genome [13] despite being from essentially the same strain of H. dujardini. Our assembly, despite having superior assembly statistics, is ~120 Mb shorter than the UNC assembly, and is congruent with values we, and others, obtained from direct measurement. We find 15,000 fewer protein-coding genes, and a hugely reduced impact of predicted HGT on gene content in H. dujardini. We find that 0.9% of H. dujardini genes have signatures of HGT from bacteria, a relatively unsurprising figure. Estimates of HGT from non-metazoan eukaryotes into H. dujardini were less easily validated, but maximally comprise ~1.5%. In Caenorhabditis nematodes, Drosophila dipterans and primates, validated, expressed HGT genes comprise 0.8%, 0.3% and 0.5% of genes respectively [28]. These mature estimates, from well assembled genomes with several closely-related sequenced representatives in each group, are reductions from early guesses, such as the initial proposal that 1% of human genes originated by HGT [47, 48]. RNA-Seq mapping suggests our filtering has not compromised the assembly by eliminating real sequence components, and we identified the few likely HGT events confirmed by Boothby et al. [13]. The remaining HGT candidates await detailed validation. Our evidence strongly suggests that the UNC assembly is compromised by sequences that derive from bacterial contaminants, and that the expanded genome span, additional genes, and the majority of the proposed HGT candidates are likely to be artefactual.
Materials and Methods
Culture of H. dujardini
H. dujardini starter cultures were obtained from Sciento, Manchester, and cloned by isolation of single females in vinyl microtitre plates. Cultures were bulked from an individual female. Tardigrades were maintained on Chlamydomonas reinhardtii algae, which was grown in 1x Bold’s medium, pelleted and resuspended in fresh spring water to be fed to the tardigrades. Cultures were maintained at 19°C and aerated continuously. DNA for sequencing was prepared from tardigrades of mixed ages from bulk cultures maintained in glass baking dishes. These were isolated from C. reinhardtii by two rounds of Baermann filtration through two layers of sterile milk filter paper and left without food until remaining green algae and darker digestion products were no longer visible in the gut (3–4 days). Tardigrades were then washed repeatedly in lab artificial freshwater by gentle centrifugation. Pelleted tardigrades were snap frozen while still alive in a minimal volume and stored at −80°C.
Genome size measurement
We estimated the size of the H. dujardini genome by propidium idodide staining and flow cytometry, using C. elegans (genome size 100 Mb), and Gallus gallus red blood cells (1200 Mb) as size controls, following published protocols [49].
RNA and DNA extraction
RNA was isolated from cleaned, pelleted tardigrades using Trizol reagent, after percussive disruption of cleaned tardigrades under liquid nitrogen. Genomic DNA was isolated by a manual phenol-chloroform method, after percussive disruption of cleaned tardigrades under liquid nitrogen.
Expressed sequence tag (EST) sequencing
A directional cDNA library was constructed in pSPORT1 using the SMART cDNA synthesis protocol and transformed into BL21 E. coli. Individual recombinant clones were picked into microtitre plates and inserts amplified using universal PCR primers (M13L and M13R). The amplified inserts were sequenced in one direction (using primer T7) after enzymatic clean-up with Exo1 and SAP, using BigDye reagents on an AB 3730 sequencer. All successful sequences were trimmed of low quality sequence and vector using trace2dbest [50] (see Table 5 for software used, version numbers and additional commands) and submitted to dbEST (see Supplemental file 3). Data were publicly released on submission in 2003–2004.
Software used
Genome survey sequencing
A 2 kb-insert H. dujardini genomic library was constructed in the pCR4Blunt-TOPO vector. Individual recombinant clones were picked to microtitre plates and inserts amplified using M13R and pBACe3.6_T7 primers and sequenced with the T3 primer. Sequences were processed with trace2seq [50] and submitted to dbGSS (see Supplemental file 4). Data were publicly released on submission in 2005.
Genome sequencing with Illumina technology
Purified H. dujardini genomic DNA was supplied to Edinburgh Genomics (http://genomics.ed.ac.uk) for Illumina sequencing. We obtained sequence from two libraries: a small insert library (~300 bp insert size, prepared with Illumina TruSeq reagents by Edinburgh Genomics) and a 4 kb virtual insert mate-pair library (constructed by CGR, Liverpool). These were sequenced on HiSeq2000 at Edinburgh Genomics to generate datasets of 101 base paired end reads. The raw data are available in the ENA under study accession PRJEB11910 (runs ERR1147177 and ERR1147178). We were granted early access to Illumina GAIIX RNA-Seq data from Itai Yanai (accession GSE70185) in advance of publication. Lars Hering granted access to assemblies of the RNA-Seq data generated for their analyses of H. dujardini opsin genes [12].
Data validation and filtering for genome assembly
We performed initial quality control on our raw Illumina data using fastqc (S. Andrews, unpublished), and used trimmomatic [51] to remove low quality and adapter sequence. We screened the quality- and adapter-trimmed data for contaminants using taxon annotated GC-coverage plots (TAGC or blobplots) using an updated version of the blobtools package (Dominik Laetsch, in prep.). The paired-end reads were normalised with one-pass khmer [52] and were assembled with Velvet [53, 54] using a k-mer size of 55, and non-normalised reads mapped back to this assembly using the CLC mapper (CLCBio, Copenhagen) or bwa mem [55]. For each scaffold, GC% was counted (ignoring N base calls) and read coverage calculated. Each scaffold was compared to the NCBI nucleotide database using BLAST megablast [56, 57] and to UNIREF90 using diamond blastx [58], and the results were filtered by the blobtools script to annotate each scaffold with the taxonomy of the highest scoring matches in these databases. Blobtools estimates taxonomic similarity of a scaffold or contig either by simply recording the taxonomy of the highest match to any segment of the sequence, or assigning taxonomy based on the sum of best match scores across of the scaffold or contig. The scaffolds were then plotted in a two dimensional scatter plot (X-axis: GC proportion, Y-axis: log coverage), coloured by putative taxon of origin based on the BLAST or diamond results. Using the blobplot we identified likely contaminant reads, and cleaned these (and their pairs) from the quality- and adapter-trimmed data. Subsequent assemblies from filtered and cleaned data were also screened using blobplots. The initial Velvet assembly was used to estimate library insert sizes so that accurate parameters could be passed to subsequent assembly steps (Supplemental File 5). The mate pair library insert distribution was not normally distributed, and the library contained many pairs that appeared to derive from non-mate fragments.
The blobtools cleaning process was repeated two more times, as newly assembled contaminants could be identified. Gaps were filled in the final assembly using GapFiller [59, 60]. The mate pair library was used to scaffold the gap-filled assembly with SSPACE [59], accepting only the information from mate pair reads mapping 2 kb from the ends of the scaffolds. The final assembly spans 135 megabases (Mb) with median coverage of 86 fold. The completeness of the genome assembly was assessed using CEGMA [43], and by mapping EST, GSS and RNA-Seq data.
Genome annotation
We annotated the assembled H. dujardini genome nHd.2.3 using a two-pass approach. We used MAKER [61] to generate a first-pass set of gene models, using the ESTs and available transcriptome data as evidence, and then used these to inform a second pass of annotation with Augustus [62]. Protein sequences were annotated using BLAST searches against UNIREF90 and the NCBI nonredundant protein database. Protein domains and motifs were predicted with InterProScan [63]. The genome sequence and annotations were loaded into an instance of BADGER [44] and made publicly available in mid-2014. The genome assembly, predicted transcriptome, predicted proteome and GFF file of annotations are available for download on the http://www.tardigrades.org website.
Comparison of H. dujardini genome assemblies
We compared the UNC H. dujardinii assembly [13], downloaded from http://weatherby.genetics.utah.edu/seq_transf/, 27 November 2015) to our raw Illumina data (quality and adapter trimmed but otherwise unfiltered) and the nHd.2.3 genome assembly. We mapped both our read data and the UNC TG-300, TG-500 and TG-800 library raw read data (from http://weatherby.genetics.utah.edu/seq_transf/, 01, 02, and 03 December 2015) to UNC and nHd.2.3 genome assemblies using bwa [55]. The resulting read mapping files, together with the results of a diamond [58] search against UniRef90 and megablast [56, 57] search against NCBI nt, were used to compute blobplots of both assemblies. We also accessed the UNC PacBio data from http://weatherby.genetics.utah.edu/seq_transf/ (03 December 2015). To explore transcription of putative HGT loci, we assessed gene expression using kallisto [64] and 351 million RNA-Seq reads, estimating expression as transcripts per million (tpm). Normalised, average RNAseq base-coverage for each scaffold in both assemblies was calculated by mapping RNAseq data using GSNAP [65, 66]. We mapped two transcriptome assemblies provided by Hering and Mayer [12]. These assemblies were based on the same raw data, assembled with CLCBio or IDBA assemblers. We screened both genome assemblies against the SILVA ribosomal RNA database [67] using BLAST.
We assessed horizontal gene transfer into H. dujardini initially by calculating a summed best diamond blastp score for every protein predicted from nHd.2.3 compared to the UNIREF90 database. From the summed scores we assessed whether the nHd.2.3.1 protein could be assigned to non-eukaryote, non-metazoan eukaryote, metazoan or unassigned origins, with assignment requiring that the taxonomic origins of ≥90% of all the hits returned by diamond were congruent. The label “unassigned” was attached to proteins that had no diamond hits, or that had conflicting hits (i.e. <90% of hits were to one taxonomic group). We also calculated assignment to metazoan versus non-metazoan within the eukaryote group using the same rule. We then assessed, for each of the 6,863 scaffolds from which we predicted proteins, the presence of proteins with different taxonomic assignments. We also classified the UNC protein predictions using this pipeline. We used the mapping file provided by UNC between the UNC PacBio assembly and the UNC genome assembly (downloaded from http://weatherby.genetics.utah.edu/seq_transf/pacbio/, 3 December 2015) and scored each potential HGT–metazoan genome junction for confirmation with these long-read data.
Availability of Supporting Data
The raw Illumina sequence read data have been deposited in SRA, and the GSS and EST data in dbGSS and dbEST respectively (see Table 6). The genome assemblies produced in Edinburgh have not been deposited in ENA, as we are still filtering the assembly for contamination, and have no wish to contaminate the public databases with foreign genes mistakenly labelled as “tardigrade”. The assemblies (including GFF files, and transcript and protein predictions) are available at http://www.tardigrades.org. The raw data from UNC, and their assembly are available from http://weatherby.genetics.utah.edu/. Our analysis intermediate files, blobDB for each TAGC plot and high-resolution versions of Figures 2, 5 and 6 are available from http://www.tardigrades.org. Code used in the analyses is available from https://github.com/drl and https://github.com/sujaikumar/tardigrade.
Raw data for H. dujardini
- Abbreviations
- UNC
- University of North Carolina
- CEGMA
- Core Eukaryotic Genes Mapping Approach
- HGT
- horizontal gene transfer
- GSS
- genome survey sequence
- EST
- expressed sequence tag
- RNA-Seq
- Transcriptomic RNA sequencing (as performed on the Illumina platform)
- PacBio
- Pacific Biosciences SMRT single-molecule real time sequencing
- BLAST
- Basic Local Alignment Search Tool
- UNIREF90
- UniProt Reference Clusters collapsed at 90% pairwise identity, a product of the UniProt database.
- GFF
- genome feature format
- GC
- guanine plus cytosine
- PCR
- polymearase chain reaction
- cDNA
- copy DNA
- TAGC
- plot taxon-annotated GC-coverage plot
- LSU
- large subunit ribosomal RNA
- SSU
- small subunit ribosomal RNA
- N50 length
- length-weighted median contig length
- Tpm
- (length-normalised) transcripts per million as estimated in kallisto
Competing interests
The authors declare no competing interests.
Author contributions
FT, JD, AA, CC and HM developed the H. dujardini culture system and prepared nucleic acids. AA supplied additional DNA samples for Illumina sequencing. The cDNA library was made by JD, and EST sequencing performed by JD and FT. The GSS library was made by CC, and GSS sequencing performed by CC and FT. JD, CC, FT and HM analysed the EST and GSS data. The initial genome assemblies were made by MB and SK, and the final assembly and annotation by GK. Analyses of the genome were performed by MB, GK, SK, DRL and AA. LS constructed the BADGER genome browser instance and managed data. The manuscript was written by MB with input from all authors.

Description of Additional Files
Acknowledgements
The Edinburgh tardigrade project was funded by the BBSRC UK (grant reference 15/COD17089). GK was funded by a BBSRC PhD studentship. DRL is funded by a James Hutton Institute/School of Biological Sciences, University of Edinburgh studentship. SK was funded by an International studentship and is currently funded by BBSRC award BB/K020161/1. LS is funded by a Baillie Gifford Studentship, University of Edinburgh. We thank Bob McNuff of Sciento for his inspired culturing of H. dujardini, Reinhardt Kristensen for guidance with microscopy and identification of H. dujardini, Sinclair Stammers of MicroMacro for videomicroscopy assistance, the University of Edinburgh COIL facility for fluorescence microscopy, Itai Yanai for pre-publication access to RNA-Seq data, Lars Hering for access to RNA-Seq assemblies, The Centre for Genomic Research for mate-pair library construction, and staff of Edinburgh Genomics (and its precursor the GenePool) for sequencing. We thank a wide community of colleagues on twitter, blogs and email for discussion of the results presented here in the three weeks since the publication of the UNC genome, and the two weeks since the first version of this article was posted.
Footnotes
Georgios Koutsovoulos (gdkoutsovoulos{at}gmail.com)
Sujai Kumar (sujai.kumar{at}ed.ac.uk)
Domink R. Laetsch (dominik.laetsch{at}gmail.com)
Lewis Stevens (lewis.stevens07{at}gmail.com)
Jennifer Daub (jennifer.daub{at}gmail.com)
Claire Conlon (claire.conlon{at}ed.ac.uk)
Habib Maroon (habibmaroon{at}hotmail.com)
Fran Thomas (fran.thomas74{at}btinternet.com)
Aziz Aboobaker (aziz.aboobaker{at}zoo.ox.ac.uk)





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}