

Abstract
Background: Low melatonin levels are a frequent finding in autism spectrum disorder (ASD) patients. Melatonin is also important for normal neurodevelopment and embryonic growth. As a free radical scavenger and antioxidant melatonin is highly effective in protecting DNA from oxidative damage. Melatonin deficiency, possibly due to low CYP1A2 activity, could be a major factor, and well a common heritable variation. ASD is already present at birth. As the fetus does not produce melatonin, low maternal melatonin levels should be involved.
Methods: We measured 6-sulfatoxymelatonin in urine of mothers of a child with ASD that attended our sleep clinic for people with an intellectual disability (ID), and asked for parental coffee consumption habits, as these are known to be related to CYP1A2 activity.
Results: 6-Sulfatoxymelatonin levels were significantly lower in mothers than in controls (p = 0.005), as well as evening coffee consumption (p = 0.034). In mothers with a second child with ASD and/or ID, 6-sulfatoxymelatonin levels were lower compared to mothers with one child with ASD (p = 0.084),
Conclusions: Low parental melatonin levels, likely caused by low CYP1A2 activity, seem to be a major contributor to ASD and possibly ID etiology.
1. Introduction
Autism Spectrum Disorder (ASD) refers to a heterogeneous group of neurodevelopmental disorders, with a complex multifactorial etiology, starting very early in life. The disease is characterized by abnormal social interactions, impaired language development, and stereotyped and repetitive behaviours. ASDs have in common that they each share one or more core symptoms of autism. But at the same time there are large differences between them, both with regard to symptomatology as to etiology (1). Intellectual disabilities (ID) and ASD, although apparently two separate disorders, co-occur much more frequent in the same patient than by chance. While in 40% of persons with ID autism is present, at the same time 70% of people with autism also has an intellectual disability (2). The frequent co-occurrence of ASD and ID, however, raises questions whether this is a true co-occurrence because both share, at least in part, similarities in their etiology. Especially since in more severe cases of ID, the risk of ASD is greater (3), as is the risk of ID in more severe cases of ASD (4).
There is a growing number of literature on the genetic basis of ASD. Over a hundred of chromosomal abnormalities have been found, however, not one of them is responsible for more than 1% of ASD cases. This means that the underlying genetic process in ASD is complex. Recent reviews (5-9) provide a good overview of the current state of knowledge.
The relative risk of a newborn child from parents having a child with ASD is 25-fold increased compared to the general population, suggesting a genetic component (6). However, in only about 10% a specific genetic disorder is diagnosed (7). It is assumed that ASD is caused by a combination of de novo inherited variation and common variation. In a recent epidemiological study in Sweden, it was possible to distinguish between both components, estimating liability of de novo variations to be 2.6% and common variation to be 49% (10). In a recent large study in twins with ASD, concordance for monozygotic male twins was 77% (females 50%), and for dizygotic male twins 31% (females 36%) (11). Based on their data they were able to calculate that the contribution of genetic factors was smaller (38%) than shared environmental factors (58%).
Epidemiological studies have identified several environmental factors that increase ASD risk (12-16) (see supplementary file). Most important factors are advanced parental age at birth, bleeding, gestational diabetes, having a mother born abroad, fetal distress, birth injury or trauma, multiple birth, maternal haemorrhage, summer birth, low birth weight, small for gestational age, congenital malformation, low 5-minute Apgar score, feeding difficulties, meconium aspiration, neonatal anaemia, ABO or Rh incompatibility, and hyperbilirubinemia. Also subnormal breast feeding increases autism risk (17). Interestingly, many of these environmental factors can be linked to melatonin deficiency.
Disturbances in melatonin turnover leading to different melatonin levels in ASD are found in several studies (18-20). Melatonin levels in ASD are negatively correlated with severity of autistic impairments (21). Melatonin is a hormone with multiple functions and it is synthesized in the pineal gland during the dark phase of the day and involved in sleep induction and circadian rhythm regulation (22). Melatonin also acts as a free radical scavenger and antioxidant and is highly effective in reducing effects of oxidative stress throughout the body (23-25). There are also indications that melatonin protects against oxidative stress-evoked DNA damage (23, 24, 26, 27).
Melatonin is synthesized from serotonin in a two step process, involving the enzymes AANAT and ASMT (see Figure 1) (28). The expression of their mRNAs encoding for these enzymes follows a day/night rhythm.
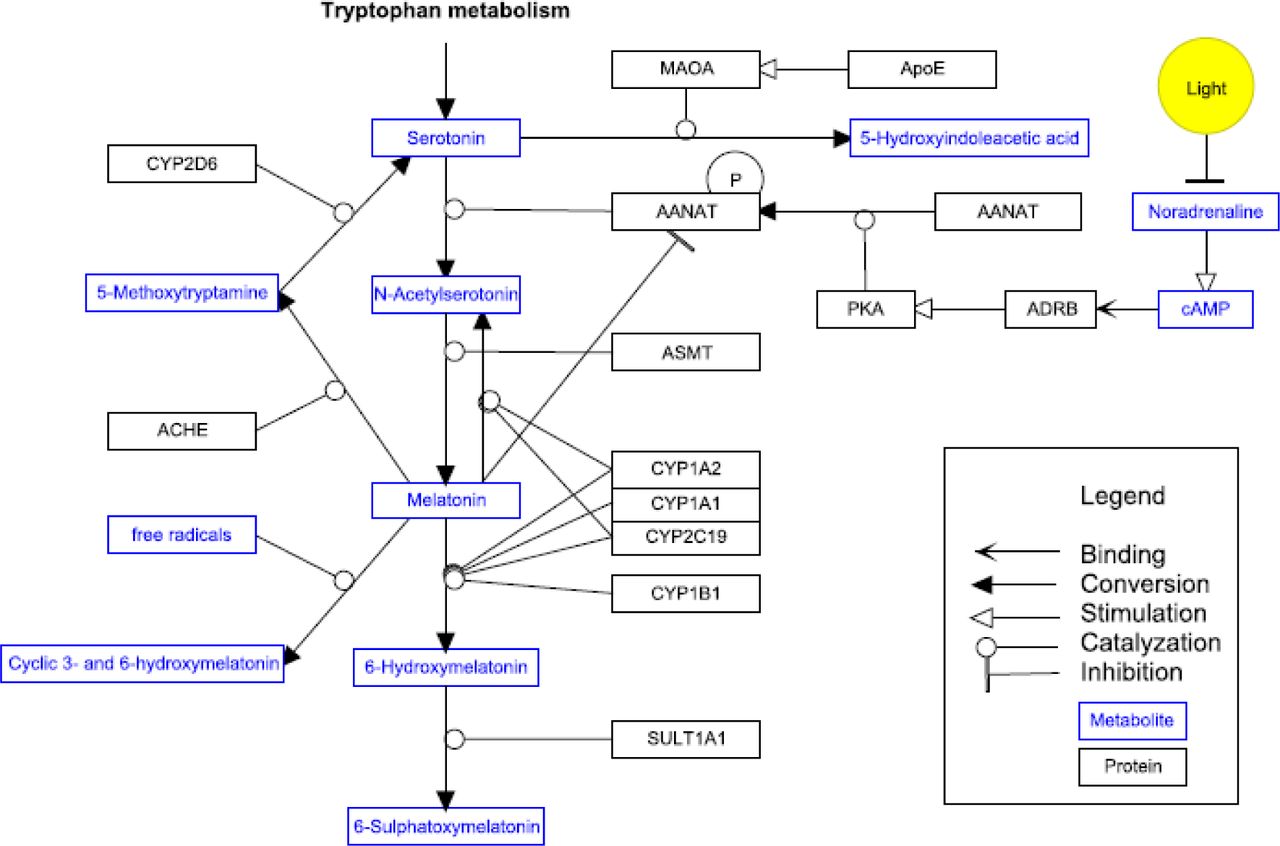
AANAT is generally considered to be the rate-limiting enzyme in the melatonin synthesis pathway. However, recent studies indicate that the rate limiting role at night is partly taken over by ASMT (29). Exposure to light rapidly decreases AANAT activity. Timing, profile and amount of melatonin secretion display large inter-individual differences, but are in healthy individuals highly reproducible from day to day like a hormonal fingerprint (22). As information on normative data of melatonin levels is scarce and internationally accepted normative data are not available, it is also not clear which melatonin levels are pathologically low. This is remarkable, given the large number of functions of melatonin. Although several positive functions are attributed to melatonin, melatonin deficiency is not regarded as a risk factor or a disease yet.
Melatonin is metabolised principally by 6-hydroxylation to 6-hydroxymelatonin, which is sulphated to 6-sulfatoxymelatonin (6-SM) and excreted in urine (30, 31) (Figure 1). CYP1A2, and to a lesser extend CYP1A1 and CYP2C19 are the main cytochrome isozymes to be responsible for the plasma clearance of melatonin in the liver (30). CYP1A2 activity in fetal and neonatal liver tissue is very low to undetectable (32). The expression of CYP1A2, as well as other members of the cytochrome P450 gene family, is regulated i.a. by the biological clock (33). Melatonin does not influence CYP1A2 activity at physiologic levels. Only at extremely high concentrations of 3 – 300 µM an inhibition was observed (34).
The half life of exogenous melatonin ranges between 28 to 126 minutes (35, 36). Differences in melatonin half-life reflect the wide inter-individual differences (10 to 200-fold) in CYP1A2 activity (37). Several reports indicate that SNPs in the CYP1A2 gene are associated with increased inducibility, decreased activity or inducibility or even loss of activity of the CYP1A2 enzyme as compared to the wild type CYP1A2*1A (38-43). The (sub)variant alleles associated with decreased of absent activity of CYP1A2 are *1C, *1K, *3, *4, *5, *6 and *7. The proportion of individuals with the slow phenotype varies among ethnic populations (41).
Caffeine clearance is considered as the gold standard for assessment of CYP1A2 activity, because more than 90% of the primary metabolism of caffeine depends on CYP1A2 (44). Caffeine related side effects, i.e. caffeine-induced insomnia, are more frequent in people who are CYP1A2 slow metaboliser (45). Several polymorphisms decrease the metabolic activity of CYP1A2 and are significantly inversely related to coffee intake, so coffee intake is influenced by CYP1A2 genotype (46, 47). In people with caffeine-induced insomnia caffeine clearance is significant slower than in people without sleep onset problems after drinking coffee in the evening (48).
Melatonin and its relation to ASD: Low levels of melatonin are found in the majority of individuals with ASD (18, 21, 49, 50). Studies on genes involved in melatonin metabolism have long been focussed on the melatonin synthesis pathway, mainly of ASMT. Interestingly no studies were performed on the melatonin catabolism by CYP1A2 until in 2013 a possible relationship between slow CYP1A2 polymorphisms in slow melatonin metabolisers and ASD was proposed (51). Even though ASD frequently is associated with low melatonin levels and suspicion exists for an involvement of the ASMT gene in the etiology of autism, several studies could not find any evidence for a significantly higher incidence of abnormalities of the ASMT gene in ASD (52, 53). And if so, ASMT abnormalities were also found in healthy controls.
Another potential link between melatonin and ASD etiology was proposed by us in an earlier report (51). In a study in 15 consecutive patients with ID and sleep problems, presenting with disappearing effect of melatonin treatment after an initial good response, extremely high (>50 pg/ml) melatonin levels at noon and 04:00 pm were found. We hypothesized that the disappearing effectiveness of melatonin treatment was associated with slow metabolisation of melatonin due to a SNP of CYP1A2 and analysed DNA for a CYP1A2 SNP. In 8 patients a SNP was found. Because 13 of 15 patients with CYP1A2 slow metabolism were diagnosed with ASD, we hypothesized that low melatonin levels in autism are caused by a yet unknown mechanism in which CYP1A2 slow metabolism is involved, and that low melatonin levels are possibly part of the mechanisms that cause ASD, rather than that low melatonin levels are the result of ASD.
The potential mechanism was investigated by Veatch et al. (2014) (54). They found a higher frequency of dysfunctional ASMT polymorphisms than earlier reported (52, 55-57). They also found substantially higher rates of CYP1A2 polymorphism in ASD patients than in our previous report (51). Based on the linkage disequilibrium structure across SNPs in ASMT and CYP1A2 in their subset of individuals, Veatch et al. predicted a potential gene–gene interaction (20).
The embryo and fetus do not secrete melatonin and are totally dependent on maternal melatonin (58, 59). Melatonin secretion begins no earlier than approximately 9 – 15 weeks after birth, depending of gestational age (59). Maternal melatonin passes the placental barrier easily and the fetal circadian melatonin rhythm follows the maternal rhythm (60). Maternal melatonin is also passing easily into breast milk, and levels are a reflection of the circadian melatonin rhythm of the mother (61). ASMT and AANAT gene activity and melatonin levels in disturbed pregnancies (intra uterine growth retardation and in severe preeclamptic women) are lower than in normal pregnancies (62, 63).
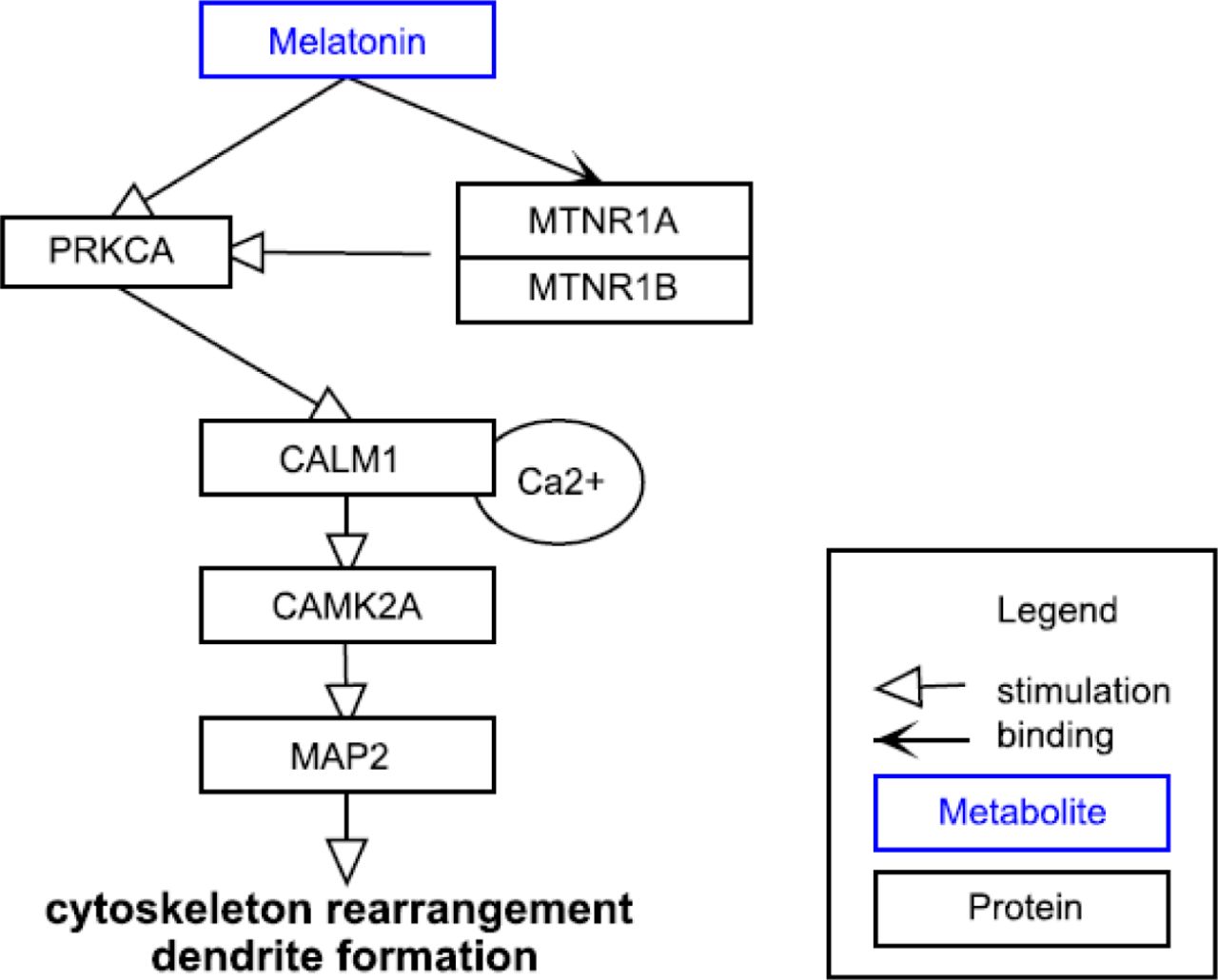
Melatonin is important for normal neurodevelopment and embryonic growth and helps regulate synaptic plasticity (18, 19, 58, 64). Melatonin is implicated in neural growth and synapse formation and it can modulate neurite outgrowth in cultured neuronal cells (56). Melatonin guides the differentiation of neuronal stem cells (in vitro) as follows (64): It stimulates the mRNA and protein levels of TH, which is a marker for dopaminergic neurons. Dopaminergic neurons are neurons which are able to release dopamine and have therefore depending on their location in the brain 1) a high impact on regulation of cognitive function, emotional behaviour, and reward system or 2) regulate motor function. In vivo, melatonin increases dendritogenesis (dendrite formation, enlargement and complexity) in the hilar and mossy neurons of hippocampus in rats (65). The accompanying in vitro results indicate that melatonin triggers CALM which stimulates MAP2 via CAMK pathway (Figure 2). Melatonin increases the total cellular level of CALM and fosters the translocation from cytosol to membrane/cytoskeleton interfaces (65).
Although the genetic basis of ASD is not disputed, no single chromosomal abnormality is responsible for >1% of ASD cases. Low melatonin levels are found in the majority of ASD cases, but is regarded as co-morbidity and not considered as part of ASD etiology. All data mentioned above can be linked directly or indirectly with melatonin. As ASD is already present at birth and the fetus does not produce melatonin, low maternal melatonin levels should also be involved. Low melatonin levels are not only caused by disturbances in melatonin synthesis, but are likely also related to low CYP1A2 activity, in view of the finding of CYP1A2 gene SNPs in ASD patients and a potential gene-gene interaction between CYP1A2 and ASMT. We therefore measured 6-SM in urine of mothers of a child with ASD that attended our sleep clinic, and asked for parental coffee consumption habits, as these are known to be related to CYP1A2 activity.
2. Material and methods
2.1. Participants
Approval was obtained through the Ethical Committee of Maastricht University (The Netherlands). Mothers of patients with chronic sleep disturbance, who visited our sleep centre for people with ID between January and July 2015, were asked to participate if her child was diagnosed with ASD. Mothers of children with a genetic ID syndrome and an autistic phenotype were also asked to participate. Out of 97 children seen in this period, 18 were not diagnosed with ASD and 13 mothers were not eligible for several other reasons (five children were adopted or foster child, four children were only seen once, with three mothers we had severe language barriers, and one mother used melatonin supplementary on a daily basis). So, 66 mothers consented to participate. The control group consisted of 15 women who work in the building where the sleep centre is located. These women all had healthy children without signs of autism and siblings of controls also did not have signs of autism.
2.2. Procedure
After explaining purpose and procedure of the study, participants received an introductory letter with information of the study and an informed consent letter to be returned by return paid mail. After receiving the signed consent letter, a questionnaire, together with a shipping container for a urine sample and instruction sheet was send to participants. Participants were instructed to empty the bladder at bedtime. Time of voiding at bedtime and first voiding in the morning were recorded. The first morning urination, plus any urine voided during the night, was collected and the total urine volume was recorded. A sample of the total urine was poured into a plastic bottle and sent to the sleep centre where it was frozen and stored at minus 20°C until measurements took place. The questionnaire (see supplementary data) contained a total of 17 questions, namely six questions on age, weight, medication use, and number and age of children (oldest and youngest child), seven questions based on the Insomnia Severity Index (66) and four questions about the use of coffee.
2.3. 6-SM concentration measurement
6-SM in urine was analysed by isotope dilution mass spectrometry using online solid phase extraction in combination with liquid chromatography and tandem mass spectrometry (XLC-MS/MS) (Van Faassen et al., manuscript in preparation). In short, 100 µl of urine was used for the analysis, deuterated 6-SM was used as internal standard. Mean intra- and inter-assay coefficients of variation were below 6.0%. Quantification limit for 6-SM was 0.2nmol/l. As advanced parental age at birth is a factor associated with autism risk (12) and melatonin levels decrease with 2.7% per year (67), we recalculated the urinary 6-SM results in mothers with an ASD child, taking into account the age at which they received this child. In 6-SM levels in controls, we based recalculating on their age at receiving their first-born child. In case controls had more than one child, we took the mean of their age receiving their oldest and their youngest child.
2.4. Statistical analysis
All statistics were performed using IBM SPSS Statistics 20 (IBM, SPSS Inc., Chicago, IL).T-tests for independent samples were conducted to test differences between ASD-groups and controls. To test for differences on sleep questions between the subgroups non-parametric tests (Fisher’s exact test and Mann-Whitney test) were used because of low cell frequencies and/or not normally distributed data. To test the relationship between sleep and melatonin bivariate correlation (Kendall’s tau-b) was used. Statistical significance was accepted at p < 0.05.
3. Results
Of 66 mothers, who consented to participate in this study, one withdrew without specifying at reason. In five cases the urine samples did not arrive. So urine samples of 60 mothers were analysed. Their mean age was 42.9 (± 5.7) years. The mean age of 15 women in the control group was 44.3 (± 9.7) years. No significant differences with respect to age (p = 0.486) were found between participant and control group. However, mean age of mothers of an ASD child at time of delivery was higher (31.2 years) than the mean age control mothers received their children (28.1 years). This difference was significant (p = 0.01). Other demographic data are shown in Table 1.
Data on etiological diagnoses of children with ASD are presented in Table 2. It is noteworthy that there are substantial groups with Angelman syndrome or Smith Magenis syndrome in the group studied. It should however be noted that both syndromes occur relatively frequently (prevalence 1:20.000) and that they are well known for their sleep problems (68, 69).
In this study we found a significant lower urinary 6-SM excretion in mothers of a child with ASD compared to control mothers (p = 0.012). Group means and SDs of the excretion of 6-SM excretion in the urine in all groups are given in Table 3. Melatonin levels are known to decline after the age of 20 year with 2.7% (67) each year and ages of mothers and children were asked for, we were able to recalculate 6-SM excretion levels in each mother to the year their child was born. Comparing urinary 6-SM levels in the year their child was born is more relevant, given our hypothesis that low melatonin levels in pregnancy are a risk factor for getting a child with ASD. As shown in Table 3 differences in recalculated 6-SM excretion levels between mothers of a child with ASD compared to control mothers were significant (p = 0.002).

Looking into subgroups, only mothers in the group with other genetic disorders and with Angelman syndrome had a significant lower urinary 6-SM excretion compared to control mothers (p = 0.012 resp. 0.042) (Table 4). Urinary 6-SM levels in mothers of a child with Smith Magenis syndrome also were lower than in controls, but these differences approached significance (p = 0.05). Five mothers of a child with ASD also had another child with ASD and/or an ID. These children, however, were not treated for sleep problems in our sleep centre. One child was known with Prader Willi syndrome, being genetically of paternal origin. Melatonin levels in the other four mothers were lower (mean 7.05 ± 2.86) compared to the other mothers with one child with ASD (mean 11.61 ± 5.57, p = 0.084).
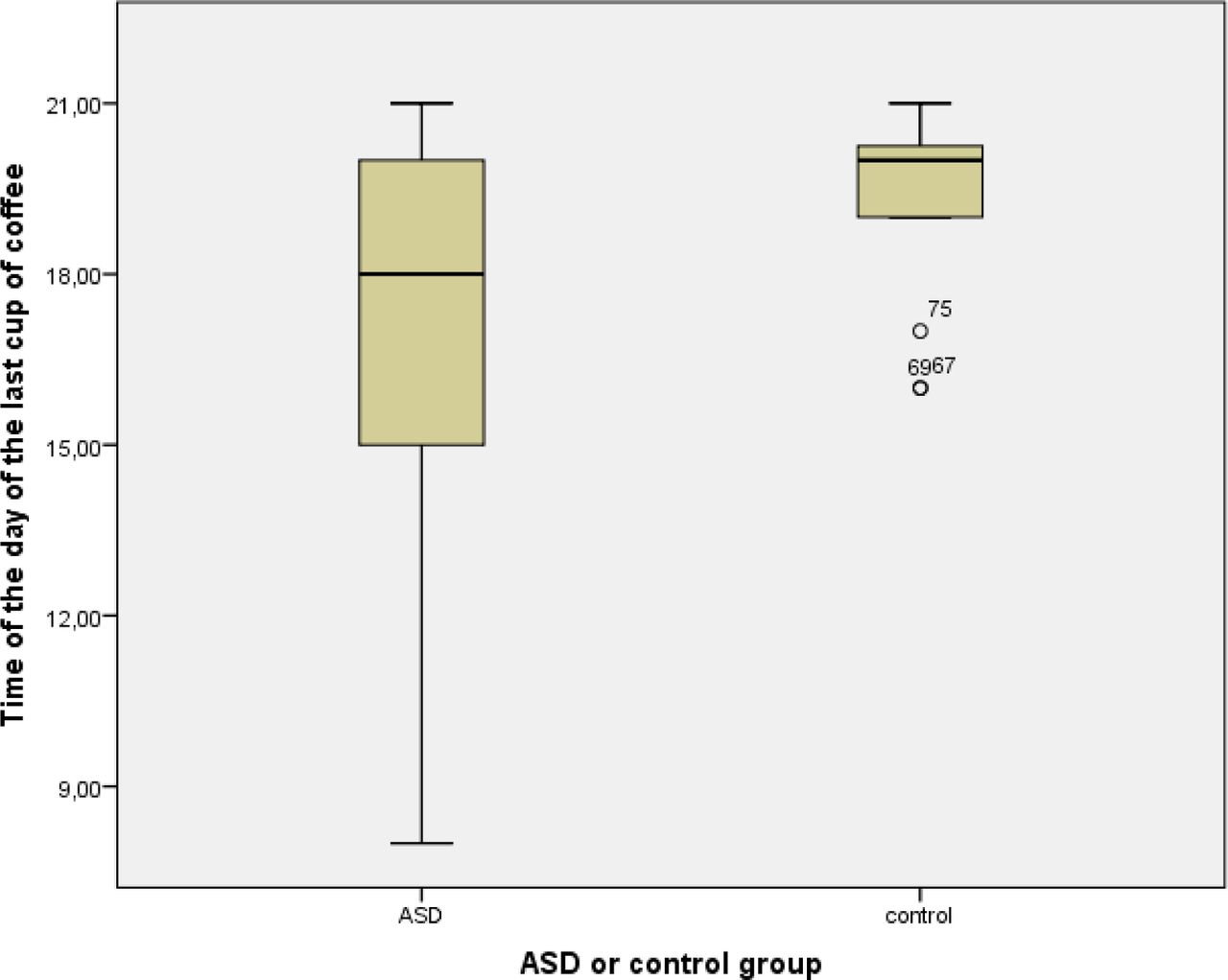
The number of mothers drinking coffee does not differ between mothers with an ASD child and controls (p = 0.375). However, more mothers with an ASD child do not drink coffee after 06:00 pm than mothers in the control group (p = 0.018). Time of the last cup of coffee was later in control mothers than in mothers with an ASD child (07:10 pm, resp. 05:05 pm: p = 0.058) (Figure 4). Urinary 6-SM levels in mothers that do not drink coffee after 06:00 pm were significantly lower (13.79±9.08) than in mothers that do drink coffee after 06:00 pm (10.39±4.90) (p = 0.039). Also more fathers with an ASD child do not drink coffee after 06:00 pm (p = 0.020).

Of 60 mothers who’s urine samples were analysed, one did not fill in the questions on sleep. So questions on sleep of 59 mothers of the patient group and 15 mothers of the control group were analysed. If a mother indicated that she did not experience difficulty with either falling asleep or staying asleep and did not have a problem with waking up too early she was asked to skip three questions and no total score on the Insomnia Severity Index (ISI) could be calculated. A cut-off score of 10 was used to indicate clinical significant insomnia (70). Of the women with an ASD child (n = 33) 67% met the cut-off score of the ISI compared to 29% of the women in the control group (n = 7). This difference was not statistically significant (p = 0.094, Fisher’s exact test).
In order to compare sleep between all mothers of the patient and the control group we also calculated a shortened ISI (sISI) based on two questions (severity of sleep problems and satisfaction with current sleep pattern) filled in by all mothers (n = 74). Higher scores indicate more severe sleep problems and less satisfaction with current sleep pattern. Scores on sISI in patient group (Mdn = 4) did not differ significantly from the control group (Mdn = 2), U = 542.50, z = 1.366, p = 0.175, r = 0.16.
Furthermore no correlation was found (tau = 0.00) between sISI and the excretion of 6-SM in the urine (p = 0.996, two-tailed).
4. Discussion
In this study we found a significant lower 6-SM excretion in mothers of a child with ASD. This difference cannot be explained by the difference in their age at birth of their child. ASD is caused by a mixture of rare and common genetic variation, as well as environmental factors. Melatonin deficiency can play a role in all three of these factors. Epidemiological studies have identified several environmental factors that increase ASD risk. Almost all of the known prenatal, perinatal and neonatal ASD risk factors, found in two meta-analyses (Gardener et al. (2009) (12) and Gardener et al. (2011) (13)) can directly or indirectly be linked to melatonin deficiency (see supplementary files). Additionally, low CYP metabolisation indicated by coffee drinking behaviour was found to be an early indicator. A correlation between ASD and low melatonin levels/slow CYP metabolisers has been found earlier in different experimental settings (see Table 5).
Finding a significant lower urinary 6-SM excretion in mothers of a child with ASD compared to control mothers, combined with the neurotrophic properties of melatonin, total dependence of the fetus on maternal melatonin and a link between already established ASD risk factors and melatonin, supports our hypothesis that low melatonin levels may be the shared “environmental factor” postulated by earlier twin studies (11).
Strong evidence points at a substantial contribution of common genetic variation to ASD (10). Low CYP1A2 activity is a common heritable genetic variation which can cause low ASMT transcript production (20) and by that low melatonin levels. Additionally, the SNP reduced enzyme kinetics of CYP1A2 may show substrate saturation or inhibition effects at much lower concentration as wild type. For CYP1A2 and melatonin this has not been measured before and remains speculative. The dual effect of melatonin (neurotropic properties and protection against oxidative DNA damage), provides an explanation for multiple questions about the complex underlying genetic process in ASD. This may explain results of a recent study in quartet families with ASD (parents and two ASD-affected sibs) whole-genome sequencing, which revealed that many (68.4%) of the affected siblings did not share the same mutations, but carried different ASD-relevant mutations (78). It also may explain why in 40% of persons with intellectual disabilities autism is present, and at the same time 70% of people with autism also has an intellectual disability (2). Our finding that in mothers of a child with ASD, who also have another child with ASD and/or an ID, urinary 6-SM excretion was significantly lower compared to mothers with one child with ASD (p = 0.023), is in line with these findings. Low parental melatonin levels can act on two seemingly independent levels: 1) directly by causing disturbances in brain development of the child and 2) indirectly by increasing risk of DNA damage in maternal and paternal germ cells. If melatonin levels in the child with autism are also low, this contributes to greater influence during the further development of the brain.
In a large study, aimed to find normative data on the melatonin excretion in urine, overnight urinary melatonin excretion values in healthy individuals ranged from 0.023 to 0.842 nmol/l and correlated inversely with age (79). There is a decrease in time with 25% in healthy adults aged 36 to 50 years, compared to those in the age group 20 to 35 years (67, 80). Kennaway et al. (67) calculated the yearly decline in melatonin plasma peak levels at night between young age groups (21 to 33 years) and old age groups (49 – 85 years) to be 2.7%, based on data in 11 studies. Melatonin levels did not have a Gaussian distribution, but showed a bimodal distribution, distinguishing two groups of individuals: low and high melatonin excretors. This is the reason why we decided to offer the measured and the recalculated data (Table 3) taking the postulated age dependant melatonin decrease into account.
Caffeine and melatonin are primarily metabolised by CYP1A2. We found that significantly more mothers with an ASD child than in the control group do not drink coffee after 6:00 h pm (p = 0.034), mostly in order to prevent coffee induced insomnia, and that 6-SM levels were also significantly lower than in mothers that do drink coffee after 6:00 h pm (p = 0.039). This is in line with the suggested mechanism that reduced CYP1A2 metabolic activity is connected with lower levels of ASMT transcript production (54) and supports our hypothesis that CYP1A2 slow metabolism due to a CYP1A2 SNP is involved in low melatonin levels in autism ethiology (51). As earlier studies already have shown that caffeine clearance by CYP1A2 is significant slower in people with caffeine-induced insomnia (48), and that metabolic activity of CYP1A2 is significantly inversely related to coffee intake (46, 47), we hypothesize that coffee drinking habits can serve as an indicator for low CYP1A2 activity as well as for low melatonin levels.
Our finding that significantly more fathers with an ASD child than in the control group do not drink coffee after 6:00 pm suggests that they also could be a CYP1A2 slow metaboliser and by that have low melatonin levels. This finding is important and can explain why 76% of the de novo mutations, found in a study in quartet families with ASD (parents and two ASD-affected sibs), were of paternal origin (78).
The limitation of our study is that we did not measure CYP1A2 activity, but relied on information on parental coffee consumption habits, as these are known to be related to CYP1A2 activity, and that our control group is not sufficiently large to draw firmer conclusions. Our pilot study needs to be replicated on a much larger scale. It should involve melatonin levels and CYP1A2 activity measurements in ASD patients, as well as both of their parents. The outcomes of this study implicate that in yet to be determined risk groups, melatonin supplementation may be a logical consequence. Nevertheless, melatonin supplementation in pregnant and new-borns that are at risk may possibly enhance brain development but needs careful investigation of possible risks.
5. Conclusion
This is to our knowledge the first experimental evidence of low melatonin levels in mothers being a risk factor for child ASD. It is a comprehensive theory on ASD etiology in which genetic and environmental factors are combined with already known ASD risk factors and results of own melatonin research. It leads to the intriguing hypothesis that low melatonin levels, caused by low CYP1A2 activity – which is again early detectable by coffee drinking behaviour – are a risk factor for ASD as well as ID in their offspring. If so, it is more intriguing that this may lead to policies to detect future parents who are at risk and to treatment strategies to reduce ASD and ID risk.
Competing interests
The authors declare that they have no competing interests.
Additional data
Questionaire (original in Dutch)
Sup. Table 1. ASD risk factors

Onderzoek melatoninespiegels in nacht-urine bij moeders
Persoonlijke informatie

Beoordeling van uw eigen slaap
Gebaseerd op de Insomnia Severity Index (Bastien, Vallières, Morin, 2001)
Heeft u moeite met in slaap vallen, doorslapen en/of problemen met te vroeg wakker worden?
nee (sla de vragen 4, 5 en 6 over)
ja (vul de volledige vragenlijst in)
Beoordeel aub de ERNST van uw huidige slaapproblemen (in de laatste 2 weken)
Hoe TEVREDEN of ontevreden bent u over uw huidige slaappatroon?
In welke mate vindt u dat uw slaapprobleem INVLOED heeft op uw dagelijks functioneren (zoals vermoeidheid overdag, al dan niet goed kunnen functioneren op werk of bij dagelijkse bezigheden, concentratievermogen, geheugen, stemming, etc)
Hoe OPVALLEND voor anderen is het volgens u dat uw slaapprobleem een negatieve invloed heeft op uw kwaliteit van leven?
In welke mate maakt u zich ONGERUST over uw slaapprobleem?
Als u iets wilt toevoegen over uw eigen slaap, dan kunt u dat hier doen.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Medicijngebruik
8. Wilt u hier aangeven welke medicijnen u gebruikt? (naam, sterkte, aantal per dag)
Koffiegebruik
9. Drinkt u (‘gewone’ cafeïnehoudende) koffie?
Ja
Nee
Indien Nee, wat is daarvan de reden (meerdere antwoorden mogelijk)?
Dat lust ik niet
Daar krijg ik last van (zoals hartkloppingen, trillerig gevoel etc)
Dan krijg ik last van mijn maag
Anders, namelijk ………………………………………………
10. Indien u (overdag) koffie drinkt, drinkt u dan ook’s avonds koffie?
Ja
Ja, maar’s avonds alleen cafeïne vrije koffie
Nee
Niet van toepassing (Ik drink overdag ook geen koffie)
11. Indien u’s avonds geen koffie drinkt, of alleen cafeïne vrije koffie, wat is daarvan de reden?
Niet van toepassing, ik drink’s avonds gewoon koffie
Ik kan niet goed in slaap vallen als ik koffie drink
Anders, namelijk ………………………………………………………
Hoe laat drinkt u uw laatste kop (cafeïne bevattende) koffie?
Om ……… uur
Niet van toepassing (ik drink geen koffie)
Hartelijk dank voor het invullen van deze vragenlijst.
Acknowledgements
This study was supported by a research grant from ‘s Heeren Loo Zorggroep Steunfonds. We thank the mothers from our patients and women working at ‘s Heeren Loo Wekerom for their participation in this study. We also thank Lars Eijssen and Egon L. Willighagen for helpful discussions and Martijn Faassen for contributions in laboratory measurements.
Abbreviations
- ASD
- autism spectrum disorder
- ID
- intellectual disability
- 6-SM
- 6-sulfatoxymelatonin
References




