

Abstract
The site frequency spectrum (SFS) has long been used to study demographic history and natural selection. Here, we extend this summary by examining the SFS conditional on the alleles found at the same site in other species. We refer to this extension as the “phylogeneticaNy-conditioned SFS” or cSFS. Using recent large-sample data from the Exome Aggregation Consortium (ExAC), combined with primate genome sequences, we find that human variants that occurred independently in closely related primate lineages are at higher frequencies in humans than variants with parallel substitutions in more distant primates. We show that this effect is largely due to sites with elevated mutation rates causing significant departures from the widely-used infinite sites mutation model. Our analysis also suggests substantial variation in mutation rates even among mutations involving the same nucleotide changes. We additionally find evidence for epistatic effects on the cSFS; namely, that parallel primate substitutions are more informative about constraint in humans when the local sequence context is similar than when there are other nearby substitutions. In summary, we show that variable mutation rates and local epistatic effects are important determinants of the SFS in humans.
Introduction
The distribution of allele frequencies across segregating sites, commonly referred to as the Site Frequency Spectrum (SFS), is a central focus of population genetics research as it can reflect a wide range of evolutionary processes, including demographic history as well as positive and purifying selection [1-8]. Until recently, the SFS was usually measured in samples of tens or hundreds of people, but advances in sequencing technology have enabled the collection of sequence data at much larger scales [9-14]. Notably, the Exome Aggregation Consortium (ExAC) recently released high quality, exome-wide allele counts for over 60,000 people [12].
Large sample sizes are valuable because they make it possible to detect many more segregating sites, and to estimate the frequencies of rare variants. For example, the recent dramatic expansion of human populations leaves little signal in the SFS in small samples [15], but is readily detected in large samples, where there is a huge excess of low frequency variants compared to model-predictions without growth [13,14,16,17]. Similarly, large samples enable the detection of deleterious variants that are held at very low frequencies by purifying selection [18-20].
In this paper, we extend the SFS by considering the SFS conditional on the observed alleles at a given site in other species (specifically, other primates in our analysis). Our original motivation was that this could allow us to measure the effects of sequence context on the selective constraint of missense variants. In general, sites with strong levels of average constraint across the mammals tend to be less polymorphic within humans [16,21], but to the best of our knowledge, there has not been extensive consideration of the joint distribution of the substitutions across other lineages and the human SFS. In particular, we hypothesized that if an identical substitution has occurred independently in a closely related species—e.g., in a great ape—then this is strong evidence that the same variant is unlikely to be deleterious in humans. However, an identical substitution in a more distantly related species may be much less informative, as substitutions at other positions within the same gene may change the set of preferred alleles due to epistatic interactions [22-26] (Figure 1). For example, it has been shown that, in a handful of cases, likely disease-causing variants in humans are actually wildtype alleles in mouse, presumably rendered harmless by parallel substitutions at interacting positions [23].

If an identical substitution occurred independently in a closely related species, then the variant is unlikely to be deleterious in humans. An identical substitution in a more distantly related species may be less informative because sequence divergence at interacting sites may change the set of preferred alleles, and hence the selective constraint at the site.
As we show below, the human SFS varies greatly depending on patterns of substitutions in other species. In part this does appear to be due to the accumulation of epistatic effects on more distant lineages; however a more important factor is mutation rate variation across sites. Under the widely-used infinite sites model, the SFS is independent of mutation rate; but in the ExAC dataset we observe a clear breakdown of this model. Mutation rates are known to vary across sites due to a variety of different mechanisms, leading to differences between CpGs, transitions and transversions, as well as additional effects that correlate with broader sequence context, replication timing, transcription, recombination rate and chromatin environment [27-30]. We show here that mutation rates are much more variable than generally appreciated, and that rates at some sites are high enough to generate substantial deviations from infinite sites predictions. A bioRxiv manuscript investigating the ExAC dataset [12,31] also recently reported that the SFS varies substantially across mutation types, and also noted that this implies departures from the infinite sites model, especially for CpGs.
In summary, our results suggest that there may be more variation in mutation rates across sites than is generally appreciated, and further that the infinite sites model provides a poor fit for population genetic analyses in large modern data sets. We also show a significant, albeit smaller, role for epistatic effects in shaping the cSFS.
Results
To investigate the properties of the human cSFS, we combined exome sequence data from 60,706 humans from ExAC version 0.2 [12,32] and orthologous reference alleles for 6 nonhuman primate species from the UCSC genome browser [32,33]. After applying several filters (see Materials and methods for details) we were left with 6,002,065 single nucleotide polymorphisms (SNPs) for which we had orthologous data in at least one nonhuman species.
We examined how the human SFS changes as we condition on various divergence patterns observed in primates. There are many possible ways to condition on variation across the nonhuman primates. We focus here on sites that are variable in human only (denoted human-private), as well as sites where exactly one other species carries the human minor allele (and all others match the human major allele); see Figure S1 for an alternative conditioning based on the most closely related species carrying the human minor allele. Throughout, we assume that the observation of the human minor allele as the reference allele in another primate implies that the mutation arose independently and fixed in that primate. This assumption may be violated for a small fraction of SNPs when comparing human to our closest relatives (notably, chimpanzee and gorilla [34]), but the overall patterns that we report here are maintained when we consider more distant species for which shared ancestral polymorphism is unlikely (see Supplementary Material for further discussion). The SFS presented here, unless otherwise stated, are constructed using minor allele frequencies.
Henceforth, we will use the term substituted species to refer to the single species in which the human minor allele is observed, and the corresponding species cSFS to refer to the human SFS conditional on a substituted-species divergence pattern. For example, “substituted-orangutan” refers to human variants for which the human minor allele is observed in orangutan, and the human major allele is observed in all other primates; “orangutan cSFS” will refer to the human SFS at these sites (Figure 2A).

(A) An example of the phylogenetic conditioning that defines what we denote as “substituted-orangutan” sites. (B) The cumulative distribution functions (CDF) of orangutan cSFS (i.e. the SFS of substituted-orangutan sites), and the SFS of phylogenetically conserved sites. The cSFS are more skewed towards common variants than the SFS of conserved sites. These skews are much more pronounced than in the comparison of synonymous and nonsynonymous sites. (C) The more closely related the substituted-species, the higher the skew of the cSFS towards common variants (only nonsynonymous mutations shown). The inset shows the rare variants slice of the CDF for each species, for both synonymous and nonsynonymous variants.
There were 5,286,937 human-private sites in the data set, and the number of substituted-species sites ranged from 22,209 (substituted-chimpanzee) to 66,254 (substituted-gibbon).
Figure 2B shows a comparison of the human-private cSFS and the orangutan cSFS for nonsynonymous and synonymous sites, respectively. Within each cSFS class, the nonsynonymous spectrum has more rare variants than the synonymous spectrum, as expected given that nonsynonymous variants are more likely to have deleterious effects. Secondly, if we compare the human-private versus orangutan cSFS at nonsynonymous sites, we see more rare variants in the human-private set. Again, this matches expectations, as the presence of a parallel substitution in orangutan implies that a substitution at this position is tolerated.
However, we were surprised to see that substituted-orangutan synonymous sites also segregate at much higher frequencies than both synonymous and nonsynonymous human-private sites. Taken at face value, this would seem to imply that a large fraction of synonymous sites are functionally constrained. While it is known that some synonymous sites play roles in functions such as splicing [35,36], it is generally believed that most synonymous variants in mammals are effectively neutral. We were thus curious to understand whether this result is primarily driven by a surprising degree of constraint at synonymous sites, or by some other factors.
Looking more broadly across the primates, we observed a clear trend of cSFS across substituted species (Figure 2C): the more closely related the substituted species, the greater the skew towards high frequency variants. Again, this is hard to explain under a simple model of selective constraint because, at least under a parsimony approximation, each cSFS has the same number of parallel fixations on the primate tree (i.e., 1). This trend is most easily noticeable in the fraction of rare variants (defined here, arbitrarily, as singletons and doubletons; Figure 2C, inset). In the following sections, we try to understand the factors driving these observations.
Effect of mutation rate variation on the human SFS
In this section we consider whether mutation rate variation may contribute to the observed trend across cSFS. Under the standard infinite sites assumption, the SFS is independent of mutation rate. However, we conjectured that in the very large sample size of ExAC, infinite sites may no longer be a good model for the data [12].
To examine this, we stratified the human SFS by mononucleotide mutation types (as well as the dinucleotide mutation type CpG->TpG), for which there are well-characterized differences in mutation rates. For this analysis we focused on intronic sites, to reduce potential effects of selective constraint. We found that the different mutation types have significantly distinct spectra. The fraction of rare variants among CpG->TpG mutations (36%) was roughly half that of non-CpG transitions (71%, see Figure 3A). Similarly, non-CpG transitions have higher mutation rates than transversions and indeed, the SFS for transitions is also skewed towards higher frequencies than transversions (Figure 3B). Overall, the fraction of rare variants in the subsample of Europeans was significantly negatively correlated with germline mutation rates (Weighted linear regression p = 4.9 · 10‒6 and see figure 4A).

All panels exhibit the SFS of derived alleles constructed from intronic sites. Except in Panel D, which is strand-specific, the notation of mutation types refers to mutations on either strand (e.g., A->C indicates an A to C change on either strand). (A) SFS stratified to mononucleotide mutation types, and CpG transitions. The fraction of rare variants in CpG transitions is nearly half that of other mutations. (B) Focusing on non-CpG mutations, transitions have an SFS significantly skewed towards common variants compared with transversions. (C) Stratification to coding and template strands revealed differences between the two for some mutation types, suggesting transcription-associated mutational mechanism also affect the SFS. CpG mutations excluded from the analysis in this panel. Points show means, lines show 95% confidence interval computed with nonparametric bootstrap. (D) SFS across chromatin states. Chromatin states in H1 human embryonic stem cells were inferred by ChromHMM. The chromatin state exhibits substantial association with the fraction of rare variants in CpG mutations, and modest association in other mononucleotide mutation types. Points show means, lines show 95% confidence interval computed with nonparametric bootstrap.

All panels exhibit the unfolded SFS (i.e., constructed using the derived alleles) of intronic sites. (A) Fit of mutational models to observed SFS. The x-axis shows previously estimated de-novo germ-line mutation rates [29]. These data illustrate that the fraction of rare variants is strongly negatively correlated with germ-line mutation rates. Lines show expectations under various mutational models: yellow – infinite sites model (SFS independent of mutation rate); teal – Jukes Cantor finite-sites model; red – Jukes-Cantor model with within-mutation-type variation (i.e., variation beyond mutation rate heterogeneity due to the type of mutation in sequence). (B) SFS subsampling and the effect of mutation rate. Dots show the fraction of rare variants in the full sample SFS of the European population in ExAC. Lines show the expected fraction of rare variants after subsampling to smaller numbers of individuals. In large samples, the SFS of CpG and non-CpG sites are very different. In smaller samples, these differences shrink. In the shaded region, the trend across mutation types is changed (the inflection point is indicated by an arrow); with these sample sizes, CpG transitions exhibit more rare variation than non-CpG transitions.
As an additional test of whether mutation rate affects the fraction of rare variants, we turned to sites in transcribed regions. It is known that in such regions, A->G and A->T mutations occur at higher rates on the template (non-coding) strand than on the non-template (coding) strand, due to the effects of transcription-coupled repair or other transcription-associated mutational asymmetries [37-39]. Indeed, as predicted from these rate asymmetries, we observed a 1% difference between the template and the coding strands in the fraction of rare variants in introns (t-test p < 2.2 · 10‒16 for A–> G,p = 6.0 · 10‒7 for A->T). C->T mutations also exhibit a small but significant difference (t-test p = 0.3 · 10‒3) between the strands, even though, to our knowledge, no previous work has observed this asymmetry (Figure 3C).
Similarly, we hypothesized that the SFS at CpG sites might also depend on chromatin environment (Figure 3D). Specifically, CpG sites experience high mutation rates only when they are methylated [40-43]. We thus examined the effect of chromatin states in H1 human embryonic stem cell lines (inferred by ChromHMM [44]) on the SFS across different mutation types. Methylation levels are expected to be low in active regions including promoters and enhancers and high in repressed regions such as heterochromatin. Indeed, we find large differences in the SFS at CpGs, consistent with this expectation: i. e., fewer rare variants in heterochromatin, where methylation levels are high. In contrast, the other mononucleotide mutations showed only modest variation across chromatin states. The SFS variation patterns across chromatin states, and across strands, illustrate that heterogeneity in mutation rates could exist within mutation types, and that it has a substantial effect on the SFS.
These observations on mutation rate variation led us to conclude that the infinite-sites model provides a poor fit for these large-sample human polymorphism data. We therefore investigated finitesites mutational models. Below, we describe the fit of various mutational models while using previously-inferred population genetic models of European demography. In particular, we eventually used a modified version of the demography suggested by Nelson et al. [14] (see Materials and Methods for the other demographic models considered).
We asked how well different finite-sites models account for the observed relationship between de-novo mutation rates and the SFS. We assumed a demographic model that fits well for sites with the lowest mutation rates. First, we considered the Jukes-Cantor model, which uses a 4 × 4 uniform mutation transition matrix [45]. Despite our observation that the fraction of variants tracks closely with mutation rate, we were surprised to find that the finite sites model barely improved the fit to the SFS across the range of estimated mutation rates (Figure 4A). In our simulations, the probability of obtaining more than 1 mutation on the genealogy of a segregating site is low enough that the finite sites SFS is similar to the infinite sites SFS, even at the relatively high mutation rate estimated for CpGs.
We hypothesized that we might achieve a better fit if some sites have higher intrinsic mutation rates than the mean for the particular nucleotide change at that site. We therefore augmented the JukesCantor model by incorporating additional variation in mutation rate across sites belonging to each mutation type (see Materials and Methods). The augmented Jukes-Cantor model with within-mutationtype variation fitted the data well, including the large difference in SFS between CpG and non-CpG sites (Figure 4A). The augmented model suggests that 3% of mutations within a mutation type have a mutation rate of over 5 times the mean rate for that type.
It is natural to wonder whether the effect of recurrent mutations has any implications for smaller samples. Small samples have the obvious disadvantage of limiting the resolution of analysis. For example, in demographic inference, larger samples are essential for detecting the signal of recent rapid growth of the human population [17,46,47]. Interestingly, we found that samples much smaller than ExAC may also create an unappreciated bias, as we describe next.
We examined the effect of subsampling the SFS of the European ExAC sample to a smaller number of individuals (see Supplementary material). SFS differences between non-CpG transitions and transversions remained roughly the same, even with a sample of a few hundred people. Conversely, the difference between CpG and non-CpG sites changed dramatically for smaller samples. For samples smaller than 1500 people, there appears to be more rare variation in CpG than non-CpG transitions (Figure 4B, Figure S3). This finding exemplifies a pitfall of analysis with a sample size much smaller than the population size: if one category of sites has substantially more rare variation in the population than a second category, the sample SFS may actually exhibit more rare variation in the second category. Large samples are therefore not only important for better resolution and reduced variance but are also essential for unbiased comparison of categories of sites.
Finally, we returned to the species trend across cSFS that we described earlier (Figure 2C). Given the previous observations on SFS differences between mutation types, we asked whether the trend across substituted-species cSFS could be explained by differing compositions of the various mutation types. Indeed, we found that more closely related substituted-species categories are enriched for mutations that are associated with higher rates (Figure S2). Importantly, the fraction of CpG transitions is strongly negatively correlated with the fraction of rare variants across substituted-species category (Pearson r = –0.997,p = 9.7 · 10‒6 for nonsynonymous mutations; r = –0.999, p = 9.9 · 10‒7 for synonymous mutations). This provides an explanation for the observed correlation between the relatedness of the substituted species and the skew of its cSFS towards common variants. We next asked whether additional causes beyond mutation rate variation might also contribute to the species trend across cSFS.
Effects of epistasis on the human SFS
A second process that could contribute to the observed pattern of cSFS differences across substituted-species is fitness epistasis [22-24,48]. It is well-known that sites that are functionally important in humans tend to be relatively conserved across the mammals [49]. However, this is not a necessary nor sufficient condition for predicting functional sites in humans, and there are some counter-intuitive examples of disease-causing mutations in humans that are annotated as the reference allele in mouse [23]. It is presumed that such cases may be explained by parallel changes at other interacting amino acids that alter the structural context of the relevant site in mouse.
We thus hypothesized that the observation of a human variant in a closely related species provides suggestive evidence that the allele may be benign for humans as well, and that this evidence would be stronger the more closely related the other species, because more-closely related species would have less time to accumulate additional epistatic interactions. An effect of this type could contribute to the trends observed in Figure 2C. In this section we test for evidence of epistatic effects of this kind.
To this end, we used a logistic regression model (see Materials and Methods). We first examined whether the probability of the variant being rare is associated with the relatedness of the substituted species. A model that included only the relatedness of the substituted species showed a perfectly correlated ordering of the two (Figure 5A). We then turned to examine whether this correlation persists after controlling for mutation rate differences between substituted-species categories. Since we do not have a direct way of measuring local mutation rate, we used the mutation type as a proxy instead. Note, however, that the distribution of other factors responsible for mutational heterogeneity could also differ across substituted-species categories, and this could contribute to differences in the fraction of rare variants (see further discussion in Supplementary Materials).

The figure shows logistic regression coefficient estimates. (A) Estimates from a simple logistic regression to the substituted species. The trend is partly due to mutational composition differences between substituted-species categories. To test whether the trend is driven solely by mutational rate differences, we estimate coefficients in a model including the variation explained by (B) mononucleotide mutation type, and (C) combinations of focal mononucleotide mutations and upstream and downstream nucleotides. Even after controlling for mutational composition with these models, a significant trend persists for nonsynonymous variants.
We controlled for the effect of mononucleotide mutation types on the probability of the variant being rare (Figure 5B). We then further refined the mononucleotide mutation types by using their two flanking nucleotides, and estimated another model with these finer mutation type categories (Figure 5C). The trend persisted even after controlling for mutation type, most noticeably for nonsynonymous sites, which likely involve the strongest purifying selection pressures (Spearman ρ = 1, p = 0.016 for the ordering of substituted-species coefficients for both models).
One explanation for the residual trend observed in Figures 5B,C is that more-related species have, on average, more similar context on which the mutation occurs. We can interpret this residual trend as support for the following hypothesis: when the sequence context of the substituted species is similar to that of humans, the fixation of the human-minor allele in the substituted species suggests that the mutation is benign for humans. As sequence context diverges, epistatic effects may come into play and change the selective effect of the mutation [25,50,51].
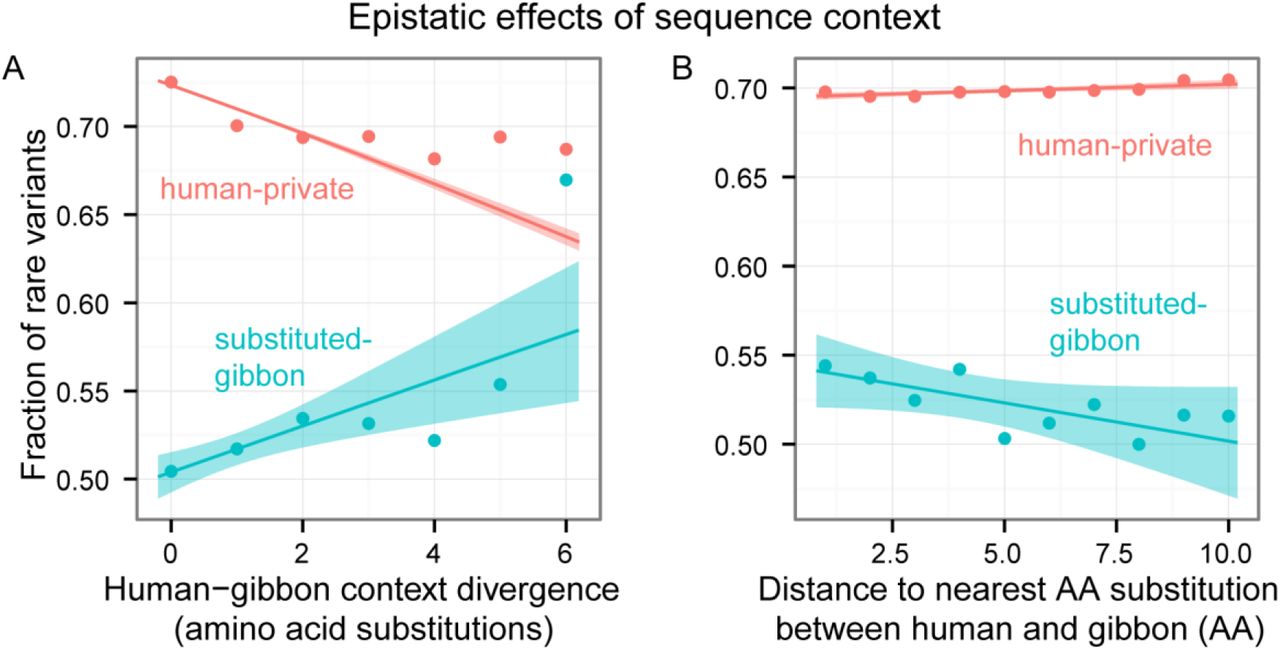
Because of the limitations of our control for mutation rate, we decided to further expand our hypothesis by examining a single substituted species at a time. In doing so, we removed the potential confounder of mutation rate differences across substituted-species categories. Within a single substituted species, we expect to observe a similar trend across sites with differing levels of context similarity. We hypothesized that the more diverged the sequence context in the substituted-species is from humans, the more deleterious the variant would tend to be in humans. The fraction of rare variants is therefore expected to increase with sequence context divergence.
To test this, we computed the amino acid sequence-context divergence between human and gibbon at all substituted-gibbon nonsynonymous SNPs. As a measure of divergence, we computed the number of mismatches between human and gibbon sequences in a window of 9 amino acids upstream and 9 downstream of the focal substituted-gibbon sites. We found that, as expected, the fraction of rare variants increased with sequence context divergence (univariate logistic regression p = 3.3 * 10‒13). As a control we examined the effect of context divergence in sites conserved across non-human primates. In these sites, the fraction of rare variants decreased with sequence context divergence from gibbon, consistent with higher regional mutation rates and lower constraint implied by higher sequence context divergence (Figure 6A). This trend reversal may suggest that sequence context, regional mutation rates and selective constraint have comparable roles in determining the trajectory of an allele. The same trends hold for all substituted-species categories (univariate logistic regression p < 3.1 * 10‒8 for each of the other four species, Figure S4). The trends also hold for a different measure of sequence context similarity, the distance to the nearest amino-acid substitution between humans and the substituted-species (t-test p < 1.8 * 10‒32 and see Figure 6B for gibbon, p < 2.2 * 10‒5 for Pearson correlation and see Figure S5 for each of the other species).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dots show means. Lines and shaded regions show simple logistic model fits to the data and associated confidence bands. The more diverged the sequence context in the substituted-species is from humans, the more deleterious the variant would tend to be in humans. The fraction of rare variants is thereby expected to increase with sequence context divergence. The two panels exhibit the same trend with different measures of sequence context divergence in a window of 9 residues upstream and 9 downstream of the SNP. In human-private sites, the trend is reversed: the fraction of rare variants decreased with sequence context divergence from gibbon, consistent with higher regional mutation rates and lower constraint implied by higher sequence context divergence.
Discussion
Our analysis showed a significant correlation between the probability that a variant is rare in humans and the relatedness of another species in which the same mutation occurred. This trend was largely driven by mutation rate variation, which we have observed to be a primary determinant of the human SFS.
The large effect that mutation rate variation has on the human SFS could have a major impact on any future work involving human polymorphism datasets with large sample sizes. For example, most demographic inference algorithms that use the SFS as a summary statistic [e.g. 6,47,52] rely on the infinite-sites model, which is evidently not a valid assumption for large samples. Adjusting demographic inference schemes to include the effects of recurrent mutations on the SFS has the potential to significantly improve inference accuracy.
We have also seen that the trend across cSFS persisted even after tri-nucleotide mutational composition was taken into account. This remaining correlation is consistent with an effect of sequence context epistasis on the fate of mutations. Our analysis of the cSFS of a single species, and its dependence on sequence context resemblance between humans and the substituted-species, provided additional support for epistatic interactions being a factor influencing human variation patterns.
Substitutions in other lineages have proven to be highly informative for understanding deleterious effects in the contemporary human genome; among numerous features that have been considered, the strongest predictors of the pathogenicity of a mutation are species divergence features [53-56]. Nevertheless, methods used to predict the deleteriousness of a mutation at a site typically rely on a single summary of how variable a site is across the phylogenetic tree. Our analysis suggests that epistatic effects can bias the inferred deleteriousness of the mutation, and that the location of a mutation on the evolutionary tree is informative of how deleterious the same mutation is for humans. It is our hope that the integration of divergence patterns and sequence context into methods that predict the fitness or health effects of human mutations could increase accuracy and predictive power.
Materials and Methods
Data
For polymorphism data, we downloaded single nucleotide polymorphism (SNP) data from version 0.2 of the Exome Aggregation Consortium database [32]. This database is a standardized aggregation of several exome sequencing studies amounting to a sample size of over 60,700 individuals and approximately 8 million SNPs. For each SNP we extracted upstream and downstream 30 nucleotides in the coding sequence of the human reference genome hg19 build. For simplicity we excluded sites that are tri-allelic (6.5% of all SNPs) or quad-allelic (0.2% of all SNPs).
For divergence data, we used the following reference genome builds downloaded from the UCSC genome browser [33]: chimpanzee (panTro4), gorilla (gorGor3), orangutan (ponAbe2), gibbon (nomLeu1), macaque (rheMac3), and baboon (papHam1). We used the UCSC genome browser's liftOver program to align each ExAC SNP along with its 60bp sequence context to the six aforementioned reference genomes. We used the baboon reference genome solely for the ascertainment of all other substituted-species categories (rather than including a substituted-baboon category in the analysis).
For gene annotations, we downloaded the refGene table of the RefSeq Genes track from the UCSC genome browser. For each SNP in our data, we extracted all gene isoforms in which the position was included. We kept all ExAC SNPs that fell in a coding exon, intron or untranslated region. We excluded from the analysis non-autosomal SNPs, SNPs that had multiple annotations corresponding to different transcript models, and SNPs with a sample size of less than 100,000 chromosomes. After applying the filters we were left with 6,002,065 SNPs.
Simulating mutational models and demographic models
To get a theoretical expectation for the fraction of rare variants under different mutational models, we used various software for computing the expected sample SFS of 33,750 diploid individuals, corresponding to the size of the non-Finnish European subsample in the ExAC dataset. For all mutational models, which we describe below, we generated predictions under various demographic models from recent literature: Gazave et al. [57] (model 2 in their work), Tennessen et al. [16] and Nelson et al. [14].
For the infinite-sites model, we computed the expected sample SFS analytically using fastNeutrino [47]. The infinite-sites model corresponds to an upper bound for the fraction of rare variants, but nonetheless predicted a fraction of rare variants much lower than that observed in data (75%-78%) for all non-CpG mutations under the Gazave et al. (59%) and Tennessen et al. (60%) demographies. The Nelson et al. model, which was inferred using a larger sample size of 11,000 people predicted 75% of biallelic polymorphisms would be rare under the infinite-sites model. In order to fit the highest observed fraction of rare variants for non-CpG sites in the ExAC data, we modified the parameters of the most recent epoch of exponential growth in Nelson et al. We estimated these parameters using fastNeutrino [47] on all A->C intronic mutations from ExAC. The inferred parameters were: current effective population size of 4,009,877 diploids, and an exponential growth onset time of 119.47 generations in the past with a growth rate of 5.38% per generation. The more ancient demographic parameters were fixed to the same values as in the model of Nelson et al.
For the finite sites model, we first simulated independent coalescent trees using ms [58] and then generated 1kb non-recombining sequences for each coalescent tree using the desired recurrent mutation rate with the 4 × 4 Jukes-Cantor model of mutation [45]. We used the program Seq-Gen [59] to drop recurrent mutations on coalescent trees drawn from ms. We used mutation rates in a uniform logarithmically spaced grid ranging from 10‒9 to 5 · 10‒6 mutations per basepair per generation per haploid. For each value of the mutation rate, we simulated enough sequence data so that at least 100,000 biallelic polymorphic sites were available to reliably estimate the expected fraction of rare variants. If we indicate whether a variant is rare by Y, then for each mutation rate μ, the expected fraction of rare variants is
 where S is an indicator variable indicating whether a site is polymorphic and, specifically, biallelic. Finally, we considered a model with additional, within-mutation-type heterogeneity in mutation rate. Specifically, we considered a model in which sites of a particular mutation type (e.g., C->A sites) have a mean mutation rate μ as before, but the mutation rate itself, M, is no longer fixed (and equal to μ), but rather a random variable with mean μ. Let f(M|S = 1;μ) be the probability density function of M in a site with mean mutation rate μ conditional on it being biallelic. Then, by the law of total expectation we have:
where S is an indicator variable indicating whether a site is polymorphic and, specifically, biallelic. Finally, we considered a model with additional, within-mutation-type heterogeneity in mutation rate. Specifically, we considered a model in which sites of a particular mutation type (e.g., C->A sites) have a mean mutation rate μ as before, but the mutation rate itself, M, is no longer fixed (and equal to μ), but rather a random variable with mean μ. Let f(M|S = 1;μ) be the probability density function of M in a site with mean mutation rate μ conditional on it being biallelic. Then, by the law of total expectation we have:

By Bayes’ rule, f(M|S = 1;μ) is determined by both the within-mutation-type distribution of mutation rates, g(M; μ), and the probability of a site with mutation rate M being biallelic, as follows:

Therefore,

For a large range of M, we have already estimated E[Y|M] as described above. From the same simulations we have estimated the probability of a site with mutation rate M being a biallelic polymorphism, P(S = 1|M). Lastly, the distribution of mutation rates due to within-mutation-type variance was modeled using a lognormal distribution:

The mean parameter in the lognormal distribution above ensures that E[M] = μ. σ was arbitrarily chosen to be 0.57 (red line in Figure 4A).
Logistic model for the probability of a variant to be rare
We tested whether the species trend across cSFS is due solely to the effect of mutation rate variation. We used a logistic regression model to examine whether a residual substituted-species trend remains after controlling for mutation type. Let Y be a binary-valued random variable indicating whether a variant is rare,  be a vector of mutually exclusive indicator (dummy) variables for each mutation type,
be a vector of mutually exclusive indicator (dummy) variables for each mutation type,  be a vector of mutually exclusive indicator variables for the divergence pattern for the variant (substituted in one of the primates or human-private) and Z be an indicator of whether the variant is nonsynonymous (we only considered coding variants). We fitted the logistic regression model
be a vector of mutually exclusive indicator variables for the divergence pattern for the variant (substituted in one of the primates or human-private) and Z be an indicator of whether the variant is nonsynonymous (we only considered coding variants). We fitted the logistic regression model
 where the parameters
where the parameters  , and
, and  were learned from the data. We tested whether the coefficients
were learned from the data. We tested whether the coefficients  exhibit a trend across s, i.e. whether the probability of the variant being rare is associated with the relatedness of the substituted species. When ignoring the mutation rate effect (i.e. fixing
exhibit a trend across s, i.e. whether the probability of the variant being rare is associated with the relatedness of the substituted species. When ignoring the mutation rate effect (i.e. fixing  , the
, the  estimates were perfectly anti-correlated with the relatedness of the substituted species to human, consistent with the observation in data (Figure 5A). We then allowed for an effect for the mutation type by estimating
estimates were perfectly anti-correlated with the relatedness of the substituted species to human, consistent with the observation in data (Figure 5A). We then allowed for an effect for the mutation type by estimating  for the different categories of mononucleotide mutation types (Figure 5B). We also estimated a model with a finer resolution of mutational categories, further partitioning the mononucleotide mutation types by their two flanking nucleotides (Figure 5C). For nonsynonymous sites, which likely involve the strongest purifying selection pressures, the trend persisted even after controlling for mutation rate variation (Spearman ρ = 1, p = 0.016 for both mononucleotide correction and for the correction including flanking nucleotides context).
for the different categories of mononucleotide mutation types (Figure 5B). We also estimated a model with a finer resolution of mutational categories, further partitioning the mononucleotide mutation types by their two flanking nucleotides (Figure 5C). For nonsynonymous sites, which likely involve the strongest purifying selection pressures, the trend persisted even after controlling for mutation rate variation (Spearman ρ = 1, p = 0.016 for both mononucleotide correction and for the correction including flanking nucleotides context).
Acknowledgements
We thank ExAC and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. We also thank Doc Edge, Ziyue Gao, Xun Lan, David Golan, Anil Raj, Kelley Harris, Yair Field, Eyal Elyashiv and Molly Przeworski for helpful comments on the manuscript and/or valuable discussions. This work was supported by grants R01MH084703 and U01HG007036 from the Howard Hughes Medical Institute (HHMI) to JKP. AB was supported by a postdoctoral fellowship from the Stanford Center for Computational, Evolutionary and Human Genomics (CEHG). The funders had no role in study design, data collection, analysis, decision to publish or the preparation of the manuscript.
References




