

Abstract
When emerging pathogens encounter new host species for which they are poorly adapted, they must evolve to escape extinction. Pathogens experience selection on traits at multiple scales, including replication rates within host individuals and transmissibility between individuals. We introduce and analyze a stochastic, multi-scale model linking pathogen growth and competition within individuals to transmission between individuals. Our analysis reveals a new factor that quantifies how quickly mutant strains increase in frequency when they initially appear in the infected host population. This cross-scale quantity combines with viral mutation rates, reproductive numbers, and transmission bottleneck width to determine the likelihood of evolutionary emergence, and whether evolution occurs swiftly or gradually within chains of transmission. Wider transmission bottlenecks facilitate emergence of pathogens with short-term infections, but hinder emergence of pathogens exhibiting cross-scale selective conflict and long-term infections. These results provide a framework for a new generation of evidence-based risk assessment of emergence threats.
Introduction
Emerging infectious diseases are rising in frequency and impact and are placing a growing burden on public health and world economies (1–4). Nearly all of these emergence events involve pathogens that are exposed to novel environments such as zoonotic pathogens entering human populations from non-human animal reservoirs, or human pathogens exposed to antimicrobial drugs (1). In these novel environments, pathogens may experience new selective forces occurring at multiple biological scales, leading to reduced replication rates within hosts or less efficient transmission between hosts. When these novel environments are sufficiently harsh, emergence only occurs when the pathogen adapts sufficiently quickly to avoid extinction. As genetic sequencing of pathogens becomes increasingly widespread, there are clear signs of such rapid adaptation (5–10), but we lack a cohesive framework to understand how this process might work. Theoretical studies over the past decade have shed important insights into circumstances under which this evolutionary emergence is possible, but have focused on the host-to-host transmission dynamics and treated within-host dynamics only implicitly (11–14). Here, we introduce and analyze a model explicitly linking these two biological scales and demonstrate how within-host viral competition, infection duration, between-host transmissibility, and the size of transmission bottlenecks determine the likelihood of evolutionary emergence. This analysis sheds new light on factors governing pathogen emergence, addresses long-standing questions about evolutionary aspects of emergence, and lays the foundation for making risk assessments which integrate outcomes from in vitro and in vivo experiments with findings from sequence-based surveillance in the field.
Recent empirical findings have highlighted the need for a new generation of theory on pathogen emergence, which addresses the current frontiers of dynamics within hosts and across scales. For most pathogens, and certainly for RNA viruses and single-stranded DNA viruses, individual hosts often are not dominated by single pathogen genotypes (15, 16). Furthermore, at the host population scale, pathogen allele frequencies at a given locus exhibit a range of dynamics from rapid selective sweeps for drug resistance or immune escape (17, 18) to gradually changing frequencies (19, 20). Together, these observations lead to the long-standing question of whether adaptive evolution of viruses occurs within single hosts by rapid fixation of beneficial mutants, or more slowly by a gradual shift of allele frequencies along chains of transmission (21, 22). A recent wave of studies tracking changes in within-host genetic diversity through chains of transmission among hosts (23–30) provide unique opportunities to address this question, but a theoretical framework is needed.
Empirical studies, together with analyses at broader population scales, have highlighted the crucial influence of the transmission process – and particularly the population bottleneck associated with transmission – in filtering viral diversity. The existence of transmission bottlenecks has long been recognized, and is hypothesized to play a critical role in pathogen evolution (31–33). Recent studies have shown that bottleneck widths vary considerably among pathogens and routes of transmission (34, 35), and perhaps across different phases of host adaptation (36). Narrow transmission bottleneck sizes of 1 to 2 virions are common for HIV-1 (37) and hepatitis C virus (38, 39). In contrast, deep sequencing data for patients infected with Ebola virus suggest that transmission bottleneck sizes are typically greater than 100 viral particles (40), and similarly wide transmission bottlenecks have been reported for natural transmission of influenza (25, 30, 41).
A major frontier in understanding viral adaptation is how the transmission process influences evolution at population scales. Past work has emphasized the potentially deleterious effect of genetic drift (31, 33), but a rising tide of studies reports direct selection for transmissibility. This can arise as a strong selection bias at the transmission bottleneck, where strains present at low or undetectable frequencies in the donor host are preferentially transmitted to the recipient (36, 42, 43), or it can be measured directly via experimental infection and transmission studies (20, 44–46)(though we emphasize that enhanced transmissibility is not inevitable, and depends on availability of suitable adaptive genotypes (47)). Overall transmission rates are thus determined by total viral loads, weighted by genotype-specific transmissibilities (43). Importantly, the transmissibility trait can vary independently from viral replication fitness within hosts, so there is potential for conflicts in selection across scales. Indeed, there is clear evidence that HIV-1 has certain genotypes that transmit more efficiently, but then the within-host population tends to evolve toward lower-transmission strains during an infection (43, 48, 49); a similar phenomenon has been reported for H5N1 influenza (42) and H9N2 influenza (50). In an extreme example, Plasmodium parasites were found to rapidly evolve resistance to an antimalarial drug, but at the cost of complete loss of transmissibility (51). Experimental evolution studies have highlighted how antagonistic pleiotropy can lead to tradeoffs between viral replication and the extracellular survival that is required for transmission (52). Together these findings contribute to a growing evidence base that cross-scale conflicts in selection may inhibit the emergence of new viral strains in many systems (reviewed in (14)).
Collectively these empirical findings highlight the need for a theory of evolutionary emergence that accounts explicitly for the within-host dynamics of competing viral strains, transmission bottlenecks, and host-to-host transmission dynamics (53). To this end, we introduce and analyze a model which integrates previous work onstochastic models of evolutionary emergence and deterministic models explicitly coupling within- and between-host dynamics (11, 13, 45, 54, 55). Our analysis allows us to address several fundamental questions about the emergence of novel pathogens: What factors limit evolutionary emergence for pathogens with different life histories? Why do some apparently ‘nearby’ adaptive mutants fail to emerge? How do bottleneck sizes influence the likelihood of emergence? Do evolutionary changes occur swiftly within individual hosts, or gradually across chains of transmission? Moreover, our analysis allows us to examine the relative importance of genetic diversity in zoonotic reservoirs versus the acquisition of new mutations following spillover into humans (56–58). Specifically, we address the long-standing question of how much is emergence risk increased if the “spillover inoculum” includes some genotypes bearing adaptive mutations for the novel host? Finally, our analysis enables us to unify findings from previous theoretical studies, and propose mechanistic interpretations of phenomenological parameters from earlier work.
Results
Our stochastic multi-scale model of evolutionary emergence follows a finite number of individuals in a large, susceptible host population exposed to a pathogen from a reservoir population (Fig 1A). Although our framework represents many types of pathogens and can be extended to any number of strains, we focus on the case of a viral pathogen with two strains: a wild-type maladapted for the novel environment and a mutant strain potentially adapted for the novel environment. Each infected individual begins with a viral load consisting of vw(0) wild-type virions and vm(0) mutant-type virions, where vw(0) + vm(0) equals N, the size of the transmission bottleneck. The viral populations increase exponentially until saturating at day Te of the infection with a maximal viral load K at which the viral replication rate is balanced by the viral clearance rate (Fig 1B). The wild and mutant strains increase exponentially at rates, rw and rm, and mutations arise with probability μ. The infectious period ends after T days. This within-host model can describe a range of viral dynamics from infections with primarily an exponential phase of viral growth to infections maintaining a stable viral load for an extended period.

The cross-scale dynamic of evolutionary emergence. An individual is initially infected from the reservoir host population (panel A) with only the wild type viral strain (in blue). Within an infected individual (panel B), the viral load increases at an exponential rate until saturating at day Te and ending after T days. Mutations ensure individuals typically have a mixed infection with wild-type (blue) and mutant (red) viral strains (panel B). The likelihood of transmission between individuals, and the composition of the infecting dose (panel C), depend on the size and composition of the infected individual’s viral load at the time of contact, and on the transmissibility of each strain. As the infection spreads in the population (panel A), the frequency of the mutant virions among infected individuals varies (panel D) and, ultimately, determines whether evolutionary emergence occurs. In D, each horizontal line marks the infectious period of an individual whose infection was initiated with that percentage of the mutant strain and the vertical arrows represent transmission events between individuals.
At the scale of the host population, the transmission dynamics along chains of hosts is stochastic. Each infectious individual encounters a Poisson-distributed number of susceptible individuals at a rate of β individuals per day. Encounters result in a transmission event with probability p(E) where E = bw vw(t) + bm vm(t) is the effective viral load at the time t of transmission, bw, bm are the transmissibilities of the viral strains, and p(E) is an increasing function of E. Our main analyses assume that the transmission function p(E) is linear, but nonlinear transmission functions yield nearly identical results (Supplementary Figs S–4 through S–7). When transmission occurs, a newly infected individual is infected with N virions (the transmission bottleneck width) sampled binomially from the source individual’s viral load weighted by the transmissibilities of the viral strains. After a transmission event, the newly infected individual’s viral load is governed by the within-host model. By explicitly modeling the cross-scale dynamics, our model simultaneously tracks the number of infected hosts and the viral loads within each infected host (Fig 1D). The structure of our model is similar to a recent model of molecular viral evolution along transmission chains (55). However, our model accounts for transmission dynamics rather than conditioning on a chain of transmission, and explicitly accounts for the dynamics of competing viral strains.
The probability of evolutionary emergence
We first focus on the scenario of a single individual in the host population getting infected by the wild-type strain. We assume that the mean number of individuals infected by this individual (the reproductive number Rw of the wild-type) is less than one. Hence, in the absence of mutations, there is no chance of a major outbreak (59). However, if the wild-type strain produces a mutant strain whose reproductive number Rm is greater than one, there is a chance for a major outbreak. The mutant strain might have a higher reproductive number than the wild-type strain because it replicates more rapidly within the host or because it transmits more effectively to new hosts (or both). We define these within-host and between-host selective advantages as s = rm − rw and r = log(bm) − log(bw), respectively.
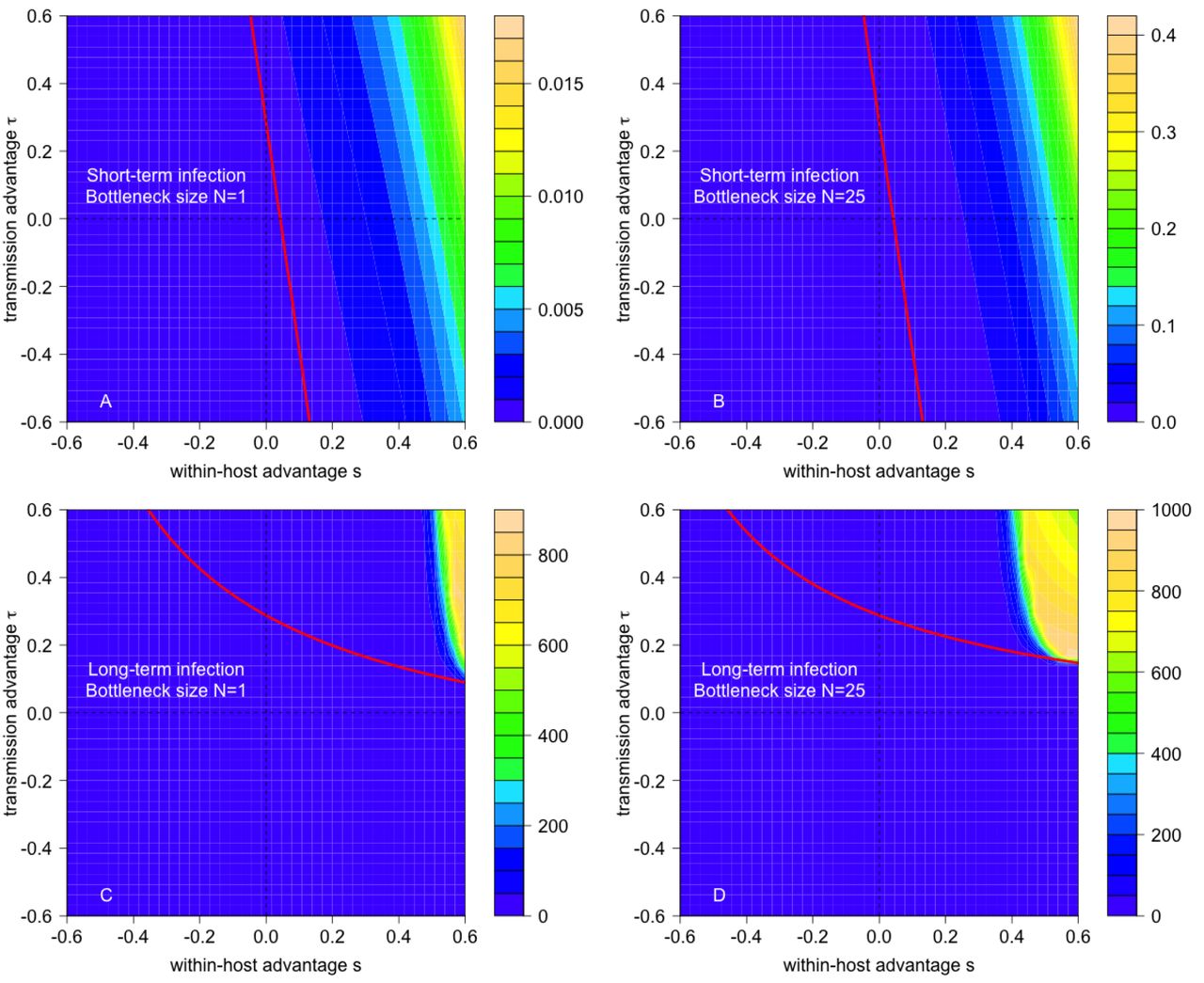
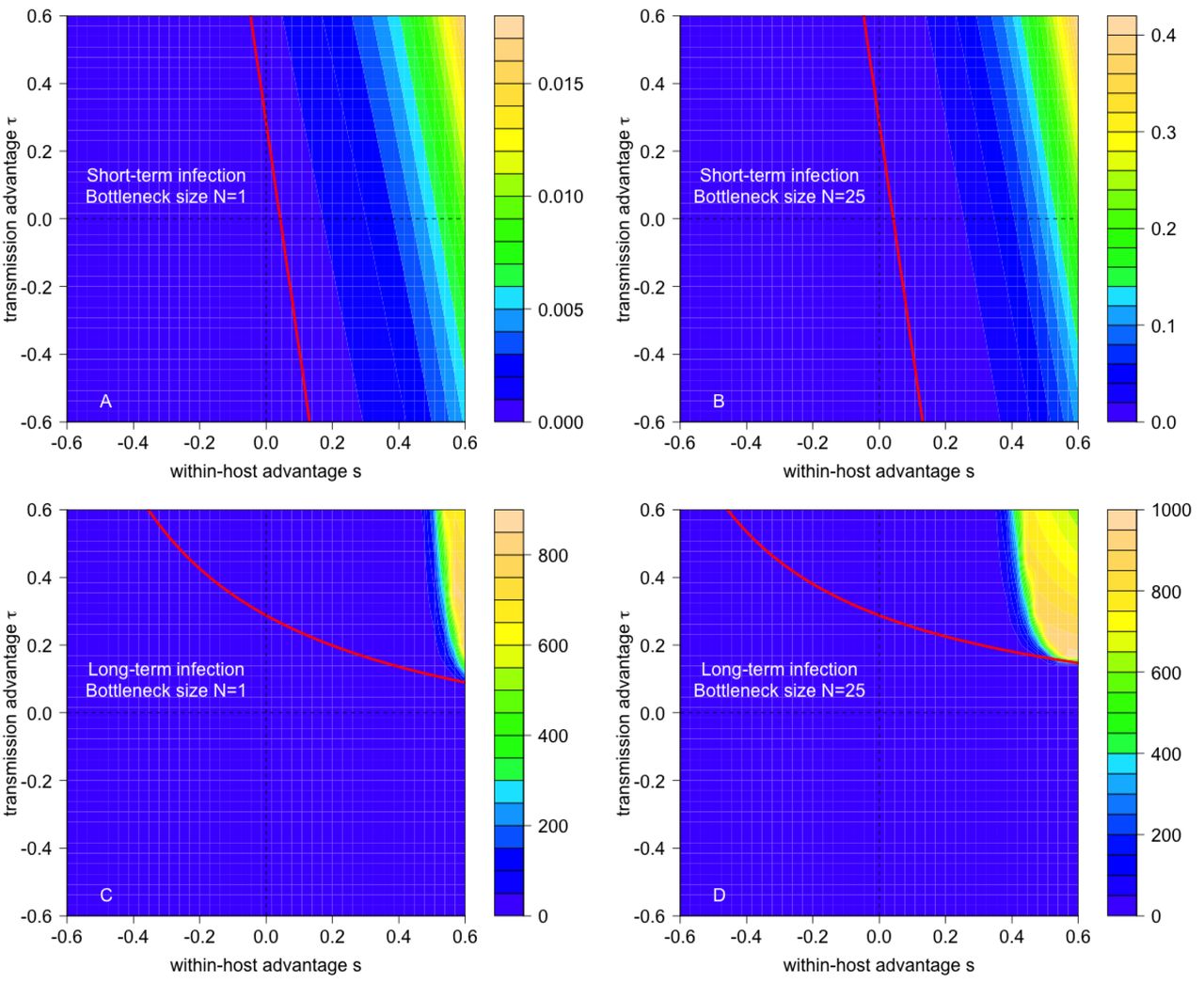
Consistent with theoretical expectations, a non-zero probability of evolutionary emergence requires the mutant’s reproductive number Rm to be greater than one (Fig 2). However, the mixture of selective advantages or disadvantages of the mutant strain that give rise to Rm > 1 depends in a complex manner on the pathogen’s life history traits, such as the duration of the infection (Fig 2A,B vs. C,D) and the transmission bottleneck width (Fig 2A,C vs. B,D). Notably, for long-term infections with a large transmission bottleneck size, the emergence probability can be effectively zero (i.e. < 10−16) for mutant strains whose reproductive number exceeds one (white region bounded by the solid red curve in Fig 2D).
To understand these complexities, we determine the conditions under which the mutant’s reproductive number Rm exceeds one, and then present analytic approximations for the emergence probability when Rm > 1.

The joint effects of within-host and between-host selective advantage of the mutant on the probability of emergence for pathogens with short-term (A,B) and long-term (C,D) infectious periods, and with transmission bottlenecks of size N =1 (A,C) and N = 25 (B,D). Colorations correspond to log10 of the emergence probability. The critical value of the mutant reproductive number Rm equaling one is drawn in solid red. Black contour lines correspond to log10 of the number of mutant virions transmitted by an individual initially only infected with the wild strain. In D, the critical value of the cross-scale mutant reproductive rate, α = 1, is shown as a red dashed line and the black asterisks correspond to the t and s values used in Fig 3. Parameters: K = 107, βT = 30, T = 7.5 (short-term infection) and T = 30 (long-term infection), bw chosen so that R0 =0.75 for wild type, rw = 1.25 and μ = 10−7.
Cross-scale selection and the mutant reproductive number Rm
The reproductive numbers of the wild-type strain (Rw) and mutant strain (Rm) can be calculated as the product of the contact rate, the average transmissibility of the strain during the infectious period, and the infection duration (see Supplementary Information). These reproductive numbers are positively correlated with the contact rate, infection duration, transmissibility, and viral per-capita growth rates. The influence of the maximal viral load K depends on the infection duration. For short-term infections, defined here as infections with a relatively short saturated phase (i.e. T − Te ≪ Te), increasing K has little effect on a strain’s reproductive number. For long-term infections, defined here as as infections with a long saturated phase (i.e. T − Te ≫ Te), reproductive numbers increase with K.
Whether a selective advantage at either scale results in the mutant reproductive number Rm exceeding one depends on the duration of the infection (Supplementary Information). For short-term infections, the mutant reproductive number satisfies

This approximation shows that a sufficiently strong selective advantage at either scale can result in the mutant reproductive number exceeding one (Rm > 1) despite a selective disadvantage at the other scale (confirmed by exact calculations in Fig 2A,B). For short-term infections where viral dynamics are dominated by the exponential phase, the longer the duration of infection, the greater the influence of the within-host selective advantage compared to the between-host selective advantage (e.g., steep contours in Fig 2A).
For long-term infections, the mutant’s reproductive number satisfies

This approximation implies that a between-host selective advantage is required for evolutionary emergence (confirmed by exact calculations in Fig 2C). When viral dynamics are dominated by the saturated phase at fixed K, a within-host selective advantage has little impact on the average viral load during the infectious period of an individual solely infected with the mutant strain and, consequently, provides a minimal increase in the mutant reproductive number.
Going beyond the mutant reproductive number
When the mutant strain has a reproductive number greater than one, there is a non-zero probability of a major outbreak that is well-approximated by the product of three terms (Supplementary Information):
 This expression, which can be viewed a multi-scale extension of earlier theory (11, 12), highlights three key ingredients, in addition to Rm > 1, for evolutionary emergence.
This expression, which can be viewed a multi-scale extension of earlier theory (11, 12), highlights three key ingredients, in addition to Rm > 1, for evolutionary emergence.
First, the size of the minor outbreak produced by the wild type determines the number of opportunities for the mutant strain to appear within a host. The average size of this minor outbreak equals  , as noted by earlier studies (11, 12). If the wild strain is badly maladapted (e.g. Rw < 1/2), then it is expected not to spread to multiple individuals (i.e.
, as noted by earlier studies (11, 12). If the wild strain is badly maladapted (e.g. Rw < 1/2), then it is expected not to spread to multiple individuals (i.e.  ) and opportunities for transmission of mutant virions are very limited. Alternatively, if the wild strain is only slightly maladapted to the new host (e.g. Rw = 0.95), then, even without any mutations, the pathogen is expected to spread to many individuals (e.g.
) and opportunities for transmission of mutant virions are very limited. Alternatively, if the wild strain is only slightly maladapted to the new host (e.g. Rw = 0.95), then, even without any mutations, the pathogen is expected to spread to many individuals (e.g.  ), thereby providing greater opportunities for evolutionary emergence. Our analysis implies that higher contact rates, within-host viral growth rates, viral transmissibility, and maximal viral loads (for long infectious periods) facilitate these larger reproductive values.
), thereby providing greater opportunities for evolutionary emergence. Our analysis implies that higher contact rates, within-host viral growth rates, viral transmissibility, and maximal viral loads (for long infectious periods) facilitate these larger reproductive values.
Second, the mutant strain must be transmitted successfully to susceptible individuals — the second term of our approximation (3). For an individual initially infected only with the wild-type strain, the mean number of transmission events with mutant virions equals the product of the contact rate, the infection duration, and the likelihood that a mutant virion is transmitted during a contact event, averaged over the full course of infection (Supplementary Information). The likelihood of transmitting mutant virions on the tth day of infection is proportional to the product of the transmission bottleneck width (N), the within-host frequency of the mutant strain, and the transmissibility bm of the mutant strain. This highlights an important distinction between short-term and long-term infections. For short-term infections, there is insufficient time for the frequency of mutants to rise within a host, so transmission events with mutant virions are rare (< 1/1, 000 for all black contour lines in Fig 2A,B). This is a key obstacle to evolutionary emergence in short-term infections. In contrast, for long-term infections where the mutant strain has a substantial within-host selective advantage, the mutant strain is transmitted frequently (e.g. the expected number of events > 1 for some contours in Fig 2C,D).
Finally, even if the mutant strain is successfully transmitted, an individual infected with the mutant strain needs to give rise to a major outbreak — the third term of equation (3). This requires the mutant strain to rise in frequency in the infected host population. A mean field analysis for larger bottleneck sizes (N > 5 in the simulations) reveals that mutant frequency initially grows geometrically by a factor a that equals the number of mutant virions, on average, transmitted by an individual initially infected with a single mutant virion and N − 1 wild type virions (Supplementary Information). We call α the “cross-scale mutant reproductive rate” as it corresponds to the number of mutant virions at the beginning of the next disease generation produced by a mutant virion in the current disease generation. If this cross-scale reproductive rate is greater than one, then each mutant virion replaces itself with more than one mutant virion in the next generation of infection and the frequency of mutant virions increases in the infected host population. If the cross-scale reproductive rate α is less than one, the frequency of mutants decreases, thereby hindering evolutionary emergence.
For short-term infections, the cross-scale mutant reproductive rate α is equal to the ratio of the reproductiv numbers:

Thus for short-term infections there is no additional condition required for emergence. Whenever the mutant reproductive number Rm exceeds one, there is a mean tendency for the mutant strain to increase in frequency once it has been successfully transmitted to susceptible individuals (i.e. α > 1 because Rm > 1 > Rw). The greater the ratio Rm/Rw, the more rapid the increase in frequency.
For long-term infections, there is sufficient time for within-host selection to change the frequency of the mutant strain within a host. Larger transmission bottlenecks increase the likelihood that these changes in frequency are transmitted between hosts. For these long infectious periods and larger bottlenecks, a within-host selective disadvantage reduces the cross-scale mutant reproductive rate α (Supplementary Information):

Hence, the cross-scale mutant reproductive rate α may be less than one even when the mutant reproductive number Rm is greater than one. This phenomenon, which arises from the interplay of dynamics at within-host and between-host scales, moderated by the transmission bottleneck width, explains the puzzling behavior about the emergence probabilities noted earlier (the white region bounded by solid and dashed red lines in Fig 2D).
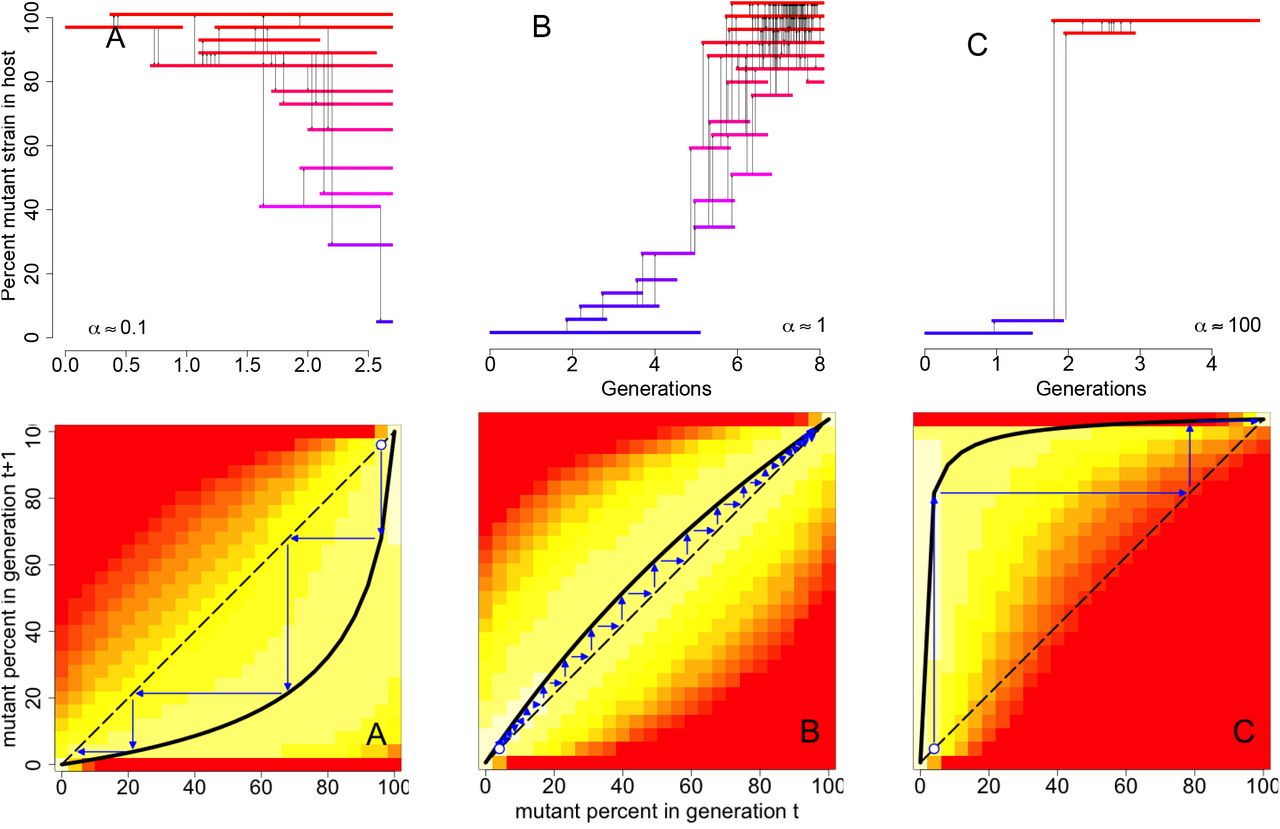
The importance of these frequency dynamics can be visualized via individual-based outbreak simulations, and cobwebbing diagrams summarizing the mean field dynamics. When the mutant reproductive number Rm is greater than one but its cross-scale mutant reproductive rate α is less than one, mutant virions may be transmitted but the resulting mixed infections are invariably taken over by purely wild-type infections (Fig 3A). Only pure mutant infections can escape this “relapse” to wild-type, and then only if the mutation rate μ is low enough that new wild-type virions are slow to appear. When the within-host selective disadvantage is weak and the between-host selective advantage is strong, the cross-scale mutant reproductive rate may be slightly greater than one and the mutant strain can drift to higher frequencies within the infected host population (Fig 3B). For large within-host selective advantages, the cross-scale mutant reproductive rate is large and the mutant strain can sweep rapidly to fixation in the infected host population (Fig 3C). Thus, in addition to revealing a new condition needed for evolutionary emergence, the cross-scale mutant reproductive rate α summarizes the conditions under which evolution occurs swiftly or gradually within chains of transmission.

Frequency dynamics of the mutant strain in the host population. For long-term infections with moderate to large transmission bottlenecks (N > 5), individual-based simulations corresponding to three values of the cross-scale mutant reproductive rate α illustrate (A) the mutant strain decreasing in frequency (despite an index case initially only infected with the mutant strain) when the cross-scale mutant reproductive rate a is less than one, (B) a gradual sweep to fixation of the mutant strain when α ≈ 1, and (C) fast sweeps to fixation for large values of α > 1. In these individual based simulations, each horizontal line marks the infectious period of an individual whose infection was initiated with that percentage of the mutant strain and the vertical arrows represent transmission events between individuals. In the bottom half of the figure, the mean field dynamics corresponding to each of the individual-based simulations are plotted as cobwebbing diagrams. The solid black curves correspond to the expected frequency of the mutant strain in the infected host population in the next generation given the frequency in the current generation. Thin blue lines indicate how the expected frequencies change across multiple generations. The colored backgrounds represent the expected number of individuals infected with a certain percentage of the mutant strain (vertical axis) by an individual with an initial percentage of the mutant strain (horizontal axis). Lighter colors correspond to higher values. Parameter values as in Fig 2D indicated with black asterisks.
The dueling effects of transmission bottlenecks
Wider bottlenecks increase the likelihood of evolutionary emergence for pathogens with a short infectious period, but can hinder or facilitate evolutionary emergence of long-term infections (Fig 4A,B). For short-term infections, evolutionary emergence is constrained primarily by the transmission of mutant virions by individuals initially infected with only the wild strain. Wider transmission bottlenecks alleviate this constraint, especially when the mutant strain is expected to increase rapidly within the infected population (α ≫ 1; Fig 4A). When the mutant strain rises slowly in the infected host population (α slightly greater than one), the emergence probability is insensitive to the bottleneck size, regardless of infection duration.

Effects of bottleneck size and mixed infections of the index case on evolutionary emergence of short-term and long-term infections. Different curves correspond to different tendencies, as measured by the cross-scale mutant reproductive rate α, for the mutant strain to increase in frequency in the infected host population. In (A) and (B), bottleneck size has negative effect on emergence when the cross-scale mutant reproductive rate α is less than one and a positive effect when α is greater than one. In (C) and (D), index cases initially infected with higher percentages of the mutant strain are more likely to lead to emergence. −∞ corresponds to numerical values of 10−16 or smaller. Parameters: K = 107, βT = 150, T = 7.5 for short-term infections and T = 30 for long-term infections, bw chosen so that R0 =0.75 for the wild strain, Rw = 1.25, τ =1, s chosen to achieve the α values reported in the legend, and μ = 10−7. N = 25 in (C) and (D).
For long-term infections for which the mutant strain’s reproductive number Rm is greater than one, but the cross-scale mutant reproductive rate α is less than one, emergence probabilities decrease sharply with bottleneck size (Fig 4B and Supplementary Information). Because a mutant reproductive number Rm greater than one requires a between-host selective advantage (τ > 0) for a long-term infection, the cross-scale mutant reproductive rate α is less than one only if there is a within-host selective disadvantage (s < 0) so that mixed infections tend to be taken over by the wild-type. Consequently, the mutant virus can start an epidemic only when a host is infected with mutant particles only, an event that becomes increasingly unlikely for larger bottleneck sizes N.
Mutant spillover events hasten evolutionary emergence
When the mutant strain is circulating in the reservoir, the index case can begin with a mixed infection which invariably makes evolutionary emergence more likely (Fig 4C,D). For short-term infections, spillover doses that contain low or high frequencies of mutants have a roughly equal impact on emergence, and the magnitudes of these increases are relatively independent of the cross-scale mutant reproductive rate α (Fig 4C). This arises because the initial production and transmission of the mutant strain is the primary constraint on evolutionary emergence for short-term infections with Rm > 1 (black contours in Fig 2A,B). Consequently, mutant spillover events of any size are sufficient to overcome this constraint.
For long-term infections, the impact of mutant spillover depends on the cross-scale mutant reproductive rate α. When α is less than one, only spillover doses with high frequencies of mutants have a significant effect on emergence (i.e. bottom three curves in Fig 4D). When the cross-scale mutant reproductive rate α is greater than one, the effect mimics short-term infections and mutant spillover events of any size can substantially increase the chance of emergence (top three curves in Fig 4C,D).
Discussion
We have presented a cross-scale model for evolutionary emergence of novel pathogens, linking explicit representations of viral growth and competition within host individuals to viral transmission between individuals. This framework integrates and extends the findings of past theory on this problem by including mixed infections, explicit transmission bottlenecks, and a distinct trait of transmissibility for each viral genotype, phenomena that are highlighted by current empirical research as essential components of viral evolution. Our work identifies four steps to evolutionary emergence (Fig 5) and four ingredients that govern these steps: (i) the reproductive number of the wild type which determines the size of a minor outbreak of this strain, (ii) the rate at which individuals infected initially with the wild-type strain transmit the mutant strain, and (iii) the cross-scale mutant reproductive rate which corresponds to the mean number of mutant virions transmitted by an individual whose initial infection only included one mutant virion, and (iv) the reproductive number of the mutant strain. Prior studies (11–14) identified the importance of the two reproductive numbers and a phenomenological ‘mutation rate’, but ingredients (ii) and (iii) are new mechanistic insights arising from the cross-scale dynamics. By analyzing these ingredients of evolutionary emergence, we show how the probability of emergence is governed by selection pressures at within-host and between-host scales, the width of the transmission bottleneck, and the infection duration. We also map the conditions under which different broad-scale patterns are observed, from rapid selective sweeps to slower diffusion of new types. While our study has focused on within-host and between-host scales of selection, it could be generalized readily to other types of cross-scale dynamics where selection may act differently at different scales, such as within-farm and between-farm scales where genetic data have given insights into the emergence of high-pathogenicity avian influenza strains (60).

The major steps and barriers for evolutionary emergence.
Previous studies of evolutionary emergence of pathogens (11–14) have assumed infected individuals are, at any point in time, infected primarily by a single pathogen strain. Consequently, shifts from infection with one strain to infection with another must occur abruptly, relative to other processes. Such abrupt shifts could correspond to within-host selective sweeps or, if mutant strains remain at low frequency, to rare events in which only the mutant strain is transmitted. The seminal studies (11, 12) showed that under these conditions the probability of emergence is proportional to the frequency of these events, which they bundled together into a phenomenological “mutation rate”.
Our cross-scale analysis identifies the mechanistic counterpart to this phenomenological “mutation rate”, which is the probability that an individual infected initially with the wild-type strain ends up transmitting at least one virion of the mutant strain (Step 3 in Fig 5). This quantity, which is approximated by the black contours in Fig 2, is governed chiefly by the ability of the mutant strain to reach an appreciable frequency within the host over the course of an infection. This is evident from the strong dependence on the strength of within-host selection—which surprisingly is much stronger than the dependence on the transmission advantage of mutant virions—and the higher values found for larger bottleneck widths, which favor transmission of low-frequency mutants through a straight-forward sampling effect. The duration of infection plays a crucial role, and our analysis showed that achieving this first transmission of the adaptive mutant is a key barrier to evolutionary emergence for short-term infections (Fig 2A,B). This aligns with the recent finding that potential immune-escape variants of H1N1pdm influenza, expected to have a strong fitness advantage, were present at surprisingly low frequencies in infected humans, and have been detected very rarely at the consensus level (i.e. they have failed to emerge) (61). While more investigation is needed to determine the relevant s and τ parameters for these strains, these data are consistent with the mechanism we identify whereby these variants may be adaptive but have insufficient time to reach high enough frequencies to avoid being lost in transmission bottlenecks.
Our analysis highlights an additional factor, the cross-scale mutant reproductive rate α, previously unrecognized in models neglecting within-host diversity and analyses centered on R0 for pure infections. Even after the mutant strain has been transmitted, it needs to increase in frequency at the scale of the infected host population (Step 4 in Fig 5). Specifically, each transmitted mutant virion, on average, needs to replace itself with more than one transmitted mutant virion in the next generation of infected hosts. When this occurs, it sets up a positive feedback along chains of infections: individuals with a higher frequency of the mutant strain tend to infect more individuals, which in turn provides more opportunities to transmit, on average, higher frequencies of the mutant strain to the next generation. Conversely, when this between-generation cross-scale mutant reproductive rate is less than one, the positive feedback leads to lower and lower frequencies of the mutant strain within the infected host population. This positive feedback mechanism is stronger for wider transmission bottlenecks (≥ 5 virions in our numerical explorations), which better preserve the mutant frequency from one host to the next.
The directionality of the positive feedback is more complex, and depends on multiple factors including the infection duration and the presence or absence of cross-scale conflicts. For long-term infections, mutant frequencies can drop deterministically within a host, and hence prevent emergence, even if the mutant strain has a reproductive number greater than one. This occurs when the mutant strain has a within-host selective disadvantage and between-host selective advantage (upper left quadrant of Fig 2D); the long infectious period allows time for the within-host disadvantage to drive the mutant strain to lower frequency and, thereby, set up the positive feedback effectively preventing evolutionary emergence. In contrast, for short-term infections the mutant strain tends to rise in frequency whenever the mutant reproductive number is greater than one, because there is insufficient time for any within-host disadvantage to act. In particular, evolutionary emergence may occur despite within-host selective disadvantages, a possibility excluded by previous theory (14). Collectively these two results imply that, in the face of cross-scale conflict and wide transmission bottlenecks, longer infectious periods can inhibit, rather than facilitate (13), evolutionary emergence (Fig 2B,D).
Our cross-scale analysis enables us to address two long-standing and interrelated questions in emerging pathogen research, regarding the influence of transmission bottleneck size on emergence probability and the importance of “pre-adapted” mutations circulating in the animal reservoir (53, 56-58, 62). In both cases, the answer depends on the cross-scale mutant reproductive rate α that governs the frequency feedback. Under most circumstances, wider bottlenecks boost the probability of emergence (Fig 4A,B), because they favor the onward transmission of mutant virions when they are rare; this is particularly vital for the first transmission of mutant virions (i.e. Step 3 in Fig. 5). The exception is for long-term infections with α < 1, such that the mutant tends to decline in frequency in the infected host population. Under these circumstances, wider bottlenecks hinder emergence by propagating reductions in the frequency of the mutant strain more efficiently from host to host (Step 4 in Fig. 5). Conventional thinking about the influence of bottlenecks on viral adaptation emphasizes fitness losses due to genetic drift and the effects of Muller’s ratchet (31–33), which become more severe for narrower bottlenecks. Contrary to these negative effects of narrow bottlenecks, our findings highlight that narrower bottlenecks can aid emergence in long-term infections with a cross-scale conflict in selection (Fig 4B). Here the adaptive gain in transmissibility at population scales can be impeded by the selective disadvantage at the within-host scale, but, intriguingly, this disadvantage is neutralized by genetic drift arising from narrow bottlenecks. Given the evidence for cross-scale evolutionary conflicts for HIV-1 (43, 48, 49), our results suggest the possibility that HIV-1’s narrow transmission bottleneck (37) could play a role in the emergence of novel strains (e.g. drug resistant strains).
Similar mechanisms dictate the influence of mutant viral strains circulating in the reservoir, particularly for long-term infections (Fig 4C,D). If the cross-scale mutant reproductive rate α is greater than one, so that the mutant frequency rises easily in the infected host population, then even low frequencies of mutants in the reservoir lead to substantial risk of emergence. Indeed, for long-term infections with α > 1, emergence becomes almost certain when there are mutants in the initial spillover inoculum. Conversely, when the cross-scale mutant reproductive rate is less than one, emergence probability scales with the proportion of mutants in the initial dose, and when α ≪ 1, the initial dose must consist almost entirely of the mutant strain in order to pose any major risk. These findings yield direct lessons for the growing enterprise of conducting genetic surveillance on zoonotic pathogens in their animal reservoirs (63–65). Generally, risk to humans increases if there is any non-zero proportion of mutant viruses in the spillover inoculum, so tracking the presence of such mutants is beneficial. Surprisingly, the quantitative frequency of mutants in the initial dose has little impact on emergence probability in most scenarios, with the one exception of long-term infections with α < 1. Collectively, these results suggest that any knowledge of the cross-scale mutant reproductive rate and mutant reproductive numbers can help to refine our goals for genetic surveillance, and that in many circumstances presence/absence detection is sufficient. Of course, a crucial prerequisite for genetic surveillance is knowledge of genotypes of concern; the integration of various research approaches to address this question, and estimate key quantities, is an on-going research challenge (66, 67).
While there are not sufficient data from past emergence events to test our model’s conclusions, recent studies combining animal transmission experiments with deep sequencing have exhibited many phenomena aligned with our findings. Moncla et al. (2016) conducted deep sequencing analyses of H1N1 influenza viruses, in the context of ferret airborne transmission experiments that examined the adaptation of avian-like viruses to the mammalian host. Their results provide in-depth insights into selection within hosts and at transmission bottlenecks, for a range of mutations on genetic backgrounds that change as adaptation proceeds (i.e. equivalent to numerous separate implementations of our model of a single mutational step). They observe a fascinating range of dynamics: some mutations appeared to have α moderately above 1, exhibiting modest increases in frequency between generations, but achieved this outcome with different traits (e.g. S113N on the HA190D225D background exhibited strong within-host selection and no evident transmission advantage, while D265V showed weak within-host selection but its frequency rises in transmission). Another mutation (I187T on the ‘Mut’ background) appeared to have α ≫ 1 and exhibited strong selection at both scales; notably, this mutation is widespread in 17/17 human-derived isolates of the post-emergence 1918 virus, consistent with the successful and rapid emergence our model would predict. Moncla et al. also present substantial evidence of cross-scale conflict in selection, as one mutation (G225D on ‘Mut’ background) exhibited declining frequencies within ferrets but rose to fixation in 2/2 transmission events, while numerous mutations in the HA2 region rose in frequency within the host but were eliminated in transmission. Another study examined a set of ‘gain-of-function’ mutations in H5N1 influenza in ferrets, and reported a slow rise in frequency when the virus was passaged between ferrets by intranasal inoculation, then rapid fixation of these mutations during airborne transmission (20); the airborne transmission data are consistent with strong between-host selection and a high α value (though we emphasize that circulating H5N1 viruses required substantial modification to the favorable genetic background used in those experiments). Intriguingly, Moncla et al. synthesized their results with those of earlier studies (35, 42, 47) to hypothesize that the ‘stringency’ of the transmission bottleneck varies systematically during the course of viral adaptation, with loose bottlenecks prevailing when viruses first encounter a new host species (and perhaps again when the virus is host-adapted (30)), and much tighter bottlenecks at the key juncture in host adaptation when a genotype with greater transmissibility is available to be selected. If this hypothesis is correct, then our findings can be applied to each adaptive step independently, and may help to identify which viral traits are most crucial to adaptive steps subject to tighter or looser bottlenecks.
Our results focus on systems where there is one major rate-limiting step to emergence, and the viral population can be represented by one wild-type and one mutant strain. This is a simplification of most viral emergence problems, but will apply directly to systems where a single large-effect mutation is the primary barrier to emergence of a supercritical strain, as for Venezuelan equine encephalitis virus emerging from rodents to horses (68). While it is possible to extend our exact computations and analysis of the cross-scale mutant reproductive rate to systems with multiple mutational steps, the present analysis already provides insights into more complex evolutionary scenarios. For evolutionary trajectories that proceed through a fixed series of genotypes, the probability of emergence can be approximated by extension of our equation (3), as in previous work (11, 12, 14). If emergence requires multiple mutational steps which pass through a fitness valley, then the scale at which this valley occurs matters. A within-host fitness valley in replication rates would hinder pathogens with long-term infections and larger bottleneck widths, more than those with smaller bottlenecks. A between-host fitness valley in transmissibility could hinder evolutionary emergence of pathogens causing long-term infections more than those causing short-term infections, unless the within-host landscape is sufficiently favorable to allow traversing the valley within a single host’s long-term infection. Recent studies have also highlighted the importance of considering the broader genotype space, which can reveal indirect paths that circumvent fitness valleys (69), alternative genotypes that yield similar phenotypes (36), and the costs of higher mutation rates arising from deleterious mutants (70).
Our analysis also focuses on a simple “logistic-like” model for within-host viral dynamics. This simplification allows us to study how evolutionary emergence is limited by different factors for pathogens dominated by exponential versus saturated phases of viral growth, while maintaining analytic tractability. Future important extensions would be to allow within-host fitness to alter the carrying capacity in the saturated phase, as well as identifying the relative contributions of stochastic within-host dynamics, immune responses, and host heterogeneity on viral emergence. We have assumed that the bottleneck width N is fixed for a given pathogen. This is broadly consistent with currently available data (37–39), but it will be important to explore the consequences of variation in bottleneck width arising from different routes of transmission, or possibly from changing viral loads. The computational and analytical framework developed here can be extended to account for these additional complexities. Other important extensions can explore the impact of clonal competition on emergence probabilities (71–73) or the potential for complementation to rescue pathogen strains from deep fitness valleys-a mechanism that depends on wide transmission bottlenecks (74).
Our cross-scale analysis opens the door for a new generation of integrative risk assessment models for pathogen emergence, which will integrate growing streams of data collected in laboratories and field surveillance programs (66). At present there is no framework, other than the intuition of individual scientists, to link together the discoveries from targeted experiments, massively parallel phenotypic screens, experimental evolution, clinical medicine, and field epidemiology and disease ecology. Mathematical and computational models that connect biological scales using mechanistic principles can make unique contributions to this transdisciplinary enterprise, by formally integrating diverse empirical findings and by identifying the crucial knowledge gaps to focus future research. The work presented here is a step on the path to realizing this potential.
Models and Methods
The cross-scale model with explicit within-host dynamics
The cross-scale dynamics are modeled as a continuous time, age-dependent, multi-type branching process (Fig 1). The “type” of individual corresponds to their initial viral load, and the “age” of an individual corresponds to the time since their initial infection. Within an infected host, the viral dynamics determine how the viral load and composition changes over time due to competition between strains and mutation events. Transmission events are determined by the viral load and composition of the host and, consequently, are age-dependent.
Due to ultimately large viral loads, the within-host dynamics are modeled with coupled differential equations where v(t) = (vw(t), vm(t)) denotes the vector of viral abundances:

At time t = 0, v(0) = (vw(0), vm(0)) corresponds to the initial viral load of an infected individual.
The number of contacts of an infected during the infectious period is Poisson distributed with mean βT. On the event of a contact at t days after becoming infected, the probability of transmission equals p(bw vw(t) + bm vm(t)). On the event of transmission, the probability of infecting an individual with a viral load of  with
with  equals
equals

Under these assumptions, during their infectious period, an infected individual of type v(0) infects a Poisson distributed number of individuals with viral load  and the mean of this distribution equals
and the mean of this distribution equals

Methods
To solve the probabilities of emergence, we use the discrete-time branching process given by censusing the infected the population at the beginning of each generation of infection i.e at times 0, T, 2T, etc. All the statistics of this process are given by the probability generating map G: [0, 1]N+1 → [0,1]N+1 where N + 1 is the number of types of initial viral loads. We index the coordinates by the initial number of mutant virions 0, 1, 2,…, N within an infected individual and have

By the limit theorem for multitype branching processes, the i-th coordinate of
 is the probability of extinction by generation t when there is initially one infected individual with initial viral load (i, N − i).
is the probability of extinction by generation t when there is initially one infected individual with initial viral load (i, N − i).
For the numerical work, we used linear, logarithmic, and saturating functions for the transmission probability function p. All gave similar results but we present the linear case as most analytical results were derived for this case. To compute the extinction probabilities, we iterated the generation map G for 2,000 generations. For the individual based simulations, we solved the within-host differential equations using matrix exponentials and renormalizing these exponentials when the viral load reached the value K. Between host transmission events where determined by a time-dependent Poisson process with rate function p(bw vw(t) + bm vm(t)) and mulitnomial sampling was used to determine the initial viral load of an infected individual.
Supplementary Information
Derivation of the Single Strain Reproductive Numbers
When the within-host dynamics only exhibit exponential growth (i.e. N exp(riT) < K) and there is a linear transmission function, the basic reproductive numbers equal

When the within-host dynamics saturate (i.e. N exp(riT) > K), the basic reproductive number equals
 where Te = log(K/N)/ri is the length of exponential phase and Ts = T − Te is the length of saturated phase.
where Te = log(K/N)/ri is the length of exponential phase and Ts = T − Te is the length of saturated phase.
We derive two approximations of Ri under the assumption that s is small, exp(riT) ≫ 1, and K ≫ N. First, assume that the infection is short-term in which case Te = T. Then provided s is sufficiently small to ensure that the mutant type doesn’t saturate, Ri are given by (S–1). The log ratio, provided exp(riT) ≫ 1, satisfies
 which yields (1) in the main text.
which yields (1) in the main text.
Now assume that the infection is long-term in which case Te < T, and that if s < 0, |s| is sufficiently small to ensure that the mutant type also saturates before time T. Then the basic reproductive numbers Ri are given by (S–2). If K ≫ N, then

Assume that |s| ≪ rw. Then

As T = Ts + Te and
 it follows that
it follows that

As log(1 + x) ≈ x for small x and |s| ≪ rw by assumption, we obtain

Equation (2) in the main text follows in the case that Ts ≫ Ts in which case the second term is approximately zero.
Derivation of the Emergence Probability Approximation
For small mutation likelihood μ, we derive a mathematically explicit version of the approximation (3) for the emergence probability from the main text. As stated in the main text, this approximation is given by the product of three terms: the expected number of secondary, wild-type cases produced during a fade-out, the mean number of individuals infected with mutant virions by an individual initially infected only with the wild-type, and the probability of emergence from an individual infected with a single mutant virion. As noted in the main text, the first term is given by  . The second term requires more work. To derive an analytic approximation for this term, notice that the mean number of individuals infected with í mutant virions by an individual only infected with the wild type equals
. The second term requires more work. To derive an analytic approximation for this term, notice that the mean number of individuals infected with í mutant virions by an individual only infected with the wild type equals
 where vw(t), vm(t) is the solution of the within host viral dynamics with vw(0) = N, vm(0) = 0, and
where vw(t), vm(t) is the solution of the within host viral dynamics with vw(0) = N, vm(0) = 0, and  is the probability of an individual with viral load (vw, vm) infecting an individual with a viral load of
is the probability of an individual with viral load (vw, vm) infecting an individual with a viral load of  where
where  . The solution (vm(t), vm(t)) is given by
. The solution (vm(t), vm(t)) is given by
 where Vw(t), Vm(t) are the solutions to
where Vw(t), Vm(t) are the solutions to

Ignoring back mutations (i.e. setting rmμ = 0 and rm(1 − μ) to rm), the solutions for Vw(t), Vm(t) are approximately
 if rw ≠ rm, and
if rw ≠ rm, and
 if rw = rm = r. Since
if rw = rm = r. Since  to first order near 0, the weighted frequency, xm(t), of mutant strain is approximately
to first order near 0, the weighted frequency, xm(t), of mutant strain is approximately
 if s ≠ 0, and
if s ≠ 0, and
 if s = 0. We have
if s = 0. We have

For ℓ ≥ 2, these terms are of order μ2 and therefore will be ignored. Hence, the only term of interest is ℓ = 1:

We also can approximate (assuming p is differentiable)

We drop the O(μ) term as it will only lead to higher order terms in the approximation.
Putting all of this together gives the following approximation for the mutant force of infection
 if s ≠ 0, and
if s ≠ 0, and
 if s = 0.
if s = 0.
In the case of a linear transmission function p(x) = x, we can write down explicit expressions for (S–3) and (S–4). There are two cases to consider. First suppose that N exp(rwT) ≤ K i.e. the infection is short-term. Then, integrating and simplifying yields the following approximation for the mutant force of infection

Assuming rw ≫ s (and thus rw/(rw + s) ≃ 1 − s/rw),
 if s ≠ 0 and
if s ≠ 0 and
 if s = 0. Now rather than writing out the entire expression for the case N exp(RwT) ≥ K, lets write down things for when the time in the saturated phase is much, much longer than the time in the exponential phase. Then, integrating and simplifying yields the following approximation for the mutant force of infection
if s = 0. Now rather than writing out the entire expression for the case N exp(RwT) ≥ K, lets write down things for when the time in the saturated phase is much, much longer than the time in the exponential phase. Then, integrating and simplifying yields the following approximation for the mutant force of infection
 if s ≠ 0, and
if s ≠ 0, and
 otherwise.
otherwise.
Putting this all together, (3) for an short-term (respectively long-term) infection with s ≠ 0 becomes the product of  , (S–5) (respectively (S–6)), and the probability of an outbreak starting with one individual infected with N − 1 wild type virions and 1 mutant type virions. The final probability term can be calculated exactly using the generating functions described in the Models and Methods section of the main text. Fig S–1 illustrates the effectiveness of this approximation, and Fig S–2 plots the the error in the approximation.
, (S–5) (respectively (S–6)), and the probability of an outbreak starting with one individual infected with N − 1 wild type virions and 1 mutant type virions. The final probability term can be calculated exactly using the generating functions described in the Models and Methods section of the main text. Fig S–1 illustrates the effectiveness of this approximation, and Fig S–2 plots the the error in the approximation.

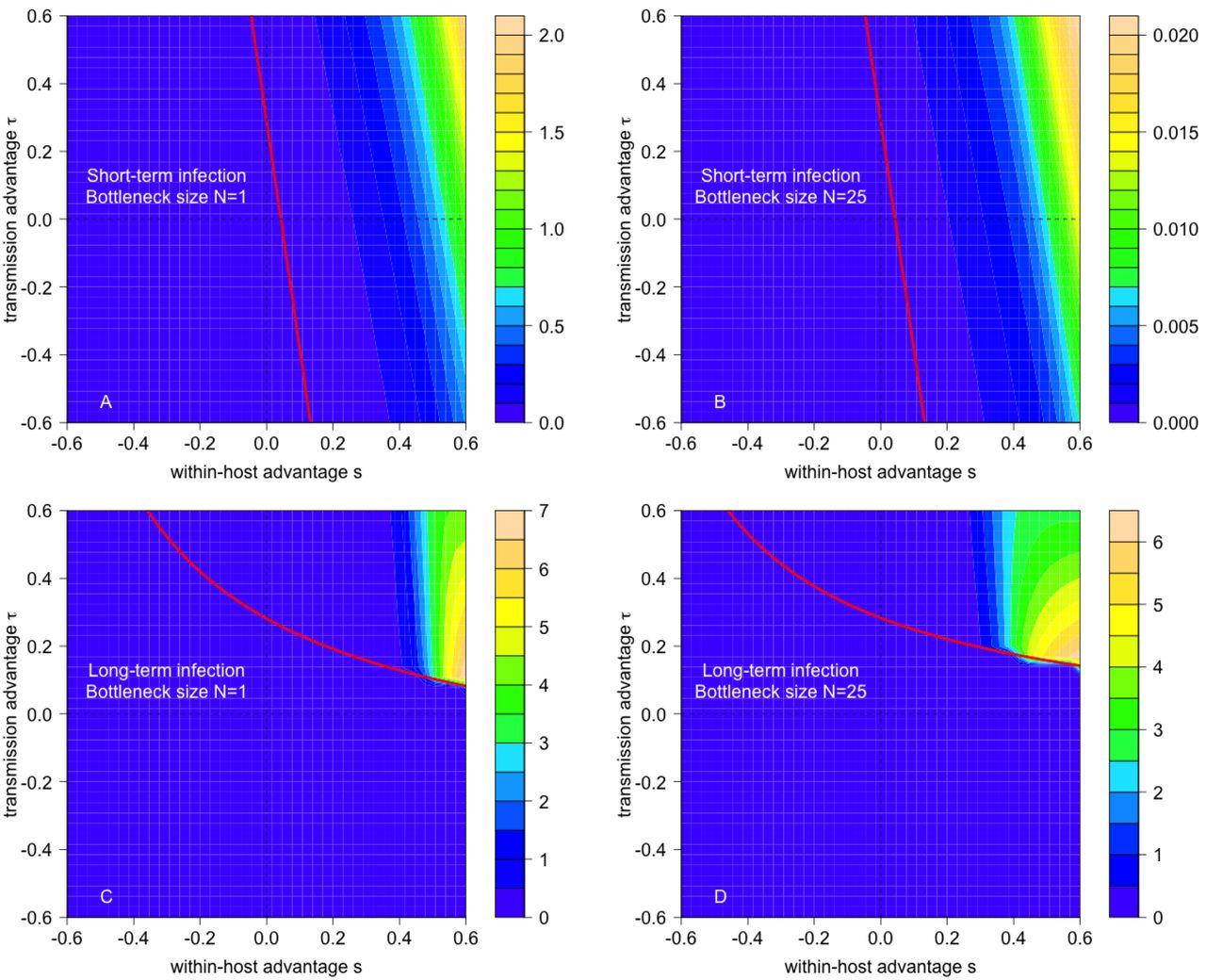
The analytic approximation based on (3) for the exact computations of the emergence probabilities in Fig 2 in the main text.
Contour plots of 1, 000 × the absolute value in the difference between the analytic approximation from (3) and the exact computations for the emergence probabilities. Parameters as in Fig 2 in the main text.
Derivation of the Mean Field Frequency Dynamics
To understand how the viral composition of infected individuals change across generations, we derive a mean field approximation for the dynamics of the mean mutant viral load at the beginning of each generation of disease spread. To this end, we define a map from h: [0, 1] → [0, 1] where x ∈ [0, 1] represents the current mean mutant viral load in the population at the beginning of the infectious period and h(x) is the mean at the beginning of infectious period in the next generation. Our derivation of this mean field dynamic is done in the limit of large N ↑ ∞ and μ ↓ 0. None-the-less, as shown by the dashed red line in Fig 2D, this approximation works quite well away from this limit.
We begin by approximating the mean initial mutant viral count in individuals infected by an individual with Vw(0) = N − ℓ and Vm(0) = ℓ. Recall, the force of infection for producing individuals initially infected with j mutant viral particles is given by
 where
where  is the within-host frequency of the mutant strain, and (vw(t), vm(t)) is the solution of the within-host viral dynamics with initial condition vw (0) = N − ℓ, Vm(0) = ℓ. Weighting this term by j and summing over j yields the expected number of mutant viral particles in an individual infected by our type (N − ℓ, ℓ) infected individual:
is the within-host frequency of the mutant strain, and (vw(t), vm(t)) is the solution of the within-host viral dynamics with initial condition vw (0) = N − ℓ, Vm(0) = ℓ. Weighting this term by j and summing over j yields the expected number of mutant viral particles in an individual infected by our type (N − ℓ, ℓ) infected individual:

Now if we let x = ℓ/N denote the initial fraction, then dividing the previous integral by the net number of viral particles infecting new individuals yields our desired update rule

Note that h(x) is a function of x as the solution of (Vw(t), Vm(t)) depends on its initial condition Vw(0) = (1 − x)N, Vm(0) = xN.
The points x = 0 and x = 1 are fixed points for h corresponding to a mutant-free and wild-type-free states.
Stability of the fixed point x = 0 is determined by
 for N ≫ 1. h′(0) corresponds to α described in the main text and the final expression has the verbal interpretation given in the main text.
for N ≫ 1. h′(0) corresponds to α described in the main text and the final expression has the verbal interpretation given in the main text.
In the special case of a linear transmission function, p(x) = x, we get the simplified expression
 where
where

Carrying out the integration, in general, is complicated by the fact that the time at which Vw(t) + Vm (t) = K has no explicit formula when s ≠ 0 and, in general, this saturation time will depend on x.
In the special case of short-term infections (i.e. there is only exponential growth), we get
 where
where

α is defined as h′(0), which here is equal to n, thus for short-term infections, α = Rm/Rw.
Since Rw < 1 by assumption and Rm > 1 is necessary for emergence, we always have α > 1 and so the frequency dependent dynamics at the scale of the host population can not significantly impede emergence.
Now, lets consider the more difficult case of a long-term infection with a saturated phase to the within-host viral dynamics. Then

For x close to 0, we have the time at which the dynamics saturate, Te, is given approximately by
 in which case
in which case

Let Ts = T − Te and assume that  where
where  is the length of the exponential phase for an individual infected only with the mutant strain. Then
is the length of the exponential phase for an individual infected only with the mutant strain. Then
 as claimed in the main text.
as claimed in the main text.
Estimating the Probability of Emergence when α < 1
When α is less than 1, the frequency of mutant virus decrease in an infected host, and consequently, even if the adapted virus may emerge, the probability of emergence is very low, and even lower when the bottleneck size, N, increases. Here, we provide an approximation for the emergence probability when α < 1, which explains why the probability of emergence decreases dramatically when N increases.
When the outbreak starts, the first individual is infected with wild-type only. When s < 0, the mutation-selection balance can be reached relatively quickly, and for s negative enough, the proportion of mutant is small. So the probability to transmit at least one mutant is roughly equal to the probability to transmit one mutant, which is N exp(τ)rwμ/|s| where rwμ/|s| is the proportion of the mutant type, and exp(τ) is its relative transmissibility. Then, if s is small enough, then the reproductive number of an individual with a mixed transmission is close to Rw of the wild-type. Thus, the number of transmissions in a wild-type outbreak can be used (Rw/(1 − Rw)). For an individual infected with a mixed infection, what will lead to emergence are the contacts for which only the mutant is transmitted. The number of such contacts is:

This can be re-written as:

As most cases of mixed infection will be cases started with a mix of one mutant and N − 1 wild-type viral particles, Vm/Vw = exp(st)/(N − 1). Thus previous expression is equal to:

Last, an individual infected with mutant viruses alone has to lead to a successful outbreak, which happens at approximately the same probability than in the case with no back mutations, with probability pm. So overall, the approximation will be:

Now we can ask, which parts of this expression depend on N? The mutant reproductive number  is independent from N, because we have chosen bw to keep Rw the same for all N values. Thus
is independent from N, because we have chosen bw to keep Rw the same for all N values. Thus  is almost independent from N. Therefore, most of the dependence of
is almost independent from N. Therefore, most of the dependence of with N stems from the dependence of f (t, N) with N. Since a ↦ a/(1 + a) is an increasing function bounded above by 1 for positive a, the expression
with N stems from the dependence of f (t, N) with N. Since a ↦ a/(1 + a) is an increasing function bounded above by 1 for positive a, the expression
 decreases whenN increases. As N ↦ (a/(1 + a))N − 1 is a decreasing function of N ≥ 1 for a > 0, we get that the probability of emergence decreases at least exponentially with the bottleneck size, as claimed in the main text. Fig S–3 illustrates that these approximations work especially when s is sufficiently negative.
decreases whenN increases. As N ↦ (a/(1 + a))N − 1 is a decreasing function of N ≥ 1 for a > 0, we get that the probability of emergence decreases at least exponentially with the bottleneck size, as claimed in the main text. Fig S–3 illustrates that these approximations work especially when s is sufficiently negative.

Approximation (S–12) (dashed lines) vs. numerical resolution of the generating maps (R code) (solid lines)
Numerics with Nonlinear Transmission Functions
To explore the robustness of our numerical results to the assumption of a linear transmission function, we redid our numerical analysis with two non-linear transmission functions p(x) = 1 − exp(−x) in Fig S–4 and p(x) = log(1 + x) in Fig S–6. Differences between the emergence probabilities for the nonlinear and linear transmission functions are shown in Figs S–5 and S–7. As these figures demonstrate, we nearly get the same results.

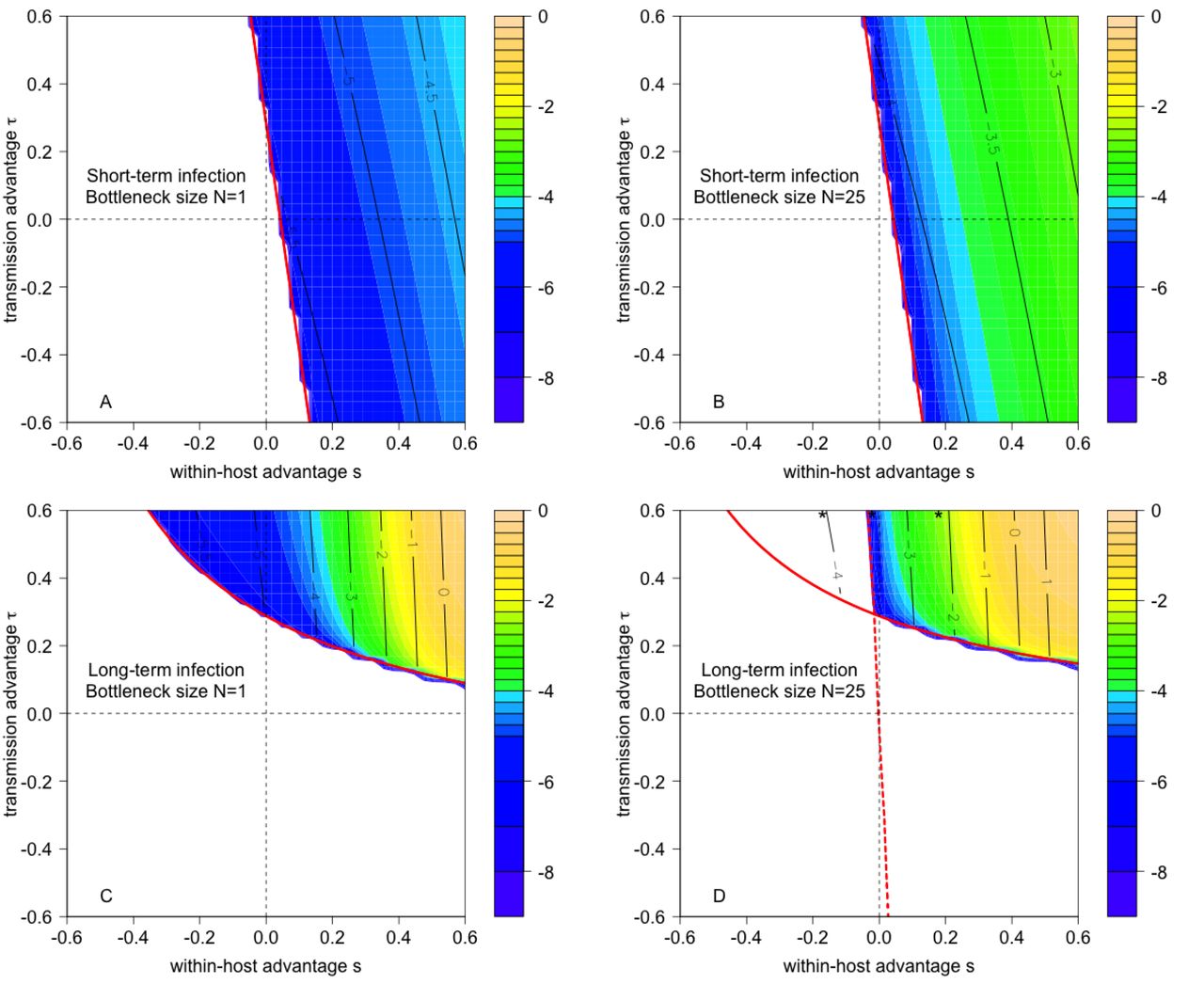
Emergence probabilities for the transmission function p(x) = 1 − exp(−x) with all other parameters as indicated in Fig 2.

Contour plots of 1, 000 × the absolute value of the difference between the emergence probabilities for the transmission functions p(x) = 1 − exp(−x) and p(x) = x. Parameters as indicated in Fig 2.

Emergence probabilities for the transmission function p(x) = log(1 + x) with all other parameters as indicated in Fig 2.
Contour plots of 1, 000 × the absolute value of the difference between the emergence probabilities for the transmission functions p(x) = log(1 + x) and p(x) = x. Parameters as indicated in Fig 2.
Acknowledgements
This work was supported by U.S. National Science Foundation Grants EF-0928987 and DMS-1313418 to SJS, and EF-0928690 to JLS.
Footnotes
↵** jlloydsmith{at}ucla.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}