

Abstract
Multiple pathogens have been detected in Alzheimer’s disease (AD) brains. A bioinformatics approach was used to assess relationships between pathogens and AD genes (GWAS), the AD hippocampal transcriptome and plaque or tangle proteins. Host/pathogen interactomes (C.albicans, C.Neoformans, Bornavirus, B.Burgdorferri, cytomegalovirus, Ebola virus, HSV-1, HERV-W, HIV-1, Epstein-Barr, hepatitis C, influenza, C.Pneumoniae, P.Gingivalis, H.Pylori, T.Gondii, T.Cruzi) significantly overlap with misregulated AD hippocampal genes, with plaque and tangle proteins and, except Bornavirus, Ebola and HERV-W, with AD genes. Upregulated AD hippocampal genes match those upregulated by multiple bacteria, viruses, fungi or protozoa in immunocompetent blood cells. AD genes are enriched in bone marrow and immune locations and in GWAS datasets reflecting pathogen diversity, suggesting selection for pathogen resistance. The age of AD patients implies resistance to infections afflicting the younger. APOE4 protects against malaria and hepatitis C, and immune/inflammatory gain of function applies to APOE4, CR1, TREM2 and presenilin variants. 30/78 AD genes are expressed in the blood brain barrier (BBB), which is disrupted by AD risk factors (ageing, alcohol, aluminium, concussion, cerebral hypoperfusion, diabetes, homocysteine, hypercholesterolaemia, hypertension, obesity, pesticides, pollution, physical inactivity, sleep disruption and smoking). The BBB and AD benefit from statins, NSAIDs, oestrogen, melatonin and the Mediterranean diet. Polymicrobial involvement is supported by the upregulation of pathogen sensors/defenders (bacterial, fungal, viral) in the AD brain, blood or CSF. Cerebral pathogen invasion permitted by BBB inadequacy, activating a hyper-efficient immune/inflammatory system, betaamyloid and other antimicrobial defence may be responsible for AD which may respond to antibiotic, antifungal or antiviral therapy.
Introduction
Multiple pathogens have been implicated in Alzheimer’s disease (AD) either via detection in the AD brain, or in epidemiological studies relating to serum antibodies. Pathological burden (cytomegalovirus, Herpes simplex (HSV-1), Borrelia burgdorferi, Chlamydia pneumoniae and Helicobacter pylori) rather than any individual pathogen may also be associated with AD [1]. Many pathogens are able to increase beta-amyloid deposition and tau phosphorylation in animal models, in vitro or in vivo and beta-amyloid itself is an antimicrobial peptide active against bacteria and fungi [2,3]and the influenza[4] and herpes simplex viruses [5,6]. These effects are summarised in Table 1 for a number of pathogens and for beta-amyloid.
The effects of diverse pathogens on beta-amyloid deposition, tau phosphorylation and their relationships with Alzheimer’s disease.
Previous studies have shown that the life cycles of several pathogens implicated in AD relate to AD susceptibility genes [7]. The proteins found in AD plaques and tangles are also enriched in those used by HSV-1 during its life cycle [8] and the HSV-1 or Toxoplasma Gondii host interactomes are also enriched in AD susceptibility genes [9,10]
Similar studies have noted significant overlaps between the Epstein-Barr viral/host interactome and diseases in which the virus is implicated, including B cell lymphoma [11]or multiple sclerosis [12]. The interactomes of oncogenic viruses also relate to cancer genes [13] suggesting important gene/environment interactions that may condition disease susceptilbility.
In this study, the host pathogen interactomes of 17 fungal, bacterial, viral and parasite pathogens were analysed in relation to 78 AD genes derived from genome-wide association studies (GWAS). The anatomical location of these genes was also queried against proteomic /genomic datasets from multiple tissues.
The host genes of the pathogen interactomes were also compared with the combined up and downregulated genes from a study of the AD hippocampus, post-mortem [14] and to the proteins found in plaques or neurofibrillary tangles. The upregulated genes from this AD hippocampal study were also compared with upregulated genes from numerous infection microarray datasets (viral, bacterial, fungal and protozoan) housed at the Molecular signatures database [15] or the Gene Expression Omnibus [16].
Pathogens have shaped human evolution, as the survivors of dangerous infections are endowed, via natural selection, with genes conveying resistance. The AD genes were also compared against a series of genome-wide association datasets related to general pathogen or protozoan diversity, viral diversity and the immune response to parasitic worms, across multiple human populations in different geographical locations. Such genes are likely to have been selected for pathogen resistance. [17–20].
The results show that host genes related to pathogens are enriched in all these AD parameters and that many AD susceptibility genes also relate to pathogens, but more likely to pathogen resistance than susceptibility. The anatomical data point to an immune function of many AD genes, while others are localised in the blood-brain barrier, which is disrupted by other environmental risk factors associated with AD.
Methods
The host/pathogen interactomes of two fungal species (Candida albicans, Cryptococcus Neoformans), the Borna virus, human cytomegalovirus, Ebola virus, Herpes simplex (HSV-1), human endogenous retroviruses HERV-W, the human immunodeficiency virus (HIV-1) (the latter from the HIV-1, human interaction database [21]
http://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions, Epstein-Barr, hepatitis C and influenza A viruses, 3 bacterial species (Chlamydia Pneumoniae, Porphyromonas Gingivalis, Helicobacter Pylori) and 2 protozoans (Toxoplasma Gondii and Trypanosoma Cruzi) were obtained by literature survey and from extant databases. These referenced interactomes can be accessed at http://www.polygenicpathways.co.uk/HPI.htm.
Genes misregulated in the AD hippocampus are those reported from a post-mortem microarray study [14]. Up-and downregulated genes (N=2879) were combined for comparison with the pathogen interactomes. These interactomes contain multiple types of interaction (protein/protein, viral microRNA, and effects on transcription etc.) and it is not possible to compare like with like for this aspect.
The upregulated genes (N= 1690) from this AD hippocampal study contain the pathways relevant to pathogens and immune activation (inflammation, complement activation and the defence response) [14] and these were chosen for comparison with upregulated genes from infection datasets at the Molecular signatures database (MSigDB) http://software.broadinstitute.org/gsea/msigdb/index.jsp. MSigDB contains several thousand microarray gene sets which can be compared against the AD input [15]. Infection related datasets, and those related to Toll-like receptor ligands, were identified using search terms (e.g. infection, virus, bacteria, TLR1, lipopolysaccharide, etc.). Microarray viral infection datasets (upregulated gene sets) from the gene expression omnibus (GEO) [22] were also downloaded from the Harmonizome database http://amp.pharm.mssm.edu/Harmonizome/ from the Ma'ayan laboratory of computational systems. [23]. For the searched gene sets, most of the data outputs were restricted at source (by MSigDb or GEO) to the top upregulated genes (usually ~ 200-300).
The proteins found in plaques or neurofibrillary tangles are from two proteomics studies yielding 488 proteins in plaques [24] and 90 in tangles [25].
Seventy eight genes associated with Alzheimer’s disease (Reported genes) were obtained from the NHGRI-EBI Catalog of published genome-wide association studies (GWAS) [26], Available at: www.ebi.ac.uk/gwas. Accessed January, 2016, version 1.0 using studies labelled as “Alzheimer’s disease” or “Alzheimer’s disease late-onset”. These genes and their relationships with pathogens or the immune system are catalogued in Supplementary Table 1. These genes are highlighted in bold throughout the text.
Genes related to general pathogen diversity, protozoan and viral diversity and to the immune response to parasitic worms are from a series of papers concerning evolutionary selection pressure relevant to pathogen resistance [17–20].
The tissue and cellular distribution of the 78 AD genes were analysed using the functional enrichment analysis tool (FUNRICH) [27]. http://funrich.org/index.html. This tool derives proteomic and genomic distribution data from >1.5 million annotations. It provides the total number of genes in datasets from each region sampled and returns the significance of any enrichment for members of the uploaded AD genes, using the hypergeometric probability test, with p values corrected using the the Storey and Tibshirani method (Q values) [27]. AD gene enrichment was also analysed in a published blood brain barrier proteome dataset of mouse cerebral arteries (6620 proteins) [28].
The presence of the AD genes in exosomes, a means of transit through cells allowing intercellular communication[29,30], was assessed using ExoCarta (http://www.exocarta.org) a manually curated database of exosomal proteins, RNA and lipids [31].The exosomal pathway is hijacked by several viruses, contributing to intercellular spread and immune evasion [32,33].
Assuming a human genome of 26846 coding genes and an interactome or other gene set of N genes one would expect N/26846 to exist in the comparator dataset. For example, when comparing 2879 misregulated AD hippocampal genes against any pathogen interactome one would expect 2879/26846 (10.7%) to figure in the pathogen interactome. This calculation was used to define expected values and the enrichment values (observed/expected) in relation to other datasets. Significance of the enrichment was calculated using the hypergeometric probability test. The resultant p values from each analysis series were corrected for false discovery (FDR) [34]. Nominally significant FDR corrected values are considered at P <0.05 and a corrected Bonferroni p value threshold is illustrated on each set of graphs. (Bonferroni P = 0.05/N, where N is the maximum number of possible comparisons for each situation (e.g. 78 AD genes or 1690 upregulated genes in the AD hippocampus).
Results
The anatomical location of the AD genes (Fig 1)

The distribution and enrichment of 78 AD genes in diverse proteomic and genomic datasets (Funrich and Exocarta data). The bars indicate the number of genes (from 78) in each tissue and the dotted line the corrected p value (q value). The maximum on this axis is set to q = 0.05. Observed/expected values, followed by the total number of genes expressed are appended after the identities of each sample. BBB refers to a separate blood brain barrier proteomics dataset. Cancer or cell line datasets are omitted and the data are limited to anatomical datasets containg more than 10 AD genes (Not all data are shown).
The AD genes are most significantly enriched in the exosome and bone marrow datasets. As noted above, exosomes are hijacked by many viruses for intercellular spread. Exosomes are prevalent in plasma [35](also enriched in AD genes) and are also the means by which intracellulary generated beta-amyloid is conveyed to the extracellular space [36]. In this context, and in relation to the antimicrobial effects of beta-amyloid, APP and gamma-secretase are highly expressed in the immune dendritic cells that scout for invading pathogens [7]. The bone marrow is the hematopoietic source of red and white blood cells and platelets [37]. B cells in the bone marrow rapidly respond to infection [38] and the bone marrow is also a source of angiogenic cells that are involved in vascular endothelial repair, a process that is disrupted in Alzheimer’s disease [39,40]. The parathyroid gland expresses many AD genes and also plays a role in hematopoesis [41,42]. Other immune related areas enriched in AD genes include the appendix, spleen, tonsils, the lymph nodes and the bronchus and neutrophils. The appendix is an important component of mucosal immune function, particularly B cell-mediated immune responses and extrathymically derived T-lymphocytes [43]. The tonsils and nasopharynx, also enriched in AD genes, play an important role in the initial defence against respiratory pathogens [44].
AD genes are enriched in the lateral ventricle, a site of the choroid plexus [45]. This provides cerebrospinal fluid (CSF) and is the location of the blood-CSF barrier, which is exploited by pathogens to gain access to the brain. The choroid plexus plays an important role in pathogen defence [46]. Post-mortem gene expression studies of the choroid plexus epithelium in AD patients show changes indicative of increased permeability of the blood-cerebrospinal fluid barrierand a reduction of macrophage recruitment [47], factors that woud favour pathogen entry and reduce their phagocytosis by macrophages. The hippocampus bulges into the temporal horn of the lateral ventricle [48] and this area, a keystone of AD pathology, is thus in close proximity to a major site of cerebral pathogen entry. AD genes are also enriched in a separate BBB dataset from mouse cerebral arteries. This is discussed in greater detail below. Other barriers in intestinal and pulmonary tissues, also enriched in AD genes (Fig 1), might also be considered as potential sites of pathogen entry. Immune systems play an important role at barrier interfaces [49].
Although AD genes are expressed in other sites, the main focus, in terms of enrichment, relates to immune and barrier systems.
A number of the 78 AD genes (referenced in supplementary Table 1) are primarily concerned with immune function (HLA-DRB1, HLA-DRB5, HMHA1, IGH) while many others with diverse primary effects also possess relevant properties in relation to the immune system (ACE, ADAMTS20, AP2A2, BCL3, BIN1, CR1, CLU, CUGBP2, DISC1, EPHA1, GAB2, INPP5D, MEF2C. MS4A3, MS4A4A, RIN3, SCIMP, SPPL2A, STK24, TREM2, TREML2, ZNF224) or pathogen defence (e.g. phagocytosis or autophagy) (ABCA7, APOC1, APOE, BCAM, CD2AP, CD33, CDON, CELF1, PAX2, PTK2B, SASH1, SQSTM1). A number of the AD genes also act as primary receptors for pathogens. These include the poliovirus receptor PVR, the HSV-1 receptor PVRL2, and complement receptor (CR1), which binds to many opsonised pathogens but which may also act as an entry receptor for Plasmodium falciparum, Legionella pneumophila and Mycobacterium tuberculosis. CD33 binds to the HIV-1 gp120 protein and to diverse forms of sialic acid which coats many pathogens. Others bind bacterial lipopolysaccharides (APOC1 and TREM2) or the Escherichia coli cytotoxic necrotizing factor 1 (BCAM). Others (AP2A2, BIN1, CD2AP, and PICALM) are involved in endocytosis, an obligate requirement for pathogen entry following binding to cognate receptors (see supplementary Table 1 for references).
Host/pathogen interactomes are enriched in AD genes (Fig 2)

The number of AD genes (of 78) overlapping with diverse host/pathogen interactomes, or with those implicated in pathogen, protozoan or viral diversity or with the immune response to parasitic worms (Helminth) (Bars). The identities on the X-axis (e.g. C. albicans (1471|5.2) are appended with the total number of genes in each interactome (1471 in this case) or genetics dataset followed by the enrichment ratio (5.2 fold). The FDR-corrected p value for enrichment, derived from the hypergeometric distribution, is shown on the right hand axis (log scale) which is set to a maximum of 0.05. Invisible points are above this value. The Bonferroni cut-off level (p=0.05/78) is also shown. The Burden data (lighter shaded bar) correspond to the combined interactomes and AD gene overlaps of the human cytomegalovirus (HCMV), HSV-1, Borrelia burgdorferi, Chlamydia pneumoniae and Helicobacter pylori. EBV= Epstein-Barr virus.
All host/pathogen interactomes, with the exception of those of the Borna virus, Ebola virus and the HERV-W retrovirus were significantly enriched in AD genes (FDR p <0.05) with all but HIV-1, the cytomegalovirus and C. pneumoniae below the Bonferroni corrected value (P=6.41E-4). Pathogen burden (cytomegalovirus, HSV-1, B. burgdorferi, C. Pneumoniae and H. Pylori) has been associated with Alzheimer’s disease [1] and the pooled interactomes of these five pathogens (3922 host genes) were significantly enriched in AD genes (p= 7.3E-6). Given the variety of pathogens reported in AD brains (Table 1) other cumulative effects might be expected for various permutations.
The most significant pathogens related to fungi (C. albicans and C. Neoformans), the gum disease pathogen P. Gingivalis and the Epstein-Barr and hepatitis C viruses. Numerous fungal species, including C.albicans, have been detected in the AD brain (Table 1), although C. Neoformans was not one of the species studied. Two case reports have demonstrated virtually complete recovery from long-term (3 years) mis-diagnosed dementia/Alzheimer’s disease following antifungal treatment for C. Neoformans infection [50,51].
The Epstein-Barr virus has been associated with AD and hepatitis C associated with dementia (table 1). In vivo studies for the Epstein-Barr and Hepatitis C viruses are however limited by their inability to infect rats or mice. Several of these pathogens including C. pneumoniae, HSV-1, cytomegalovirus and the Epstein-Barr and hepatitis C viruses or H. pylori and B. Burgdorferri [52–59], periodontitis and P.Gingivalis [57]have also been associated wiith atherosclerosis, an important endophenotype in AD [60].
Apart from APOE4 no AD genetic variants seem to have been studied in relation to effects on pathogens and it is impossible to note whether the variants favour or oppose their destructive potential. The apolipoprotein E (APOE4) variant protects against hepatitis C [61], but favours the cerebral entry of HSV-1 [62]and enhances the attachment of C. pneumoniae elementary bodies to host cells [63].
AD genes overlap with those implicated in pathogen, protozoan or viral diversity or with the immune response to parasitic worms (Fig 2)
The AD genes are enriched in a series of genome-wide and global-wide datasets related to general pathogen diversity, protozoan or viral diversity (the number of different pathogens in a geographic region) or with the immune response to parasitic worms, most significantly so for general pathogen and protozoan diversity (FDR p < 0.05). The overlaps in relation to viral diversity or the response to parasitic worms exceeded the Bonferroni cut-off.
In evolutionary terms, these pathogen-related genes likely reflect pathogen resistance rather than susceptibility [17–20].
It has also been noted that genes related to inflammatory diseases [64] or to the AD gene network [65]are subject to positive selection pressure. While many pathogens have been implicated in AD, the selection of AD genes for pathogen resistance rather than susceptibility seems logical in relation to several considerations, as already proposed [66,67]. Firstly, the old age of AD patients indicates survival from the many infectious diseases that are among the principal causes of death in adults and children. In the USA, the leading non-accidental causes of death in adults (2013 figures) include heart disease; cancers; chronic lower respiratory diseases; cerebrovascular diseases; Diabetes mellitus; Influenza and pneumonia; nephritis, nephrotic syndrome and nephrosis [68].
Certain viruses, helminths and bacteria are oncogenic and it has been estimated that 15-20% of cancers are due to infections [69]. The inverse association between the incidence of cancer and Alzheimer’s disease [70] suggests that AD genes might well be cancer protective (but also that death due to cancer precludes AD). Inflammatory heart diseases [71] and atherosclerosis, cerebrovascular disorders and stroke have also been linked to infection [72,73]. Enteroviruses have been implicated in Type 1 diabetes mellitus [74].
The leading non-accidental causes of infant deaths were congenital malformations, deformations and chromosomal abnormalities; disorders related to short gestation and low birth weight, not elsewhere classified; newborn affected by maternal complications of pregnancy; sudden infant death syndrome; newborn affected by complications of placenta, cord and membranes; bacterial sepsis of newborn; respiratory distress of newborn; diseases of the circulatory system; and neonatal haemorrhage. Again, many of these relate to infections. In evolutionary terms, pandemics and infectious diseases have been, and in poorer countries still are, associated with high mortality.
In relation to Alzheimer’s disease, the apolipoprotein E (APOE4) variant protects against malaria [75] and hepatitis C [61], although APOE4 favours cerebral entry of the herpes simplex virus [62]and enhances the attachment of Chlamydia pneumoniae elementary bodies to host cells [63]. Malaria and hepatitis C are both associated with high mortality [76,77] and the protective effects of APOE4 would encourage its maintenance in the population, to the detriment of infection by the less virulent agents.
The APOE4 variant is also associated with enhanced immune/inflammatory responses. For example, Toll-like receptor activation (TLR3, 4) in microglia induces cyclooxygenase-2 (PTGS2), microsomal prostaglandin E synthase (PTGES), and prostaglandin E2, an effect exaggerated in APOE4/APOE4 mice [78]. APOE4 is also associated with enhanced in vivo innate immune responses in human subjects. Whole blood from healthy APOE3/APOE4 volunteers induced higher cytokine levels on ex vivo stimulation with Toll-like receptor (TLR2, 4 or 5) ligands than blood from APOE3/APOE3 patients [79]. Gain of function also applies to AD variant forms of complement receptor CR1, which are better able to bind complement component C1q or C3B [80]. C1q and C3B are opsonins that interact with complement cell-surface receptors (C1qRp, CR1, CR3 and CR4) to promote phagocytosis (including that of infectious agents) and a local pro-inflammatory response [81]. TREM2 variants in AD are also associated with enhanced inflammatory responses (upregulation of proinflammatory cytokines) [82]. In presenilin (PSEN1) mutant knockin mice, microglial challenge with bacterial lipopolysaccharide results in enhanced nitric oxide and inflammatory cytokine responses, relative to normal mice [83]. For these genes at least, this gain of immune/inflammatory function concords with selection for pathogen resistance.
It has also been noted that unaffected offspring with a parental history of AD have an enhanced inflammatory response in lipopolysaccharide-stimulated whole blood samples, producing higher levels of interleukin 1beta, tumor necrosis factor alpha and interferon gamma in response to LPS. This effect was independent of the APOE4 variant [84] suggesting that other AD genes are also endowed with gain of function in relation to the immune/inflammatory system. Monocyte-derived dendritic cells from Alzheimer’s disease patients also produce more interleukin 6 than those from healthy controls. AD monocytes stimulated with LPS also show a higher induced expression of the pro-inflammatory ICAM-1 adhesion molecule than controls [85]. Beta-amyloid also stimulates cytokine production in peripheral blood mononuclear cells (PBMC) and the production of the chemokines, RANTES, MIP-1beta, and eotaxin as well as that of CSF2 (colony stimulating factor 2 (granulocyte-macrophage)) and CSF3 (colony stimulating factor 3) is greater than controls in AD-derived PBMC stimulated with beta-amyloid [86].
Given the antimicrobial properties of beta-amyloid, any genetic variant that increase its production, at least in the periphery, might also be considered as desirable, in evolutionary terms, in relation to pathogen defence. A high percentage of AD GWAS genes are involved in APP processing [87]. The AD genetic variant of ABCA7 results in increased secretion of beta amyloid and raised beta-secretase activity in CHO-and HEK cells with the Swedish APP mutation [88], but the effects of late-onset AD variant genes on the beta-amyloid response to pathogens remain to be determined.
Host/pathogen interactome enrichment in misregulated genes of the Alzheimer’s disease hippocampal transcriptome (Fig 3)

The number of genes misregulated (combined up and down) in a microarray study of the AD hippocampus overlapping with diverse host/pathogen interactomes. The identities on the X-axis (e.g. C. albicans (1471|5.2) are appended with the total number of genes in each interactome (1471 in this case) or genetics dataset followed by the enrichment ratio (5.2 fold). The p value for enrichment, derived from the hypergeometric distribution, is shown on the right hand axis (log scale) which is set to a maximum of 0.05. The Bonferroni cut off (1.74E-05) is also shown.
All pathogen interactomes, most notably relating to influenza, C. Neoformans and Hepatitis C were highly enriched in genes relating to this microarray dataset (combined up and downregulated genes). The significance level of the interactome enrichment for most pathogens was several orders of magnitude below the Bonferroni cut off (p=1.74E-05) (Fig 3). 14/78 AD genes appear in this microarray dataset (FDR p = 0.001). Two case reports have demonstrated virtually complete recovery from long-term (3 years) mis-diagnosed dementia/Alzheimer’s disease following antifungal treatment for C. Neoformans infection [50,51]. Regarding the influenza data, bronchopneumonia, often caused by influenza, is a common final cause of death in dementia patients [89] and such recent infections close to death may well influence the data.
Regardless of the rank order, it is clear that many diverse pathogen interactomes affect several hundred genes of the 2879 misregulated in the AD hippocampus and/or that these misregulated AD genes represent a substantial percentage of the individual pathogens' interactomes (Fig 3).
Kegg pathway analysis of these misregulated hippocampal genes using the consensus path database [90] showed that many infection-related pathways were also significantly enriched (FDR p < 0.05). These included (pathogen with N genes followed by the FDR corrected p value): Epstein-Barr virus infection (74,5.5E-7); Salmonella infection (36,0.0001); Tuberculosis (57,0.0009); Epithelial cell signaling in Helicobacter pylori infection (28,0.00097); Shigellosis (27,0.001); Influenza A (54,0.003); Herpes simplex infection (56,0.0036); Vibrio cholerae infection (21,0.0089); HTLV-I infection (71,0.0096); Toxoplasmosis (37,0.013); Hepatitis B (43,0.018); Pathogenic Escherichia coli infection (20,0.02); Bacterial invasion of epithelial cells (26,0.02); Measles (38,0.04).
Upregulated genes in the AD hippocampus are enriched in genes upregulated by multiple viral, bacterial and fungal pathogens or Toll-like receptor ligands
Numerous infection-related microarray datasets exist in the Molecular signatures database or in the Gene expression omnibus (see methods), using blood cells taken from infected patients, or cells or tissues infected under laboratory conditions.
The hippocampal genes upregulated in Alzheimer’s disease were significantly enriched in upregulated genes in datasets from multiple viral species and to double stranded RNA and the viral mimic/TLR3 agonist, Polyinosinic:polycytidylic acid (poly I:C) (Fig 4). The viruses ranged from the benign (e.g. the rhinovirus that causes the common cold) to the highly malignant (e.g. the ebolavirus, rabies virus or HIV-1). They include common human infectious agents (e.g. adenovirus 5, influenza, Epstein-Barr virus, herpes simplex virus (HSV-1), measles or the Norwalk virus). Apart from HSV-1, the human cytomegalovirus, HIV-1 or hepatitis C (See Table 1) none of these have been implicated in Alzheimer’s disease or dementia. Most microarray experiments related to immunocompetent blood cells (B cells, T cells, dendritic cells, monocytes and macrophages) or to cultured cell lines. No infection-related datasets were found for microglia, the brain resident immunocompetent cells, but significant enrichment of the AD upregulated genes was observed for genes upregulated by interferon gamma in microglial cells (Fig 4). Interferon gamma plays an important role in the response to viral, bacterial and parasitic infections [91].

The number of upregulated genes (bars) from the AD hippocampal transcriptome that overlap with upregulated genes in viral infection datasets from the Molecular signatures database or the Gene expression omnibus (see methods). The effects of the mimic poly(I:C) are also shown, as is the effect of interferon gamma on gene expression in microglial cells. For each datapoint, the name of the virus is shown, followed by the cell type and the total number of upregulated genes in the viral datasets (limited by MSigDb or GEO). The significance of enrichment (right axis) represents the FDR corrected p value from the hypergeometric test. All values are below the Bonferroni corection (0.05/300 = 1.67E-04). Because the number of downloaded genes is mostly limited to 300, this is the maximum number of possible overlaps. The pale bar represents the microglial response to interferon gamma.
Tissue/cell abbreviations; A549= adenocarcinomic human alveolar basal epithelial cells; ABL = Akata Burkitt’s lymphoma cells; B2B/16HBE, BE(2)C or BEAS-2B = human bronchial epithelial cells; BroLav = human bronchial lavage; Calu-3 = Cultured Human Airway Epithelial Cells; DC = dendritic cells; GRE = glioma cell line; HAE = human airway epithelial cells; HBEC = Human Bronchial Epithelial Cells; HEK = human embryonic kidney cells; HeLa = cervical cancer cell line; HuH-7 = hepatocarcinoma cell line; Macro = macrophage; Mgli = microglia; Mono =monocytes; NES = human nasal epithelial scrapings; NK = natural killer cells; PBMC = peripheral blood mononuclear cells; PLC/PRF/5 cells = human liver hepatoma cells; Trach epi = Tracheal epithelial cells
Viral abbreviations (Reading from the left): HIV= human immunodeficiency virus, Cox B3 =Coxsackie B3 virus; RSV = respiratory syncytial virus; LCMV= Lymphocytic Choriomeningitis Virus; HMPV= Human metapneumovirus; Ebola= Ebola virus; Influenza = Influenza A virus; Sendai = Sendai virus, HCoV = human coronavirus; IFNG = interferon gamma; SARS = severe acute respiratory syndrome coronavirus; HCMV = human cytomegalovirus; MCMV = mouse cytomegalovirus; Dhori = Dhori virus; EBV = Epstein-Barr virus; HepC = hepatitis C virus; KSHV= Kaposi’s sarcoma-associated herpesvirus; HSV-1 = herpes simplex; Norwalk = Norwalk virus (Norovirus); Ad5 = adenovirus 5; SIV= Simian immunodeficiency virus; poly(I:C)= Polyinosinic:polycytidylic acid (a viral mimic stimulating Toll-like TLR3 receptors); NDV = Newcastle disease virus; WestEq = Western equine encephalomyelitis virus; LASV = Lassa virus; dsRNA = double stranded RNA; HEV = hepatitis E virus.
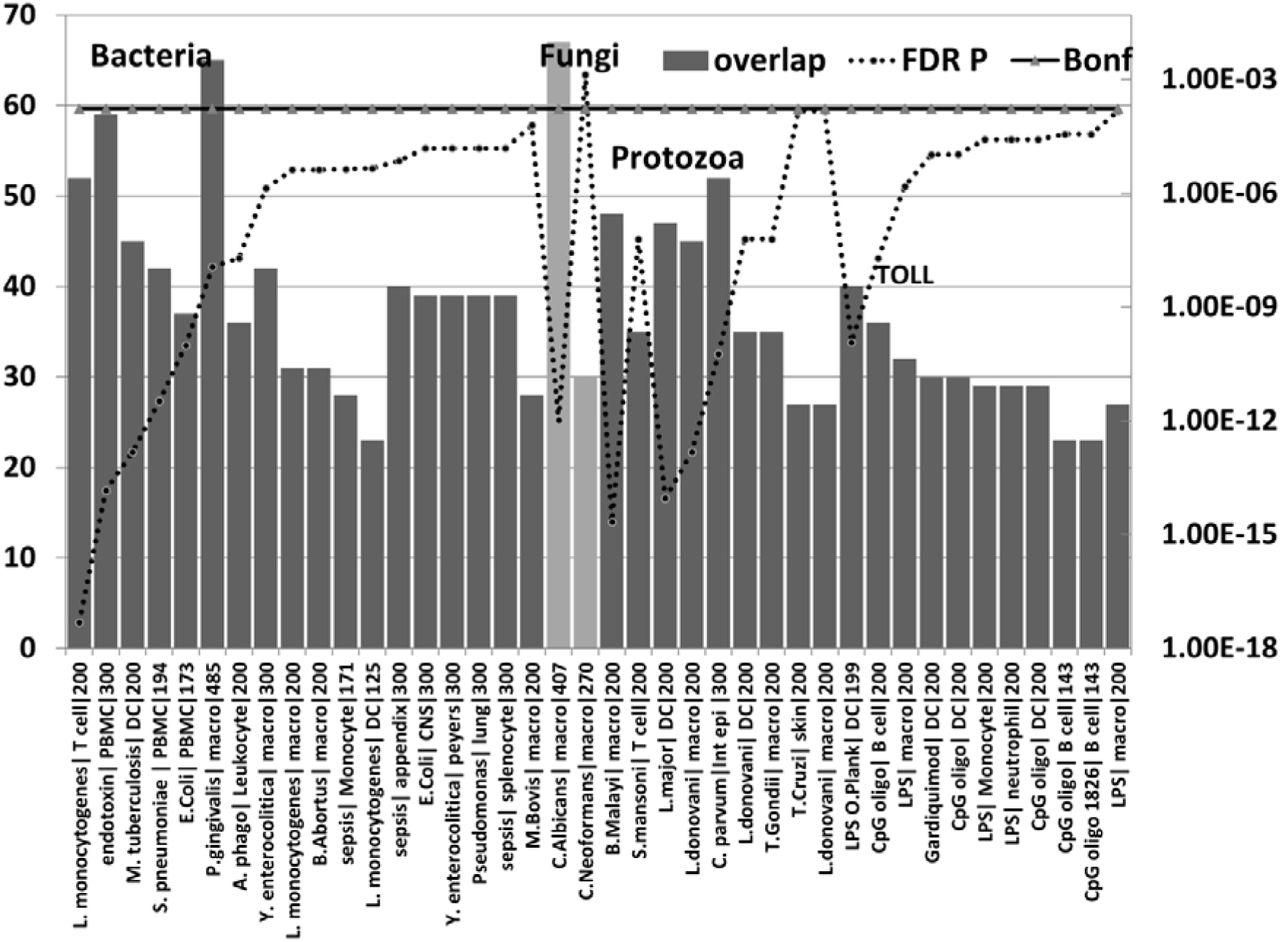
The upregulated hippocampal genes in AD were also enriched in infection datasets for numerous bacteria as well as to fungal species (C. albicans and C.neoformans) and in those related to bacterial endotoxin or sepsis and to nematode/trematode or protozoan infection datasets (FDR p < 0.05) (Fig 5). This also applied to diverse lipopolysaccharide datasets and responses to Toll-like receptor ligands, CpG oligonucleotide (a ligand for TLR9, which mediates cellular response to unmethylated CpG dinucleotides in bacterial DNA (definition from Refseq)) and R848 (a ligand for TLR7/TLR8 both of which recognize RNA released from pathogens that enter the cell by endocytosis [92]) (Fig). With the exception of H.Pylori, P.Gingivalis and Borrelia burgdorferi and C.albicans or C.Neoformans, none have been implicated in AD.
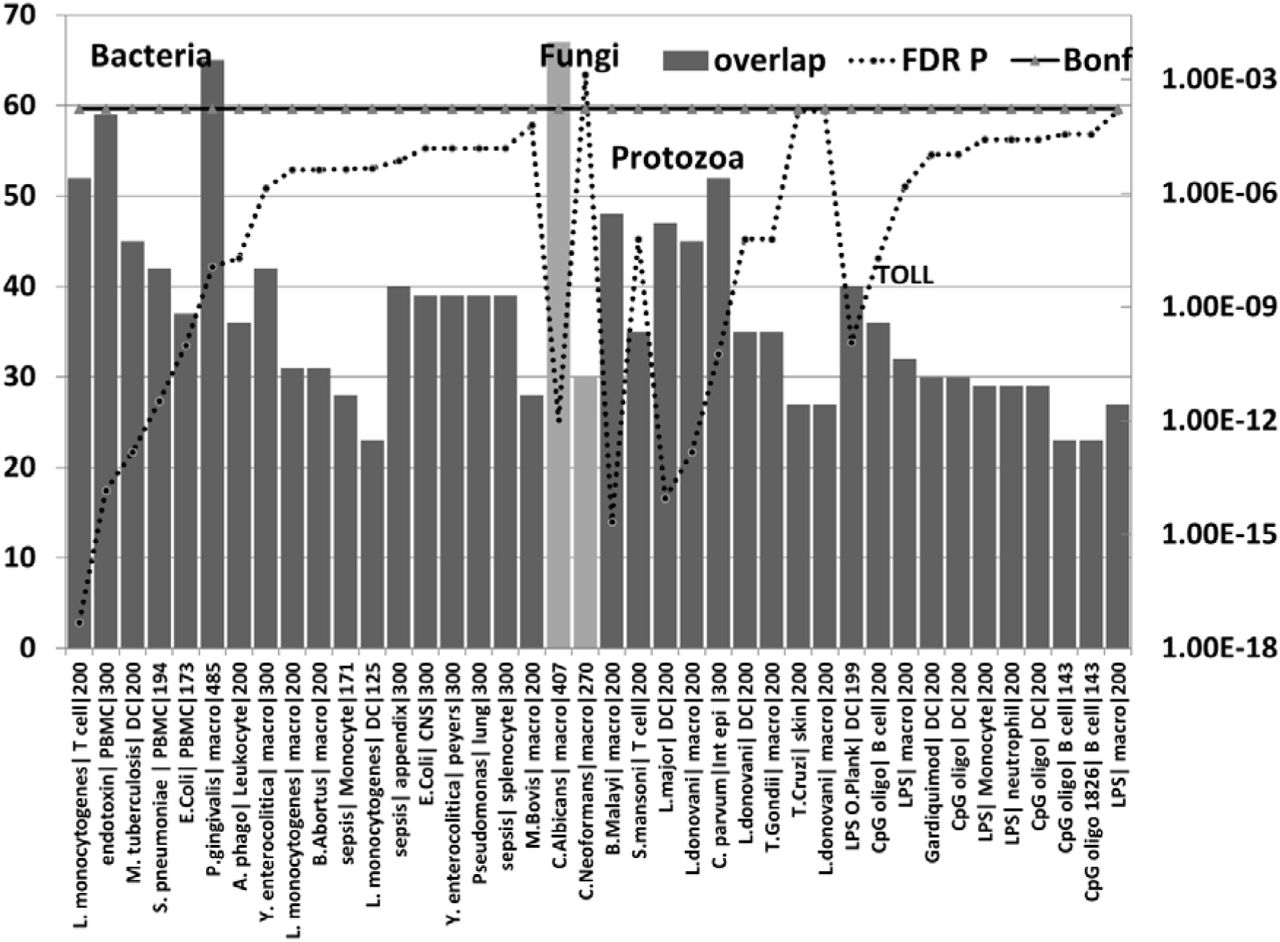
The number of upregulated genes (bars) from the AD hippocampal transcriptome that overlap with upregulated genes in bacterial (first batch), fungal (pale bar =C.albicans, C.Neoformans), nematode (B.Malayi) /trematode (S.Mansoni), or protozoan microarray datasets (see methods). The effects Lipopolysaccharides and other Toll receptor ligands are also shown.
For each datapoint, the name of the pathogen or ligand is shown, followed by the cell type and the total number of upregulated genes in the comparator datasets (limited by MSigDb or GEO). The significance of enrichment (right axis) represents the FDR corrected p value from the hypergeometric test. All values except for C.Neoformans are below the Bonferroni correction level.
Pathogen or ligand abbreviations (from left) L. monocytogenes = Listeria monocytogenes; endotoxin = gram-negative bacterial wall component; S.pneumoniae = Streptococcus pneumoniae; E.Coli = Escherichia coli; P. gingivalis = Porphyromonas gingivalis; A.phago = Anaplasma phagocytophilum; Y. enterocolitica = Yersinia enterocolitica; M.Bovis = mycobacterium bovis; C.albicans = Candida albicans, C.Neoformans = Cryptococcus neoformans, B.Malayi = Brugia malayi (filarial parasite causing elephantiasis); S.mansoni = Schistosoma mansoni; L. donovani = Leishmania donovani; C. parvum = Cryptosporidium parvum; L. Major = Leishmania major; T.Gondii = Toxoplasma Gondii; T. Cruzi = Trypanosoma Cruzi; LPS = lipopolysaccharide; LPS O.Plank = Oscillatoria Planktothrix (cyanobacteria lipopolysaccharide) CpG oligo = CpG Oligodeoxynucleotide (TLR9 ligand); Gardiquimod = TLR7 ligand;
Cell type abbreviations as for Fig 4. CNS = central nervous system; peyers = peyers patch; Int epi = intestinal epithelial cells;
Together these data suggest a significant parallel between the upregulated genes in the AD hippocampus and the responses to multiple and diverse infectious agents with little overall discrimination between viral, bacterial, fungal or protozoan types of infection. Multiple pathogens have been detected in the AD brain (see Table 1) and the diversity of these infection related overlaps with the AD hippocampal transcriptome suggests that many other pathogens could induce similar pathological transcriptome changes. Microbiome studies in the AD brain and periphery will help to elucidate the role of multiple pathogens.
Pathogen interactomes are enriched in the proteins found in AD amyloid plaques and neurofibrillary tangles (Fig 6)

Host pathogen interactome enrichment in a set of 488 proteins isolated from amyloid plaques in the AD brain or from 90 proteins isolated from neurofibrillary tangles. The identities on the X-axis are appended with the total number of genes in each interactome followed by the enrichment ratio. The FDR p value for enrichment, derived from the hypergeometric distribution, is shown on the right hand axis (log scale) which is set to a maximum of 0.05.
All pathogen interactomes were significantly enriched in proteins found in plaques and all except HERV-W and B.Burgdorferi interactomes significantly enriched in tangle proteins (below the Bonferroni cut-off level). The Borna virus and HIV-1 ranked highly in both cases. There is only one publication relating to Borna virus effects on beta-amyloid and none could be found for tangles. The microglial activation produced by the virus reduced brain parenchymal, but increased cerebral vascular beta-amyloid deposition, in APP transgenic mice [93]. The top agents relating to plaques were predominantly viral, while those relating to tangles were mostly viral, but included the parasites, T. Cruzi and T.Gondii.
Beta-amyloid is an antibacterial, antifungal and antiviral agent (Table 1). It has been shown that it binds to C.albicans and S.Typhimurium [2] and presumably to other microbes. Such microbes may well have sequestered host proteins specific to their particular life cycles during their passage to the cell, and this would partly explain the interactome enrichment. In addition to the plaque proteins relating to pathogen life cycles (for example receptor binding, endocytosis and transport between intracellular compartments or nuclear entry and subsequent translation in the case of HSV-1), the proteins found in plaques and tangles contain many related to the immune system, inflammation and autophagy, all of which play a general role in pathogen defence [8,24,25] as does beta-amyloid.
Viruses are transported via the microtubule network [94], which is also exploited by C.Pneumoniae, T.Cruzi and T.Gondii to reorganise cellular organelles to the pathogens' advantage [95,96]. Phosphorylated tau is a hallmark of neurofibrillary tangles and is induced by many pathogens (Table 1). Tau phosphorylation can also be induced by interferon gamma, an effect related to disinhibition of glycogen synthase kinase [97]. It is not clear whether or how such effects could influence the pathogens.
AD genes are localised in the Blood brain barrier
30/78 AD genes are expressed in the BBB proteome dataset of mouse cerebral arteries [28] (Fig 1). The list below indicates the 30 BBB genes, annotated with the number of pathogen interactomes with which they overlap. Most BBB expressed genes interact with none or few pathogens (5 or less of the 17 studied), suggesting a subdivision of mainly BBB and mainly pathogen related. This could of course be confounded by missing data, as several of these genes are poorly characterised in terms of function. These 30 genes (N interactomes in brackets) are:- PCNX (0), ABCA7 (1), ADAMTS20 (1), ATXN7L1 (1), TREML2 (1), AP2A2 (2), BCAM (2), CNTNAP2 (2), ECHDC3 (2), FRMD4A (2), GRIN3B (2), PAX2 (2), PICALM (2), DISC1 (3), LUZP2 (3), RELN (3), TTLL7 (3), FERMT2 (4), HMHA1 (4), MSRA (4), PPP1R3B (4), SASH1 (4), BIN1 (5), SORL1 (5), PVRL2 (7), MMP12 (8), CLU (9), PTK2B (10), BCL3 (13), SQSTM1 (13).
The BBB location of a high proportion of AD genes indicates an important function in relation to AD. Several studies have reported that disruption of the blood brain barrier is an important feature of AD [98–101]. Cerebral microbleeds and cortical siderosis (an increase in blood-derived iron deposition) are a feature related to BBB disruption in AD patients [102–104]. Many bacteria depend upon the availability of free iron and such effects may contribute to their sucessful colonisation in AD [105].
Other environmental risk factors in AD disrupt the blood-brain barrier and BBB integrity is maintained by beneficial factors
AD susceptibility genes might have been selected for pathogen resistance rather than susceptibility (see above). In which case, what are the factors, in the aged, that nevertheless permit the cerebral invasion of a large variety of pathogens? (See Table 1) Certain viruses (e.g. HSV-1) can enter the brain via the olfactory or other neural routes, exploiting an ability to use the axonal transport system [106]. Some parasites [107] and bacteria (e.g. C. Pneumoniae [108,109]) have also found ways to circumvent the barrier systems that usually protect the brain.
Aging itself leads to blood brain barrier dysfunction [110] and immunosenescence is also a feature of ageing and AD. However, while immunosenescence can increase susceptibility to pathogens due to immunodefficiency, it is also accompanied by an increase in the pro-inflammatory activity of monocytes and macrophages which can lead to chronic low grade inflammation, termed “inflammageing”[111,112]. This increased inflammatory function also applies to microglia, the macrophage-like cells in the brain [113]. Certain AD gene variants are associated with enhanced pro-inflammatory responses (see above) and cerebral pathogen entry would thus be met with a doubly vigorous inflammatory response related to both immunosenescence and genetic variation. Persistently activated monocyte/macrophages have been observed in the blood of patients with early AD [114] and increased activation of microglia/macrophages, colocalized with the area of heavy beta-amyloid concentration, is also observed in the brains of AD patients [115].
Apart from pathogens, many other environmental risk factors have been reported in AD. These include diabetes, midlife hypertension or obesity, smoking and physical inactivity [116]. Other contributory factors include previous head injury [117], exposure to toxic metals (aluminium [118,119] or copper[120]), pesticides (organochlorine and organophosphate insecticides) [121,122], industrial chemicals (flame retardants) and air pollutants (particulate matter and ozone [123–126]). High levels of cholesterol or homocysteine [127–130]and low levels of folic acid [131,132] have also been associated with AD. In relation to cholesterol, atherosclerosis of the carotid arteries or of leptomeningeal vessels and in the circle of Willis has also been observed in AD. Such atherosclerotic effects can lead to chronic cerebral hypoperfusion [60,133,134].Sleep disruption or obstructive sleep apnoea are also associated with AD risk [135,136].
Factors reported to be of benefit, or that reduce the incidence of AD include the use of non-steroidal anti-inflammatories (NSAIDs) [137,138], and the early use of statins [139–141]. Statins also have antimicrobial effects against oral microorganisms including Aggregatibacter actinomycetemcomitans and P. Gingivalis, and against most dental plaque bacteria, including Streptococcus mutans. They possess antiviral properties against the human cytomegalovirus, HSV-1, hepatitis B and C viruses, and antifungal properties against Candida albicans, Aspergillus fumigatus, and Zygomycetes species [142].
Beneficial dietary factors in AD include caffeine [130], chocolate (versus cognitive decline in the nondemented aged)[143]) and the Mediterranean diet [144–146]. Melatonin [147,148], estrogen [149–151]and memantine [152,153] also have reported benefits in AD.
The environmental risk factors associated with AD disrupt the BBB, and BBB integrity is maintained by the beneficial factors (Table 2). While infections are random uncontrollable events, many of the other environmental risk factors are modifiable by lifestyle changes, for example diet, obesity, smoking and exercise, and it has been estimated that addressing such modifiable risk factors might result in a significant reduction in the incidence of AD [116]. Amelioration of BBB disruption has already been proposed as a potential therapy in AD, and several drugs including angiotensin receptor blockers, etodolac (NSAID), granisetron (5HT3 serotonin receptor antagonist) or beclomethasone (corticosteroid) [154,155] as well as other NSAIDS, statins and other drugs referenced in Table 2 might be considered as suitable candidates.
The effects of Alzheimer’s disease environmental risk factors and beneficial agents on blood brain barrier function.
Diverse pathogen sensors and defenders relating to bacteria, viruses, parasites and fungi are upregulated in the AD brain, blood or CSF
We have evolved numerous pathogen detectors whose activation leads to stimulation of the immune system and to the production of defensive mechanisms, including inflammation and free radical attack. Multiple pattern recognition receptors including Toll-like receptors, C-type lectin receptors and nucleotide-binding oligomerization domain-like receptors (NOD-like) sense motifs in bacterial, viral, fungal and parasite proteins or other compounds or respond to foreign bacterial or viral DNA or RNA in cellular locations where host DNA or RNA should not exist [156–159].
Infection also activates inflammasomes, which trigger the maturation of proinflammatory cytokines, activating innate immune defences [160].
In addition to this, a large number of defensins and other antimicrobial peptides exist, targeting bacteria, fungi, parasites and viruses [161]. Beta-amyloid is one such [3].
EIF2AK2 (eukaryotic translation initiation factor 2 alpha kinase 2) better known as pkr, is activated by viral double stranded RNA and to bacterial RNA. This phosphorylates eif2alpha, leading to the arrest of the protein translation that is needed for viral replication. Pkr stimulation also leads to the production of interferon and to activation of the inflammasome [162–165]. Other viral RNA-sensors include RIG-I (coded by retinoic acid-inducible gene 1= DDX58), MDA5 (Melanoma Differentiation-Associated protein ; coded by IFIH1) and LGP2 (coded by DEXH-box helicase 58= DHX58) [92].
Indoleamine 2,3-dioxygenase 1 (IDO1) diverts tryptophan metabolism to N-formyl-kynurenine, (away from serotonin production). IDO1 upregulation is an important defence mechanism against pathogenic bacteria, many of which rely on host tryptophan. It is involved in antimicrobial defence and immune regulation, and its effects are not restricted to bacteria This IDO1 response is also deleterious to other pathogens and parasites, including T.Gondii, and to a number of viruses, including herpes simplex virus and other herpes viruses [166]. Kynurenine and kynurenic acid produced by IDO1 activation, are ligands for the aryl hydrocarbon receptor (AHR), which plays an important role in antimicrobial defence and immune regulation [167].
The function of these players with respect to the main pathogens studied above is reviewed in Supplementary Table 2, which also reports expression data in the Alzheimer’s disease brain, blood or CSF. Data derived from this table are illustrated in Figs 7 (viral) and 8 (bacteria, fungi and parasites).
A survey of the roles of diverse microbial sensors and defensive proteins. Their expression levels in the Alzheimer’s disease brain, blood, cerebrospinal fluid or other defined cells etc. are also reviewed.

Fig 7 and 8. Viral (Fig 7) and fungal or bacterial (Fig 8) defenders and sensors and their expression (^ = upregulated; down = downregulated) in the brain, blood or cerebrospinal fluid of Alzheimer’s disease patients. CP = choroid plexus; CSF= cerebrospinal fluid; GVS= granulovacuolar degeneration; HPC = hippocampus; lympho = lymphocytes; macro = macrophages; mcyt=monocytes; mgli = microglia; PBMC = peripheral blood mononuclear cells; Plaq = amyloid plaques; Ser = serum; tang = tangles;
αdefs or βdefs= unspecified alpha or beta defensins: AGER= advanced glycosylation end product-specific receptor (also known as RAGE); APCS= amyloid P component, serum ; CAMP = cathelicidin antimicrobial peptide (LL-37); Calpro= Calprotectin (S100A8/S100A9 dimer);CHI3L1 = chitinase 3 like 1 (aka YKL-40); C-type lectin = CLEC’s; CRP = C-reactive protein; DEAD box proteins = DDX’s; Defensins = DEFA’s, DEFB’s: EIF2AK2 = eukaryotic translation initiation factor 2 alpha kinase 2 (pkr); ELANE = elastase, neutrophil expressed ; IAPP = islet amyloid polypeptide (Amylin); IDO1= indoleamine 2,3-dioxygenase 1; Interferons = IFNA1, IFNA5, IFNB1, IFNG; LCN2 = lipocalin 2; LGALS3 = lectin, galactoside binding soluble 3; LTF = lactotransferrin; MAC = membrane attack complex (complement components C5b-C9); MRC1 = mannose receptor, C type 1; NAIP = NLR family, apoptosis inhibitory protein ; NLRP1 and 3 = NLR family pyrin domain containing 1 and 3; NOD1 and NOD2 = nucleotide binding oligomerization domain containing (1 and 2) ; RARRES2 and 3 = retinoic acid receptor responder (2 and 3) : S100’s= S100 calcium binding protein; Toll-like receptors = TLR1 to 10; ZBP1 Z-DNA binding protein 1. Gene = gene related to the respective pathogen in association studies or with Alzheimer’s disease (Gene AD). mod sens = modified senstitivity; The strikethrough’s (e.g. TLR1) represent a pathogen’s ability to inhibit or overcome the combative effects of the defensive or sensor protein. ? = unknown
Borna = Borna virus; CMV = human cytomegalovirus; EBV = Epstein-Barr virus; HepC = Hepatitis C; HSV-1= Herpes simplex; Influ= Influenza A virus; Borrel= Borrelia burgdorferi; C.Alb= Candida albicans; C.Neo = Cryptococcus neoformans; C. Pneu = Chlamydia pneumoniae; H.Pyl= Helicobacter pylori; P.Ging = Porphyrymonas gingivalis; T.Gon = Toxoplasma Gondii
Those shaded in black are those most implicated in Alzheimer’s disease (Table 1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig 7 and 8. Viral (Fig 7) and fungal or bacterial (Fig 8) defenders and sensors and their expression (^ = upregulated; down = downregulated) in the brain, blood or cerebrospinal fluid of Alzheimer’s disease patients. CP = choroid plexus; CSF= cerebrospinal fluid; GVS= granulovacuolar degeneration; HPC = hippocampus; lympho = lymphocytes; macro = macrophages; mcyt=monocytes; mgli = microglia; PBMC = peripheral blood mononuclear cells; Plaq = amyloid plaques; Ser = serum; tang = tangles;
αdefs or βdefs= unspecified alpha or beta defensins: AGER= advanced glycosylation end product-specific receptor (also known as RAGE); APCS= amyloid P component, serum ; CAMP = cathelicidin antimicrobial peptide (LL-37); Calpro= Calprotectin (S100A8/S100A9 dimer);CHI3L1 = chitinase 3 like 1 (aka YKL-40); C-type lectin = CLEC’s; CRP = C-reactive protein; DEAD box proteins = DDX’s; Defensins = DEFA’s, DEFB’s: EIF2AK2 = eukaryotic translation initiation factor 2 alpha kinase 2 (pkr); ELANE = elastase, neutrophil expressed ; IAPP = islet amyloid polypeptide (Amylin); IDO1= indoleamine 2,3-dioxygenase 1; Interferons = IFNA1, IFNA5, IFNB1, IFNG; LCN2 = lipocalin 2; LGALS3 = lectin, galactoside binding soluble 3; LTF = lactotransferrin; MAC = membrane attack complex (complement components C5b-C9); MRC1 = mannose receptor, C type 1; NAIP = NLR family, apoptosis inhibitory protein ; NLRP1 and 3 = NLR family pyrin domain containing 1 and 3; NOD1 and NOD2 = nucleotide binding oligomerization domain containing (1 and 2) ; RARRES2 and 3 = retinoic acid receptor responder (2 and 3) : S100’s= S100 calcium binding protein; Toll-like receptors = TLR1 to 10; ZBP1 Z-DNA binding protein 1. Gene = gene related to the respective pathogen in association studies or with Alzheimer’s disease (Gene AD). mod sens = modified senstitivity; The strikethrough’s (e.g. TLR1) represent a pathogen’s ability to inhibit or overcome the combative effects of the defensive or sensor protein. ? = unknown
Borna = Borna virus; CMV = human cytomegalovirus; EBV = Epstein-Barr virus; HepC = Hepatitis C; HSV-1= Herpes simplex; Influ= Influenza A virus; Borrel= Borrelia burgdorferi; C.Alb= Candida albicans; C.Neo = Cryptococcus neoformans; C. Pneu = Chlamydia pneumoniae; H.Pyl= Helicobacter pylori; P.Ging = Porphyrymonas gingivalis; T.Gon = Toxoplasma Gondii
Those shaded in black are those most implicated in Alzheimer’s disease (Table 1)
These figures show that sensors and defenders relating to multiple pathogens are upregulated in the AD brain, blood or CSF. These involve reactions to many different classes (bacteria, viruses, fungi and parasites) and there appears to be no discrimination, or focus on any particular type. This would concord with the multiple and diverse pathogen species that have been detected in the AD brain (Table 1) and with the relationship between the AD genes or the hippocampal transcriptome with multiple pathogen species.
Caveats
This analysis is based on overlapping gene symbols rather than on specific polymorphisms. There is thus no indication of the physiological weight or importance of any gene/pathogen interaction, some of which will be more important than others. Pathogen effects may also be strain-dependent, and the size of the interactomes also varies widely. Within any large interactome there will be deleterious, neutral and beneficial effects. While HSV-1 infection causes beta-amyloid deposition and neurodegeneration [168], in its latent form, the virus can have neuroprotective effects. For example the viral latency transcript inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells [169], protects neuronal C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-Cell killing [170] and also protects trigeminal neurones from apoptosis [171]. The Bornavirus is capable of promoting hippocampal degeneration in Man [172]. In rats Borna virus infection decreases choline acetyltransferase activity in the cerebral cortex, horizontal diagonal band of Broca, hippocampus and amygdala [173] a situation similar to that observed in Alzheimer’s disease [174] but the inflammation and microglial activation it produces can also reduce beta-amyloid immunoreactivity in the brain parenchyma of Tg2576 mutant beta-amyloid mice [93]. Chronic, adult acquired T. Gondii infection causes neurologic and behavioural abnormalities secondary to inflammation and neuronal loss, in a strain-dependent manner [175]. T.Gondii infection in BALB/C mice induces neuroinflamation and learning and memory deficits. It also potentiates the toxic effects of low doses of intracerebrally administered beta-amyloid[176], but chronic infection can also increase beta-amyloid phagocytosis and clearance by recruited monocytes [177].
Dementia or neurodegeneration, in the absence of amyloid plaques is, by current clinical definition, not considered as Alzheimer’s disease, but as already noted, there is no inherent biological reason for this [178,179]. Such divergent effects might also be relevant to findings relating to the presence of amyloid plaques in the absence of dementia, as observed in the Nun study [180,181] or to diagnosed Alzheimer’s disease in the absence of beta-amyloid. A recent report showed that ~15% of patients clinically diagnosed with AD do not have amyloid deposits as indexed by positron emission tomography [182]. While some amyloid-negative patients could be re-diagnosed (~50%), the clinical follow-up using other criteria in other amyloid-negative patients continued to support the definition of Alzheimer’s disease.
There are also many inter-pathogen interactions relevant to this relatively small sample of the potential microbiome. For example HSV-1 infection activates replication of the Epstein-Barr virus, [183]. Gingipains or other proteases secreted by P. Gingivalis degrade multiple complement components [184] as well as alpha-and beta defensins [185],immunoglobulins, IgG1 and IgG3 [186] and interleukin-12, preventing its ability to stimulate interferon production [187]. Such effects enable the pathogen to counteract immune defence and would also impinge on the viability of many other pathogens.
HIV-1 is immunosuppressant and has been associated with many opportunistic pathogens including tuberculosis, toxoplasmosis, cytomegalovirus encephalitis and Cryptococcal brain invasion [188,189]. The human cytomegalovirus is also immunosuppressant via an ability to target MHC class I molecules for degradation [190]and to inhibit MHC class II antigen presentation [191]. Parasites, which maintain a long-term, if unwelcome presence in the host have also developed immunosuppressant and anti-defensive strategies[192,193]. In addition, the success of most pathogens depends upon their ability to subvert the defensive armoury of the host in some way.
The AD genes affect human processes relevant to the disease itself, but given that they are also part of pathogen interactomes, polymorphisms therein are also likely to affect pathogen life cycles or the ability of pathogens to promote diverse effects within the host. Apart from APOE4 there are no studies relating to the effects of the AD gene variants on pathogens or their effects.
For these and many other reasons, it is perhaps unwise to rank the pathogens by order of importance in relation to their enrichment or p value in any of the data described above. Suffice it to say that diverse pathogens have been detected in the AD brain and all of the bioinformatics data presented above, whether related to genes, transcriptomes, plaques or tangles implicate multiple species of pathogens across viral, bacterial, fungal and protozoan classes.
While there are statistical limitations to this type of analysis, correction for false discovery followed by the Bonferroni correction has been conservatively applied. The relationship of AD to pathogens is supported by experimental observation (Table 1) and by the antimicrobial effects of beta-amyloid. This study also relies on multiple and diverse in silico bioinformatic analyses linking AD GWAS genes, plaques and tangles as well as the hippocampal transcriptome to multiple pathogen interactomes, and the upregulated AD hippocampal genes to multiple infection datasets from diverse pathogen species. Polymicrobial involvement is also supported by the diversity of bacterial, viral and fungal sensors and defenders that are upregulated in the AD brain, blood or CSF. Each comparison relates to single pathogens but given the diversity of pathogens detected in AD such effects are likely to be cumulative.
Discussion
Multiple and diverse pathogens (bacteria, viruses, fungi and spirochetes) have been detected in the AD brain and many cause neurodegeneration, increase beta-amyloid deposition and tau phosphorylation or are killed/incapacitated by beta-amyloid, an antimicrobial peptide that is part of the innate immune defence system. Representatives of these pathogens target multiple AD GWAS genes, and their interactomes are enriched in genes related to the AD hippocampal transcriptome and to the proteins found in AD plaques and tangles. The upregulated genes of the AD hippocampal transcriptome also correspond to those upregulated by multiple species of viral, bacterial, fungal and protozoan pathogens or by interferon gamma and Toll-like receptor ligands.
The AD genes are preferentially localised in the bone marrow and other immunocompetent tissues, and in exosomes that are hijacked by pathogens for intercellular spread. They are also localised in the lateral ventricle and the hippocampus which abuts this area, a prime site of pathogen invasion via the choroid plexus and the blood/csf barrier.
The AD genes are enriched in global GWAS datasets relating to pathogen diversity, suggesting that some have been selected for pathogen resistance rather that susceptibility. This is supported by the old age of AD patients, indicating survival from the many infections that contribute to mortality in the younger population. APOE4 variants protect against malaria and hepatitis C, and immune/inflammatory gain of function applies to APOE4, CR1, TREM2 and presenilin variants, supporting this contention. Logically, any gene variant increasing the production of the antimicrobial peptide beta-amyloid in response to pathogens might also be considered as beneficial in these evolutionary terms. Apart from APOE4, there is however little data examining the effects of AD gene variants on pathogen life cycles or that relate specifically to pathogen responses.
Many AD genes are also localised in the blood brain barrier. This should provide an effective shield against many infections but it is disrupted by multiple environmental risk factors implicated in Alzheimer’s disease and protected by several factors reported to be beneficial in relation to Alzheimer’s disease, including NSAIDs, statins, oestrogen, memantine, melatonin, and components of the Mediterranean diet.
The relationship between pathogens and Alzheimer’s disease has a long history coupled with a degree of scepticism, perhaps related to an inability to fulfil Koch’s postulate. For example, the same pathogen is not always found in all AD brains, or in different laboratories. Laboratory confirmation in animal models may be impossible for certain pathogens, for example the Epstein-Barr or hepatitis C virus, that do not infect rodents. Nevertheless, the diversity of pathogens able to promote neurodegeneration, beta-amyloid deposition or to mimic the effects observed in the hippocampal AD transcriptome suggests that many candidates, alone or severally, could be involved in the pathogenesis of AD. A polymicrobial involvement seems likely given the multiple species detected in the AD brain. Evidently, this could be assessed by microbiome studies in the periphery or in postmortem brains.
Recent work suggests that the production of the antimicrobial/antiviral peptide beta-amyloid is an expected consequence of infection in general [2,3]. In the context of the amyloid hypothesis [194], this places pathogens upstream of the production of this toxic peptide, and logically as causal, both in terms of beta-amyloid production and in relation to Alzheimer’s disease.
Two separate case reports have shown remission from dementia or mis-diagnosed Alzheimer’s disease in patients subsequently diagnosed with and treated for Cryptococcus neoformans infection [50,51].
In a Greek study, H. Pylori-infected AD patients receiving the triple eradication regime (omeprazole, clarithromycin and amoxicillin) showed improved cognitive and functional status parameters where bacterial eradication was successful [195].H. Pylori eradication in AD patients with peptic ulcer was also associated with a decreased risk of AD progression in a Taiwanese study [196].
Taking all of the above into consideration the combined data suggest that polymicrobial brain invasion, enabled by environmentally-induced blood-brain barrier defects may be responsible for Alzheimer’s disease. This could essentially be mediated via activation of a hyper-efficient inflammatory network, including the call-up of beta-amyloid that, as a consequence, causes massive neuronal destruction in a tissue incapable of regeneration. The role of the innate immune system and the inflammatory response in neurotoxicity has recently been reviewed, and innate surveillance mediated cell death has been suggested as a plausible common pathogenic pathway responsible for many neurodegenerative diseases, including AD [197].
It is therefore not unreasonable to suggest that antibiotic, antifungal and antiviral agents, possibly in combination, tailored to the individual, might be able to halt, delay or perhaps even provide remission in patients with Alzheimer’s disease.
The author reports no funding and no conflict of interest.
Acknowledgements
Thanks are due to the many authors who have sent reprints and supplementary datasets that made this work possible. Thanks also to David Eby from the Broad Institute of MIT and Harvard for his help with the structure of the Molecular Signatures database.
References
- [1].↵
- [2].↵
- [3].↵
- [4].↵
- [5].↵
- [6].↵
- [7].↵
- [8].↵
- [9].↵
- [10].↵
- [11].↵
- [12].↵
- [13].↵
- [14].↵
- [15].↵
- [16].↵
- [17].↵
- [18].
- [19].
- [20].↵
- [21].↵
- [22].↵
- [23].↵
- [24].↵
- [25].↵
- [26].↵
- [27].↵
- [28].↵
- [29].↵
- [30].↵
- [31].↵
- [32].↵
- [33].↵
- [34].↵
- [35].↵
- [36].↵
- [37].↵
- [38].↵
- [39].↵
- [40].↵
- [41].↵
- [42].↵
- [43].↵
- [44].↵
- [45].↵
- [46].↵
- [47].↵
- [48].↵
- [49].↵
- [50].↵
- [51].↵
- [52].↵
- [53].
- [54].
- [55].
- [56].
- [57].↵
- [58].
- [59].↵
- [60].↵
- [61].↵
- [62].↵
- [63].↵
- [64].↵
- [65].↵
- [66].↵
- [67].↵
- [68].↵
- [69].↵
- [70].↵
- [71].↵
- [72].↵
- [73].↵
- [74].↵
- [75].↵
- [76].↵
- [77].↵
- [78].↵
- [79].↵
- [80].↵
- [81].↵
- [82].↵
- [83].↵
- [84].↵
- [85].↵
- [86].↵
- [87].↵
- [88].↵
- [89].↵
- [90].↵
- [91].↵
- [92].↵
- [93].↵
- [94].↵
- [95].↵
- [96].↵
- [97].↵
- [98].↵
- [99].
- [100].
- [101].↵
- [102].↵
- [103].
- [104].↵
- [105].↵
- [106].↵
- [107].↵
- [108].↵
- [109].↵
- [110].↵
- [111].↵
- [112].↵
- [113].↵
- [114].↵
- [115].↵
- [116].↵
- [117].↵
- [118].↵
- [119].↵
- [120].↵
- [121].↵
- [122].↵
- [123].↵
- [124].
- [125].
- [126].↵
- [127].↵
- [128].
- [129].
- [130].↵
- [131].↵
- [132].↵
- [133].↵
- [134].↵
- [135].↵
- [136].↵
- [137].↵
- [138].↵
- [139].↵
- [140].
- [141].↵
- [142].↵
- [143].↵
- [144].↵
- [145].
- [146].↵
- [147].↵
- [148].↵
- [149].↵
- [150].
- [151].↵
- [152].↵
- [153].↵
- [154].↵
- [155].↵
- [156].↵
- [157].
- [158].
- [159].↵
- [160].↵
- [161].↵
- [162].↵
- [163].
- [164].
- [165].↵
- [166].↵
- [167].↵
- [168].↵
- [169].↵
- [170].↵
- [171].↵
- [172].↵
- [173].↵
- [174].↵
- [175].↵
- [176].↵
- [177].↵
- [178].↵
- [179].↵
- [180].↵
- [181].↵
- [182].↵
- [183].↵
- [184].↵
- [185].↵
- [186].↵
- [187].↵
- [188].↵
- [189].↵
- [190].↵
- [191].↵
- [192].↵
- [193].↵
- [194].↵
- [195].↵
- [196].↵
- [197].↵
- [198].
- [199].
- [200].
- [201].
- [202].
- [203].
- [204].
- [205].
- [206].
- [207].
- [208].
- [209].
- [210].
- [211].
- [212].
- [213].
- [214].
- [215].
- [216].
- [217].
- [218].
- [219].
- [220].
- [221].
- [222].
- [223].
- [224].
- [225].
- [226].
- [227].
- [228].
- [229].
- [230].
- [231].
- [232].
- [233].
- [234].
- [235].
- [236].
- [237].
- [238].
- [239].
- [240].
- [241].
- [242].
- [243].
- [244].
- [245].
- [246].
- [247].
- [248].
- [249].
- [250].
- [251].
- [252].
- [253].
- [254].
- [255].
- [256].
- [257].
- [258].
- [259].
- [260].
- [261].
- [262].
- [263].
- [264].
- [265].
- [266].
- [267].
- [268].
- [269].
- [270].
- [271].
- [272].
- [273].
- [274].
- [275].
- [276].
- [277].
- [278].
- [279].
- [280].
- [281].
- [282].
- [283].
- [284].
- [285].
- [286].
- [287].
- [288].
- [289].
- [290].
- [291].
- [292].
- [293].
- [294].
- [295].
- [296].
- [297].
- [298].
- [299].
- [300].
- [301].
- [302].
- [303].
- [304].
- [305].
- [306].
- [307].
Reference List
- [1].
- [2].
- [3].
- [4].
- [5].
- [6].
- [7].
- [8].
- [9].
- [10].
- [11].
- [12].
- [13].
- [14].
- [15].
- [16].
- [17].
- [18].
- [19].
- [20].
- [21].
- [22].
- [23].
- [24].
- [25].
- [26].
- [27].
- [28].
- [29].
- [30].
- [31].
- [32].
- [33].
- [34].
- [35].
- [36].
- [37].
- [38].
- [39].
- [40].
- [41].
- [42].
- [43].
- [44].
- [45].
- [46].
- [47].
- [48].
- [49].
- [50].
- [51].
- [52].
- [53].
- [54].
- [55].
- [56].
- [57].
- [58].
- [59].
- [60].
- [61].
- [62].
- [63].
- [64].
- [65].
- [66].
- [67].
- [68].
- [69].
- [70].
- [71].
- [72].
- [73].
- [74].
- [75].
- [76].
- [77].
- [78].
- [79].
- [80].
- [81].
- [82].
- [83].
- [84].
- [85].
- [86].
- [87].
- [88].
- [89].
- [90].
- [91].
- [92].
- [93].
- [94].
- [95].
- [96].
- [97].
- [98].
- [99].
- [100].
- [101].
- [102].
- [103].
- [104].
- [105].
- [106].
- [107].
- [108].
- [109].
- [110].
- [111].
- [112].
- [113].
- [114].
- [115].
- [116].
- [117].
- [118].
- [119].
- [120].
- [121].
- [122].
- [123].
- [124].
- [125].
- [126].
- [127].
- [128].
- [129].
- [130].
Reference List
- [1].
- [2].
- [3].
- [4].
- [5].
- [6].
- [7].
- [8].
- [9].
- [10].
- [11].
- [12].
- [13].
- [14].
- [15].
- [16].
- [17].
- [18].
- [19].
- [20].
- [21].
- [22].
- [23].
- [24].
- [25].
- [26].
- [27].
- [28].
- [29].
- [30].
- [31].
- [32].
- [33].
- [34].
- [35].
- [36].
- [37].
- [38].
- [39].
- [40].
- [41].
- [42].
- [43].
- [44].
- [45].
- [46].
- [47].
- [48].
- [49].
- [50].
- [51].
- [52].
- [53].
- [54].
- [55].
- [56].
- [57].
- [58].
- [59].
- [60].
- [61].
- [62].
- [63].
- [64].
- [65].
- [66].
- [67].
- [68].
- [69].
- [70].
- [71].
- [72].
- [73].
- [74].
- [75].
- [76].
- [77].
- [78].
- [79].
- [80].
- [81].
- [82].
- [83].
- [84].
- [85].
- [86].
- [87].
- [88].
- [89].
- [90].
- [91].
- [92].
- [93].
- [94].
- [95].
- [96].
- [97].
- [98].
- [99].
- [100].
- [101].
- [102].
- [103].
- [104].
- [105].
- [106].
- [107].
- [108].
- [109].
- [110].
- [111].
- [112].
- [113].
- [114].
- [115].
- [116].
- [117].
- [118].
- [119].
- [120].
- [121].
- [122].
- [123].
- [124].
- [125].
- [126].
- [127].
- [128].
- [129].
- [130].
- [131].
- [132].
- [133].
- [134].
- [135].
- [136].
- [137].
- [138].
- [139].
- [140].
- [141].
- [142].
- [143].
- [144].
- [145].
- [146].
- [147].
- [148].
- [149].
- [150].
- [151].
- [152].
- [153].
- [154].
- [155].
- [156].
- [157].
- [158].
- [159].
- [160].
- [161].
- [162].
- [163].
- [164].
- [165].
- [166].
- [167].
- [168].
- [169].
- [170].
- [171].
- [172].
- [173].
- [174].
- [175].
- [176].
- [177].
- [178].
- [179].
- [180].
- [181].
- [182].
- [183].
- [184].
- [185].
- [186].
- [187].
- [188].
- [189].
- [190].
- [191].
- [192].
- [193].
- [194].
- [195].
- [196].
- [197].
- [198].
- [199].
- [200].
- [201].
- [202].
- [203].
- [204].
- [205].
- [206].
- [207].
- [208].
- [209].
- [210].
- [211].
- [212].
- [213].
- [214].
- [215].
- [216].
- [217].
- [218].
- [219].
- [220].
- [221].
- [222].
- [223].
- [224].
- [225].
- [226].
- [227].
- [228].
- [229].
- [230].
- [231].
- [232].
- [233].
- [234].
- [235].
- [236].
- [237].
- [238].
- [239].
- [240].
- [241].
- [242].
- [243].
- [244].
- [245].
- [246].
- [247].
- [248].
- [249].
- [250].
- [251].
- [252].
- [253].
- [254].
- [255].
- [256].
- [257].
- [258].
- [259].
- [260].
- [261].
- [262].
- [263].
- [264].
- [265].
- [266].
- [267].
- [268].
- [269].
- [270].
- [271].
- [272].
- [273].
- [274].
- [275].
- [276].
- [277].
- [278].
- [279].
- [280].
- [281].
- [282].
- [283].
- [284].
- [285].
- [286].
- [287].
- [288].
- [289].
- [290].
- [291].
- [292].
- [293].
- [294].
- [295].
- [296].
- [297].
- [298].
- [299].
- [300].
- [301].
- [302].
- [303].
- [304].




