

Abstract
Age-dependent loss of cardiac tissue homeostasis largely impacts heart performance and contributes significantly to cardiovascular diseases later in life. Cellular quality control machinery, such as autophagy/lysosome system, plays a crucial role in maintaining cardiac health and preventing age-induced cardiomyopathy and heart failure. However, how aging alters autophagy/lysosome system to impact cardiac function remain largely unknown. Here using Drosophila heart as a model system, we show that activin signaling, a member of TGF-beta superfamily, negatively regulates cardiac autophagy and promotes age-dependent decline of cardiac functions. We found that cardiac-specific knockdown of Daw, an activin-like protein in Drosophila, increased cardiac autophagosome number and prevented age-induced cardiac arrhythmias and diastolic dysfunction. Conversely, cardiac-specific expressed activin type I receptor Babo results in pre-matured cardiac dysfunction at young ages. Although Daw positively modulates mTOR signaling (Mechanistic target of rapamycin, the major negative regulator for autophagy), activation of mTOR through the knockdown of Tsc1 (Tuberous sclerosis protein 1) did not rescue the autophagy and cardiac aging phenotypes in Daw RNAi flies. On the other hand, inhibition of autophagy via chloroquine feeding or Atg1 knockdown attenuated the beneficial effects of Daw RNAi on age-related cardiac arrhythmias. Finally, reduction in cardiac activin signaling prolonged lifespan and improved climbing ability during aging. Thus, our findings highlight the emerging role of activin signaling in mTOR-independent regulation of autophagy and cardiac aging.
Introduction
Aging is associated with an exponential increase of the incidence of cardiovascular diseases (CVD) [1,2]. Age-related changes in cardiovascular structure and output have been linked to increased risk of coronary heart disease, sudden cardiac death and stroke in aging population [3]. During normal aging, the left ventricular wall of human hearts becomes thickened and the diastolic filling rate of left ventricle gradually decreases. On the other hand, the left ventricular systolic performance at rest remains less or shows no change with age [3]. Several mechanisms underlying age-associated changes in cardiovascular structure and function are proposed, for example changes in growth factor signaling, decreased cellular quality control, altered calcium handling, elevated extracellular matrix deposition or fibrosis, increased mitochondria damage, and the production of reactive oxygen species (ROS) [2,4]. Resolving the contributing mechanisms of age-dependent decline of cardiovascular function is a critical for the development of biomedical interventions for the treatment of cardiovascular diseases.
Cellular quality control systems, such as macroautophagy (hereafter autophagy), are essential to maintain tissue homeostasis during aging [5,6]. Autophagy is a highly conserved process that maintains tissue and cellular homeostasis by degradation and recycling of damaged organelles, protein aggregates and other cytoplasmic substances. The autophagic process initiated with the isolated membrane, or phagophore that elongates to form a double-membrane structure, the autophagosome. Then the autophagosome fuses with a lysosome to form an autolysosome to degrade the enclosed materials along with autophagosomal membrane [7]. Disruption of autophagy pathways by knocking out Atg5 in the mouse heart accelerates cardiac aging, including an increase in left ventricular hypertrophy and decrease in factional shortening [8]. Induction of autophagy by rapamycin inhibition of mTOR (Mechanistic target of rapamycin) protected mouse cardiomyocytes from antimycin A-induced oxidative stress [9]. Although many longevity interventions activating autophagy can greatly preserve cardiac function during aging [2], no evidence has indicated that autophagy activation alone can delay the aging process in animal hearts. Furthermore, how aging negatively impacts and modulates autophagy is largely unknown [5]. In general, autophagy activity declines with age [8,10,11]. However, some studies suggest that autophagy activity remains no change with age [12-15]. The age-dependent alternation of autophagy and the vital role of autophagy in cardiac protection need to be further investigated.
Autophagy can be activated by various stress, such as dietary restriction, hypoxia, oxidative stress, and pathogen infection [16]. Autophagy activity is tightly controlled many signaling pathways, in particular the nutrient signaling like mTOR and AMPK (AMP-activated protein kinase) [17]. These pathways are often involved in longevity control. We recently identified activin, a TGF-beta family protein, as a pro-aging hormonal factor regulating autophagy and proteostasis in fight muscle of Drosophila [18]. As in mammals, TGF-beta family in Drosophila has two branches, bone morphogenetic protein (BMP) and activin signaling pathways [19]. In both pathways, signaling starts with ligand binding to a receptor complex composed of type I and type II receptor kinases, followed by phosphorylation of receptor-activated Smad (R-Smad). There are three ligands in Drosophila activin signaling pathway, Activin-beta (Act-beta), Dawdle (Daw), and Myoglianin (Myo). We showed that reduced expression of Daw, but not Act-beta and Myo, prolongs lifespan and increases autophagosome formation [18].
It is known that TGF-beta factors promotes cardiac fibrosis during aging [20]. However, it is unclear whether TGF-beta family protein, in particular activin, regulates cardiac homeostasis and function through autophagy. Given that the cardiac development and aging are remarkably conserved between Drosophila and mammal, as well as the advanced genetic tools and cardiac movement detection methodologies in Drosophila [21], we have applied fly heart model to investigate the role of activin signaling in the regulation of cardiac autophagy and cardiac aging. Here we show that activin signaling acts on hearts to regulate autophagy and age-induced cardiomyopathy in Drosophila. The regulation of cardiac aging by activin signaling is ligand-dependent. RNAi against activin-like protein Daw preserved cardiac function with age, while reduction in growth differentiation factor 11 (GDF11)/myostatin-like protein Myo promotes cardiac aging. Reduction of activin signaling in the heart prolongs fly lifespan and improves climbing ability during aging. In addition, we found that Daw negatively regulates cardiac autophagy, while autophagy inhibition attenuates the positive effects of Daw RNAi on cardiac aging. Interestingly, despite the positive relationship between Daw and mTOR, we found that activation of mTOR through TSC1 RNAi did not block the beneficial effects of Daw knockdown during cardiac aging. Our findings suggest that Drosophila activin signaling may regulate autophagy and age-induced cardiomyopathy through novel mTOR-independent mechanisms.
Results
Heart-specific knockdown of Daw slows down cardiac aging
Our previous study demonstrated that activin-like protein Daw regulates tissue homeostasis (especially in flight muscle) and longevity in Drosophila [18]. The expression of Daw and other activin family ligands (Myo and Act-beta) are also detected in fly heart from previous transcriptome analyses [22-24]. To verify the expression of these activin ligand in the heart, we performed a tissue-specific ribosome profiling using Ribo-Tag technology. We found that both Daw and Myo expressed highly in the heart compared to Act-beta (Fig. 1A). To investigate the role of activin signaling in cardiac aging, we first knocked down the expression of Daw specifically in fly hearts with a binary GAL4/UAS system by crossing Daw RNAi lines to a cardiac driver (Hand-gal4). We then analyzed cardiac performances of young and old flies using a high-speed video imaging system (semi-automatic optical heartbeat analysis, SOHA) [25] (Fig. 1B). Consistent with previous studies, cardiac arrhythmia, heart period, and diastolic interval of control flies increase with age (Fig. 1 C, 1D, 1E). There is no significant change in systolic intervals (Fig. 1F). In contract, flies with cardiac-specific knockdown of Daw attenuates age-induced arrhythmia, heart period, and diastolic interval (Fig. 1 C, 1D, 1E). To avoid the potential off-target effects in knockdown experiments, we tested three different Daw RNAi fly lines and all of them produce similarly phenotypes.

(A). Actively translated mRNA profiling using Ribo-tag qPCR showed that all three activin ligands have detectable expression in fly heart, while the mRNA expression of Daw and Myo are relatively higher than Act-beta. Data are represented as mean ± SE of three trials. Statistical significance is assessed by one-way ANOVA followed by Tukey multiple comparisons test (* p<0.05, ** p<0.01, *** p<0.001). (B). Representative M-mode traces (8 seconds) showing age-dependent movement of heart wall in control (or Hang-gal4>wild-type/WT) and heart-specific Daw knockdown flies (Hang-gal4>UAS-Daw RNAi). (C-E). Age-induced arrhythmia index, heart period, and diastolic interval are blocked by heart-specific knockdown of Daw. (F). No significant change in systolic interval between young and old control/WT and Daw RNAi flies. Flies were cultured at 40% relative humidity. Hand-gal4 driver was used to knock down gene expression specifically in cardiac tissues (cardiomyocytes and pericardial cells). Results from three independent UAS-Daw RNAi lines are shown (RNAi-1: Bloomington Drosophila Stock Center (BDSC) #34974, RNAi-2: Vienna Drosophila Resource Center (VDRC) #105309, RNAi-3: BDSC #50911). Statistical significance is assessed by two-way ANOVA followed by Tukey multiple comparisons test (* p<0.05, ** p<0.01, *** p<0.001, ns = not significant). N=7∼20. The interaction between genotype and age is statistically significant for heart period (P = 0.0041) and diastolic interval (P = 0.0243).
Because Hand-gal4 drives gene expression in both cardiomyocytes and pericardial cells (or nephrocytes) [26]. To test whether Daw acts on cardiomyocytes to regulate cardiac arrhythmia, we crossed Daw RNAi into a cardiomyocyte-specific driver Tin-gal4 [27]. Similar to the results from Hand-gal4 (Fig. 2A), cardiomyocyte-specific knockdown of Daw prevented age-related increases of cardiac arrhythmia (Fig. 2B). In addition, Daw knockdown preserved cardiac myofibrillar structure with age (Supplemental Figure S1A). We also tested whether Daw acts on pericardial cells to regulate cardiac aging using a pericardial cell driver Dot-gal4 [28]. Interestingly, knockdown of Daw in pericardial cells prevented age-induced arrhythmia (Fig. 2C), but not diastolic interval (Fig. 2D). Thus, Daw appears to promote cardiac aging, in particular age-induced arrhythmia and diastolic dysfunction, through cardiomyocyte-specific regulation.

(A-B). Knockdown of Daw using both Hand-gal4 (cardiomyocytes and pericardial cells) and Tin-gal4 (cardiomyocytes) prevents age-induced cardiac arrhythmia. Student t-test (* p<0.05, ** p<0.01, *** p<0.001). N=25∼31. (C-D). Knockdown of Daw using Dot-gal4 (pericardial cells) prevents age-induced cardiac arrhythmia, but not diastolic interval. Student t-test (* p<0.05, ** p<0.01, *** p<0.001). N=13∼26.
Cardiomyocyte-specific knockdown of activin receptor Babo, but not ligand Myo, delays cardiac aging
To investigate whether Daw acts through its receptor Babo to modulate cardiac aging, we first asked if Babo plays a similar role in preventing age-related increases of cardiomyopathy. Consistently, we found that cardiomyocyte-specific knockdown of Babo blocked the age-induced cardiac arrhythmia, heart period, and diastolic interval (Fig. 3A, 2B, 2C). Two Babo RNAi lines were used and both gave similar results. In a reciprocal experiment, we used a GeneSwitch heart driver to express a constitutive active form of Babo (Babo-Act) in adult hearts. We found that over-expression of Babo significantly induced cardiac arrhythmia and diastolic interval at young age (Fig. 3E, 3F; Supplemental Figure S1B). Interestingly, over-expression of Babo also increased the diastolic diameter of the heart tube, suggesting a role of Babo in dilated cardiomyopathy of the fly heart (Supplemental Figure S1C).

(A-D). Age-induced arrhythmia index, heart period, and diastolic interval, but not systolic interval, are delayed by cardiomyocyte-specific knockdown of Babo (Tin-gal4). Results from two independent UAS-Babo RNAi lines are shown (RNAi-1: BDSC #25933, RNAi-2: BDSC #40886). One-way ANOVA followed by Tukey multiple comparisons test (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=15∼30. (E-F). Over-expression of constitutive activate form of Babo (Babo-Act) induces cardiac arrhythmia and diastolic interval at young age (2- week-old). Hand-GS-gal4 was used to induce adult-onset Babo activation. RU: RU486, Mifepristone. Flies were cultured at 40% relative humidity. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05). N=6∼9.
In Drosophila, there are three Babo isoforms and isoforms of Babo exhibit tissue-specific expression pattern and response differently to different activin ligands [29]. We found that all three Babo isoforms showed enriched expression in fly heart (about three fold) (Fig. 4A). Since both Daw and Myo are highly expressed in the heart, we examine the differential regulation of cardiac aging by the two activin ligands. Notably, knockdown of Myo did not block the age-induced arrhythmia, rather it further increased the arrhythmia at old age (Fig. 4B). These results suggest that Daw and Myo differentially regulate cardiac aging, and Daw may directly regulate age-related cardiomyopathy through its receptor Babo-c.

(A). Ribo-tag PCR shows that all three Babo isoforms are expressed in dissected adult hearts (1-week-old). Student t-test (* p<0.05). N=3. (B). Two activin ligands, Daw and Myo, play opposite roles in controlling cardiac arrhythmia. Knockdown of Daw prevents age-induced cardiac arrhythmia, while knockdown of Myo promotes arrhythmia. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=25∼29.
Daw regulates autophagosome formation and promotes cardiac mTOR activities
Autophagy is one of the key mechanisms in maintaining tissue hemostasis and its activity is thought to decline with aging [6,8]. Our previous studies showed that activin signaling regulates autophagosome formation and Atg8a transcription [18]. Here, we examined the potential role of Daw in the regulation of cardiac autophagy. Using immunostaining and an antibody against Drosophila Atg8a protein (fly homolog of mammalian LC3/GABARAP family proteins), we observed an increased Atg8a-positive autophagosome in the heart of Daw RNAi flies (Fig. 5A). Interestingly, flies with heart-specific Daw knockdown maintained high levels of cardiac autophagosome number at old age, whereas in control flies autophagosome number decreased with age (Fig. 5A). Because autophagy is a dynamic process, the increased autophagosome number could result from increased autophagy activities, or blockage of lysosomal degradation [30]. To examine the role of Daw in controlling autophagy flux (the turnover of autophagosome), we exposed semi-intact fly hearts to a V-ATPase inhibitor bafilomycin A1 (Baf A1), which blocks both lysosomal acidification and autophagosome-lysosome fusion [31]. Notably, both young and aged hearts in control flies maintained active autophagy flux (or autophagosome turnover activity), as indicated by an increase of autophagosome number after one hour of Baf A1 treatment (Fig. 5B). Daw RNAi flies showed less changes in autophagosome number after Baf A1 treatment at both young and old ages, although the total autophagosome numbers per heart in Daw RNAi flies are higher than control flies (Fig. 5B). Thus, both control and Daw RNAi flies maintain active cardiac autophagy with age and Daw knockdown resulted an induction of autophagosome formation.

(A). Quantification of the fluorescence signaling of mcherry-Atg8a reporter in young and old control and cardiomyocyte-specific Daw knockdown flies. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=4. (B). Quantification of autophagy flux by comparing the changes in mcherry-Atg8a signaling before and after lysosomal inhibitor Baf A1 treatment. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=4. (C). Immunostaining of the phosphorylation of 4E-BP (p4E-BP) from the dissected cardiomyocytes of cardiac-specific knockdown of Daw and Myo flies. Phalloidin was used to stain F-actin of the cardiomyocytes. Scale bar: 10 μm. (D). Western blots showing p4E-BP levels in Kc167 cells after the incubation with conditioned media containing Daw and Myo proteins. ß– Actin was used as a loading control.
It is known that nutrient-sensing pathways, such as mTOR, negatively regulate the autophagy initiation and autophagosome formation [32]. We then tested whether Daw regulates autophagosome formation through mTOR signaling. Cardiomyocyte-specific Daw knockdown decreased the phosphorylation of 4E-BP, a major target of mTOR signaling (Fig. 5C). On the other hand, incubation of conditioned media from Daw-expressing Drosophila Kc167 cells induced the phosphorylation of 4E-BP, while conditioned media from Myo-expressing cells did not increase p4E-BP levels (Fig. 5D). Taken together, these results suggest that Daw positively regulates mTOR activity.
Activation of mTOR does not attenuate the autophagosome induction and delayed cardiac aging in Daw RNAi flies
Given that Daw promotes mTOR signaling, a negative regulator of autophagy, we predict that Daw negatively regulate autophagosome formation through mTOR. To test this possibility, we first generated a double knockdown flies by combining UAS-daw RNAi and UAS-Tsc1 RNAi. Tsc1 (Tuberous sclerosis protein 1) is a negative regulator upstream of mTOR (in particular mTOR Complex 1, mTORC1). Cardiomyocyte-specific knockdown of Tsc1 significantly induced p4E-BP levels (Supplemental Figure S2A, S2B). Surprisingly, the number of Atg8a-positive autophagosome remains no changed between Daw RNAi and Daw; Tsc1 double RNAi flies (Fig. 6A, 6B). In addition, activation of mTOR via Tsc1 RNAi (two independent RNAi lines) did not change the cardiac arrhythmia (Fig. 6C) and showed reduced diastolic interval at young age (Fig. 6D), which is very different from cardiac-specific over-expression of activin receptor Babo (Fig. 3E, 3F). Furthermore, Tsc1 RNAi did not attenuate the effects of Daw knockdown on age-induced cardiac arrhythmia (Fig. 6E). Daw; Tsc1 double RNAi flies did not show age-dependent increase of arrhythmia, similar to Daw RNAi alone. These results suggest that Daw negatively regulates autophagosome formation and promotes cardiac aging independent of mTOR activation.

(A). Immunostaining of autophagosome in cardiomyocytes from Daw RNAi and Daw; Tsc1 double knockdown flies using Atg8a antibody. Scale bar: 20 μm. (B). Quantification of autophagosome staining in Panel A. (C-D). Cardiac arrhythmia and diastolic interval of cardiomyocyte-specific Tsc1 knockdown flies. Two Tsc1 RNAi used (RNAi-1: BDSC #52931, RNAi-2: BDSC #54034). One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=16∼18. (E). Cardiomyocyte-specific knockdown of Tsc1 did not rescue Daw-regulated cardiac arrhythmia to the wild-type level. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=7∼30.
Intriguingly, Tsc1 knockdown flies (with high mTOR activities) did not exhibit age-induced cardiac arrhythmia (Fig. 7A) and diastolic dysfunction (Fig. 7B). This observation is somewhat inconsistent with the notion of mTOR being a pro-aging factor. To further confirm the unexpected role of mTOR in the regulation of cardiac aging, we tested other key factors involved in mTOR signaling. Reptor (Repressed by TOR) is a recently identified bZIP transcription factor downstream of mTORC1 signaling in Drosophila [33]. The transcriptional activities of Reptor is negatively regulated by mTORC1 and rapamycin. Similar to Tsc1 RNAi, cardiomyocyte-specific knockdown of Reptor blocked age-induced arrhythmia (Fig. 7C) and diastolic interval (Fig. 7D). Furthermore, over-expression of Rheb (Ras Homolog Enriched in Brain), a positive regulator of mTORC1, resulted in decreased diastolic interval, while no changes in cardiac arrhythmia (Fig. 7E, 7F). The positive role of mTORC1 signaling in preventing age-induced arrhythmia is unexpected, and somewhat inconsistent with the pro-aging role of mTORC1 as suggested by previous studies [34-36]. However, we should point out that mTORC1 might regulate tissue homeostasis and cardiac arrhythmia through distinct mechanisms. Interestingly, we observed a significant increase of cardiac arrhythmia in cardiomyocyte-specific knockdown of PTEN (Phosphatase and tensin homologue deleted on chromosome ten), a major negative regulator of PI3K/AKT signaling, suggesting activation of PI3K signaling is detrimental to cardiac function (Fig. 7G, 7H). Taken together, we found that activation of both activin and PI3K signaling pathways specifically in cardiomyocytes negatively impacts on heart function, while increased mTOR activity prevents age-related increase of cardiac arrhythmia in Drosophila.

(A-B). Cardiomyocyte-specific knockdown of Tsc1 prevents age-induced arrhythmia and diastolic interval. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=14∼18. (C-D). Cardiomyocyte-specific knockdown of Reptor prevents age-induced arrhythmia and diastolic interval. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=13∼28. (E-F). Cardiomyocyte-specific over-expression of Rheb reduced diastolic interval, but not arrhythmia index. Student t-test (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=21∼31. (G-H). Cardiomyocyte-specific knockdown of PTEN reduced diastolic interval, but not arrhythmia index. Student t-test (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=21∼31.
Autophagy inhibition accelerates cardiac aging and attenuates the effects of Daw on age-related decline of cardiac function
Since we observed an induction of autophagosome formation in Daw knockdown flies, we next tested whether autophagy activity is required for Daw-regulated cardiac aging. Cardiac-specific (Hand-gal4 and Tin-gal4) knockdown of Atg1, a serine/threonine-protein kinase essential for autophagy initiation, increased cardiac arrhythmia and diastolic interval at young age (Fig. 8A, 8B), similar to what we observed in Babo over-expression flies.

(A-B). Both arrhythmia and diastolic interval are induced in heart-specific Atg1 knockdown flies. Two driver used (Hand-gal4 and Tin-gal4). (C). Chloroquine treatments (20 mM, 24 hours) increases cardiac arrhythmia of 6-week-old Daw RNAi flies to the wild-type level. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=14∼38. (D). Cardiac-specific knockdown of Atg1 attenuates the effects of Daw on age-induced arrhythmia. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=16∼35.
To test whether autophagy plays any role in Daw-regulated cardiac aging, we first fed control and Daw RNAi flies with lysosomal inhibitor chloroquine (CQ) for 24 hours. As predicted, CQ treatment increased cardiac arrhythmia of Daw RNAi flies at advanced age (Fig. 8C). To further test the role of cardiac-specific autophagy in Daw-mediated cardiac aging, we generated a double RNAi line targeting both Daw and Atg1 simultaneously. Consistent with CQ results, cardiac-specific knockdown of both Daw and Atg1 showed age-dependent increase of cardiac arrhythmia that is similar to control flies (Fig. 8D). Thus, these results suggest that autophagy is required for Daw-regulated cardiac aging.
Cardiac-specific activin signaling regulates systemic aging
The important roles of several tissues (such as adipose, muscle, and hypothalamus) in systemic aging control have been previously reported [10,37,38]. However, whether heart can serve as an aging control center remains unclear. Here we test the effects of cardiac-specific knockdown and over-expression of activin signaling in longevity control. Knockdown of Daw in the heart using two cardiac tissue drivers (Hand-gal4 and Tin-gal4) significantly prolongs lifespan (Fig. 9A, 9B, Supplemental Figure S3). On the other hand, knockdown of Babo slightly extended median lifespan (Fig. 9C, Supplemental Figure S3), while over-expression of Babo shortened median lifespan and increased mortality early in life (Fig. 9D, Supplemental Figure S3).

(A). Survival curve showing the lifespan of heart-specific (Hand-gal4) Daw knockdown and control flies. Log-Rank test, p<0.01. (B). Lifespan of cardiomyocyte-specific (Tin-gal4) Daw RNAi flies. Log-Rank test, p<0.01. (C). Lifespan of cardiomyocyte-specific Babo RNAi flies. Log-Rank test, p<0.05. (D). Lifespan of cardiomyocyte-specific Babo over-expression flies. Log-Rank test, p<0.05. (E). Heart-specific knockdown of Daw prevents age-related decline of climbing ability. One-way ANOVA (*** p<0.001, ** p<0.01, * p<0.05, ns = not significant). N=5.
To test if long-lived Daw RNAi flies show slow aging in non-heart tissues, we performed climbing assays as an indirect functional assessment for both flight muscle and neuromuscular junctions. Consistent with prolonged lifespan, cardiac-specific knockdown of Daw prevented age-dependent decline of climbing ability (Fig. 9E) Taken together, our data suggest that reduction in cardiac activin signaling improves cardiac function, prolongs lifespan and healthspan in Drosophila.
Discussion
TGF-beta signaling plans vital roles in many human diseases, including cancer, kidney and cardiovascular diseases [39]. Originally identified as a reproductive hormone, TGF-beta subfamily member activin has become an emerging target of therapy for the treatment of many human diseases [40]. In the present study, we investigated the role of activin in cardiac aging using a Drosophila heart model. Similar to its role in flight muscle aging [18], reduction of activin signaling prevents age-related decline of cardiac function (e.g. diastole interval and arrhythmia). Activin negatively regulates cardiac autophagy and blockade of autophagy process attenuates the effects of activin on cardiac aging, suggesting the autophagy is essential for activin-regulated cardiac aging. Interestingly, despite its positive regulation of mTOR, fly activin-like protein Daw controls autophagy and cardiac arrhythmia independent of mTOR signaling. Although we did not directly examine the role of mTORC1 key components (like Raptor) in cardiac aging and the genetic interaction between Raptor and Daw, the consistent results from multiple mTORC1 regulators or effectors (Tsc1, Rheb, and Reptor) suggest that mTORC1 may not contribute to Daw-mediated autophagy and cardiac aging control.
There is a positive correlation between activin A mRNA expression and heart failure [41,42]. Activin A promotes cardiac fibrosis and myocardial damage after ischemia reperfusion [43], while heterozygous mutant of activin receptor-like kinase 4 (ALK4) protects mouse heart from pressure overload-induced fibrosis and dysfunction [44]. Our findings support the protective role of reduced activin signaling in cardiomyopathy, in particular age-induced cardiac dysfunction. Interestingly, we also found a ligand-dependent regulation of cardiac aging by two different activin-like ligands. We show that knockdown of activin-like protein Daw delayed cardiac aging, while reduction in GDF11/ myostatin-like protein Myo promotes cardiac aging.
Although GDF11 can bind to activin type II receptor with similar affinities to that of activin A, two different ligands may activate very different downstream cascades and cellular processes. Similarly, TGF-beta family protein GDF11 was identified as an anti-aging circulating factor that regulates age-related cardiac hypertrophy [45], although the anti-aging effects of GDF11 is still under debating [46,47].
It has been shown that activin signaling play important roles in tissue homeostasis [40,48]. Our findings suggest that fly activin-like protein Daw regulates cardiac aging through autophagy inhibition. It remains to be determine how Daw regulates autophagy. We found that Daw knockdown induced the number of autophagosome, whereas the autophagy flux (autophagosome turnover) remains no change. These results suggest that Daw regulates early step of autophagy to promote autophagosome formation, rather than the late step (lysosome degradation). While basal autophagy activity in fly hearts is relatively low (based on the number of autophagosome under normal condition), we found that autophagy is still active in old fly hearts and autophagy flux remain no change during aging. These observations are not quite surprising, because several previous studies also found that autophagy/lysosome system remains active in aged animals [12,13,15,49]. In addition, Daw seems to only modulate the autophagosome formation, but not autophagy flux. Reduced Daw expression maintains high autophagosome formation activity throughout the life. These findings raise the possibility that the cardioprotection by Daw inhibition may be achieved by increasing the autophagosome cargo, without modulation of the degradation/turnover rate.
It is well known that mTOR is a major autophagy regulator and it inhibits autophagosome formation and autophagy flux by increasing the phosphorylation of the serine/threonine protein kinase complex Atg1 (ULK1, ULK2 in mammals), the initiator of the autophagy machinery [7]. Surprisingly, mTOR activation did not repress the high autophagosome formation in Daw knockdown flies, suggesting unknown mTOR-independent mechanisms involved in Daw-regulated autophagy. mTOR-independent autophagy regulation has been observed in a previous genome-wide screen [50]. This study identified an important role of type III PI3 kinases in autophagy control. They also find that growth factor signaling pathways, such as MAPK-ERK and AKT, negatively regulate autophagy by inhibiting the type III PI3 kinase cascade. Since it is known that TGF-beta/actin tightly links to AKT and MAPK signaling pathways, it is possible that Daw regulates autophagy via AKT/MAPK-regulated type III PI3 kinases.
It is known that mTOR promotes age-dependent increase of cardiac dysfunction in Drosophila [35,36]. Cardiac-specific over-expression of mTOR increases stress-induced heart failure, whereas over-expression of Tsc1 and Tsc2 prevents age-related increase of stress-induced heart failure [35]. However, our studies suggest that mTOR activation prevents age-induced cardiac arrhythmia, rather than accelerates it. The inconsistency may be due to the distinct roles of mTOR in the regulation of cardiac arrhythmia and electrical pacing-induced heart failure. Interestingly, the protective effects of mTOR was previously observed in a mouse model with cardiac-specific over-expression of mTOR (mTOR-Tg) [51]. The mTOR-Tg mice show attenuated inflammatory response and cardiomyopathy upon left ventricular pressure overload induced by transverse aortic constriction. Thus, the specific role of mTOR in stress- and aging-induced cardiac dysfunction remains to be further examined.
During normal aging, the heart undergoes complex phenotypic changes such as progressive myocardial remodeling, declined myocardial contractile capacity, increased left ventricular wall thickness and chamber size, prolonged diastole as well as increased arrhythmia [3]. All of these biological changes can gradually alter the cardiac functions and confer vulnerability of the heart to various cardiovascular stresses, thus increase the chance of developing cardiovascular disease dramatically. The present study uncovered an important role of TGF-beta/activin signaling pathway in the regulation of autophagy and cardiac aging in Drosophila. It is likely that autophagy might not be the only process controlled by activin signaling during cardiac aging. Therefore, future research is needed to explore the role of activin in other key homeostatic regulatory mechanisms and stress response pathways. The advanced understanding of TGF-beta/activin signaling pathway and its role in tissue homeostasis will help the development of therapeutic interventions targeting activin for the treatment of age-related cardiovascular diseases.
Materials and methods
Fly husbandry and stocks
Flies were maintained at 25°C, 60% relative humidity and 12-hour light/dark. Adults were reared on agar-based diet with 0.8% cornmeal, 10% sugar, and 2.5% yeast (unless otherwise noted). Fly stocks used in the present study are: UAS-Daw RNAi (BDSC #34974, BDSC #50911, and VDRC #105309), UAS-Babo RNAi (BDSC #25933, BDSC #40886), Babo-Act (or Babo-1A3) [52], UAS-Myo RNAi (BDSC #31114), UAS-Tsc RNAi (BDSC #52931, #54034), UAS-Reptor RNAi (BDSC #25983), UAS-PTEN RNAi (BDSC #33643), UAS-Rheb (BDSC #9688), UAS-Atg1 RNAi (BDSC #26731), Hand-gal4 (or Hand4.2-gal4) [26], and Tin-gal4 (or TinCΔ4-gal4) [27], Dot-gal4 [28], Hand-GS-gal4 [23], UAS-RpL13A-FLAG (gift from Dr. Pankaj Kapahi).
Daw RNAi (BDSC #34974), Babo RNAi (BDSC #25933), and Myo RNAi (BDSC #31114) were backcrossed into ywR background for 5∼7 generations, and ywR flies were used as control/wild-type flies in the experiments. For other UAS-RNAi lines, y1 v1; P[CaryP]attP2 (BDSC #36303) and y1 v1; P[CaryP]attP40 (BDSC # 36304) were used as control/wild-type. Female flies were used in all experiments.
Ribo-tag and quantitative RT-PCR
For Ribo-tag analysis, about 200 female flies (Hand-gal4>UAS-RpL13A-FLAG) were homogenized in lysis buffer containing 1% NP-40 (Thermo Fisher Scientific, Waltham, MA, USA). FLAG-tagged polyribosomes were immunoprecipitated by incubating tissue lysate (input samples) with anti-FLAG antibodies and SureBeads Protein G Magnetic Beads (Bio-Rad, Hercules, CA, USA). Actively translated mRNAs were extracted from bead-antibody-ribosome complex using RNeasy Micro kit (Qiagen, Hilden, Germany). Isolated mRNA was reverse transcribed to cDNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA).
For Babo isoform expression analysis, Total RNA from 12∼15 dissected adult hearts (ywR) was extracted using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA). DNase-treated total RNA was quantified and about 50∼100 ng of total RNA was reverse transcribed to cDNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). QRT-PCR was performed with a Quantstudio 3 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). mRNA abundance of each gene was normalized to the expression of ribosomal protein L32 (RpL32 or rp49) by the method of comparative CT. Primer sequences are listed in Supplementary Table S1.
Fly heartbeat analysis
To measure cardiac function parameters, semi-intact Drosophila adult fly hearts were prepared according to previously described protocols [25]. High-speed 3000 frames movies were taken at a rate of 100 frames per second using a Hamamatsu ORCA-Flash4.0 digital CMOS camera (Hamamatsu Photonics, Hamamatsu, Japan) on an Olympus BX51WI microscope with a 10X water immersion lens (Olympus, Waltham, MA, USA). The live images that contain heart tube within abdominal A3 segment were processed using HCI imaging software (Hamamatsu Photonics, Hamamatsu, Japan). M-modes and cardiac parameters were generated using SOHA, a MATLAB-based image application as described previously [25,53]. The M-mode provides a snapshot of movement of heart wall over time. Four cardiac parameters were analyzed, including heart period, diastolic interval, systolic interval, and arrhythmia index. Diastolic interval (DI) is the duration for each heart relaxation phase (diastole). Heart period (HP) is the pause time between the two consecutive diastole. Systolic interval (SI) is calculated as the HP minus the DI. Arrhythmia index (AI) is calculated as the standard deviation of all HP in each fly normalized to the median HP.
Antibodies and immunostaining
Anti-Atg8a polyclonal antibodies were generated in rat against a recombinant Atg8a protein (1-60 aa) (Iowa State University Hybridoma Facility) and affinity purified using CNBr-Activated Sepharose 4B (GE Healthcare Life Sciences, Marlborough, MA, USA). Other antibodies for immunostaining included: phospho-4E-BP1 antibody (1:300) (#2855, Cell Signaling Technology, Danvers, MA, USA), Alexa Fluor 488-conjugated Phalloidin for F-actin staining (Thermo Fisher Scientific, Waltham, MA, USA), all fluorescence-conjugated secondary antibodies from Jackson ImmunoResearch, West Grove, PA, USA). For immunostaining, adult female flies were collected and dissected in PBS. Hearts were fixed in 4% paraformaldehyde for 15 min at RT. After washing in PBS with 0.1% Triton X-100 (PBST), the fixed hearted were blocked in 5% normal goat serum diluted in PBST for 1 hour at RT. Hearts were then washed with PBST and incubated overnight at 4°C with primary antibodies diluted in 5% NGS. After washing with PBST, the samples were incubated for 2 hours at RT with appropriate fluorescence-conjugated secondary antibodies. Hearts were mounted in ProLong Gold anti-fade reagent (Thermo Fisher Scientific, Waltham, MA, USA) before imaged using an epifluorescence-equipped BX51WI microscope (Olympus, Waltham, MA, USA). For imaging analysis, fluorescence images were first processed using the deconvolution module in Olympus cellSens demensions package, and then the number/area of positive immunostaining was measured with the “Measure and Count” module provided by cellSens software (Olympus, Waltham, MA, USA).
Western blotting
Antibodies for western blot included: beta-Actin antibody (1:2000) (#4967, Cell Signaling Technology), phospho-4E-BP1 antibody (1:1000) (#2855, Cell Signaling Technology), and HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Protein samples were homogenized in lysis buffer with leupeptin, benzamidine, antipain, PMSF and 2-Mercaptoethonal. Supernatant was denatured at 95°C for 5 min. About 30 μg of denatured protein was separated on Mini-PROTEAN precast gels (Bio-Rad, Hercules, CA, USA) and then transfer to 0.2 μm PVDF membrane. Following incubation with primary and secondary antibodies, the blots were visualized with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA).
Bafilomycin A1 and Chloroquine Treatment
For Bafilomycin A1 treatment, semi-intact hearts were incubated with 100 nM of Bafilomycin A1 (Alfa Aesar, Haverhill, MA, USA) in artificial hemolymph (buffer receipt in [25]) for two hours at room temperature prior to the appropriate immunostaining. DMSO was used as a control.
For chloroquine treatments, 100 μl of 20 mM chloroquine diphosphate salt (Sigma-Aldrich, St Louis, MO, USA) was added onto the fly food. Flies were fed with chloroquine for at least 24 hours prior to the appropriate cardiac analysis.
Cell culture and Daw activity assay
A cell-based Daw activity assay was previously described [54]. Briefly, Drosophila Kc167 S2 cells were transfected with FLAG-tagged Daw for three days (Transfection with empty vector was served as a control). Conditioned media were harvested and incubated with a fresh flash of Kc167 cells. Three hours after incubation, cell lysate was analyzed by western blotting.
Demography and survival analysis
Newly enclosed female flies were allowed to mate for two days, then separated from males and assigned to replicate one-liter demography cages at a density of 100-125 flies per cage. Three independent cages were set-up per genotype. Food was changed every two days, at which time dead flies were removed from the cage and counted. Survival analysis was conducted with JMP statistical software (SAS Institute, Cary, NC, USA), and data from replicate cages were combined. Mortality distributions were compared by Log-rank test.
Climbing assay
Climbing ability was measured by tapping flies to the bottom of an empty vial and counting (with video recording) flies that climbed 8 vertical centimeters within 20 seconds. Thirty females (10 per vial) were scored once every other week for each genotype.
Statistical analysis
GraphPad Prism (GraphPad Software, La Jolla, CA) was used for statistical analysis. To compare the mean value of treatment groups versus that of control, either student t-test or one-way ANOVA was performed using Dunnett’s test for multiple comparison. The effects of mutants during aging was analyzed by two-way ANOVA, including Tukey multiple comparisons test. In SOHA analysis, the outliers were identified using Robust regression and Outlier removal (ROUT) method (Q=1%) prior to the data analysis.
Author Contributions Statement
Conceived and designed the experiments: RB KO HB. Performed the experiments: KC PK YL KH HB. Analyzed the data: KC PK YL KH ET HB. Wrote the paper: KC RB KO HB. All authors reviewed and approved the manuscript.
Competing Interests
The authors declare that no competing interest exists.
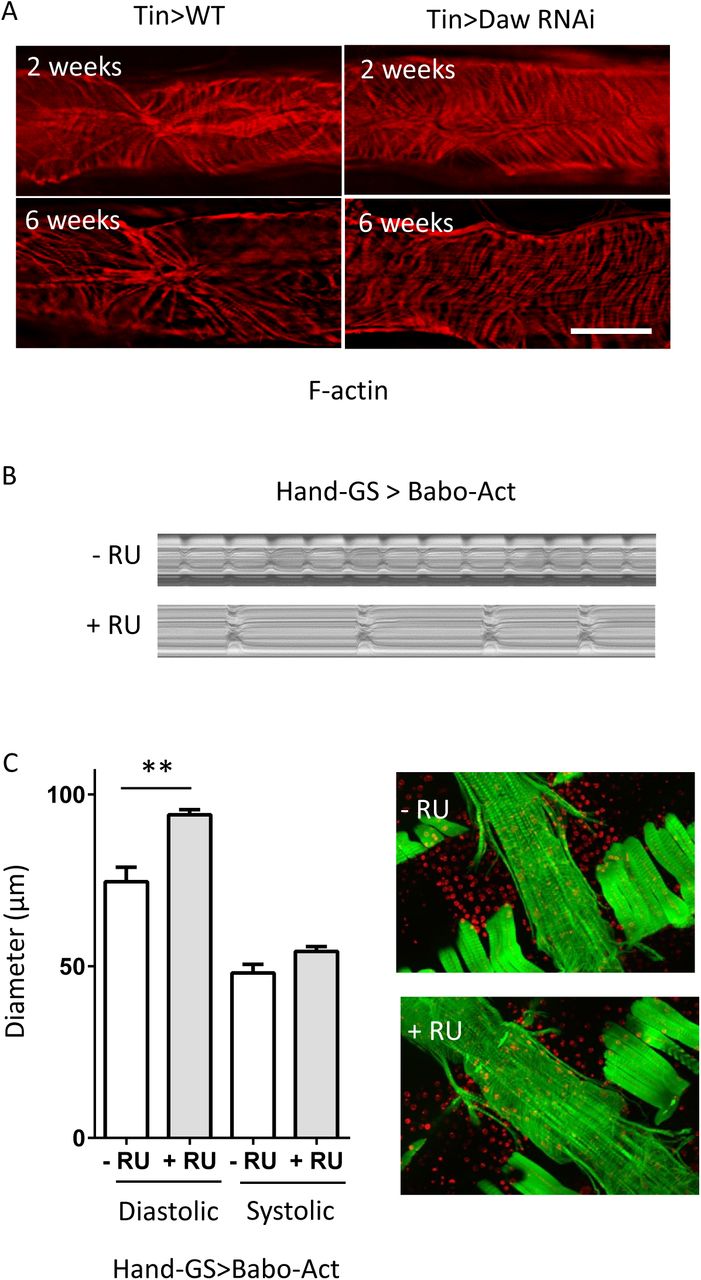
(A). F-actin staining showing cardiomyocyte structure in young and old control and cardiomyocyte-specific Daw knockdown flies. (B). M-mode of heat-specific Babo over-expression. RU: RU486. (C). Over-expression of Babo increases diastolic diameter. N=7∼8.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A). Immunostaining of p4e-BP in cardiomyocyte-specific knockdown of Daw or Tsc1. (B). Quantification of p4E-BP staining of Panel A.
Acknowledgements
We thank Bloomington Drosophila Stock Center, Harvard Medical School, and Drosophila Genomics Resource Center for fly stocks and cDNA clones. We thank Michael O’Connor for the kind advice, activin reagents, and fly lines, Dr. Pankaj Kapahi for Ribo-tag protocols and fly lines, ISU Hybridoma Facility for Atg8a antibody production.
References




