

Molecular dynamics (MD) simulations employ a potential energy function, referred to as a force field, in order to sample the free energy landscapes of biomolecular systems. Due to the intractable complexity of biological systems, the force field is commonly of an approximate classical form, and is fit using quantum mechanical and experimental data (1, 2). As a result, the accuracy of these force fields has tracked with advances in computational hardware and methodology, as well as the increased availability of high resolution experimental data (3). The current state-of-the-art protein force fields demonstrate high accuracy in their ability to describe the protein native state and its equilibrium behavior: these models are all able to describe ensemble averaged properties of proteins with highly populated native states within experimental error (4). Of greater discrepancy is their description of the denatured state ensemble. As such, one of the major frontiers in protein force field development is the accurate description of proteins away from equilibrium.
Protein folding is a strong validation test of a protein force field (5, 6). This is because as the protein folds and unfolds, it samples beyond the native state. Performing protein folding simulations using multiple force fields allows for the comparison of their denatured state ensembles. Furthermore, we can make force field-agnostic conclusions from an aggregated dataset. A popular system for such a task is the ultrafast folding miniprotein, CLN025. This protein folds within timescales accessible to computation into a highly stable beta-hairpin. At room temperature, the native state is almost exclusively populated (7). As temperature increases, the denatured state population increases. Experiments probing relaxation kinetics over a range of temperatures have shown that there is a critical break in the folding mechanism of this protein at 308 K (8). Above this temperature, folding can no longer be described using a two-state model. Because the experimental description of this system is both detailed and nontrivial at high temperature, we have benchmarked a set of popular protein force fields in their ability to describe the conformational dynamics of CLN025 at its experimental melting temperature of 340 K (7).
The folding of CLN025 is of additional interest due to its beta-hairpin structure. Neither experimentalists nor theorists have reached a consensus on the mechanism or rate-determining step of beta hairpin folding (9–11). In this work, we use our aggregated MD dataset to facilitate the understanding of beta-hairpin formation. We first enumerate the force fields studied and discuss the Markov state model (MSM) framework used to analyze our MD datasets. Next, we examine the thermodynamics and kinetics of folding for the three force fields investigated and note that only the AMBER-FB15 model (4) exhibits melting behavior at the simulation temperature. Lastly, we analyze the three MD datasets simultaneously to interrogate the mechanism and rate-determining process of CLN025 folding. Through this analysis we find that the CLN025 folding mechanism comprises a downhill hydrophobic collapse followed by the slower formation of the hairpin turn over a barrier. The order of these conformational changes is consistent with a recent experimental study of CLN025 (8).
Models
The force field combinations used in this study are:
The CHARMM22* and AMBER ff99SB-ILDN parameter sets were developed by (12) and (14), respectively, as augmentations to previous generations of CHARMM and AMBER parameter sets. The AMBER-FB15 parameter set, developed by (4), was built via a complete refitting of the bonded parameters of the AMBER ff99SB force field (17) with training data taken from RI-MP2 calculations using augmented triple-zeta and larger basis sets (18). Notably, the training set contained complete backbone and side chain dihedral scans for all (capped) amino acids. During force field validation, it was found that parameter optimization yielded improved melting curves for both CLN025 and Ac-(AAQQAA)3-NH2. We suspect that the improved thermal dependence could be attributed to improved description of the dihedral barrier heights (19–21). Note that each protein force field was simulated using its corresponding water force field. The dataset for model (a) was obtained from D.E. Shaw research, and was generated as described in their seminal fast folding protein study (22). The datasets for models (b) and (c) were generated via the distributed computing platform Folding@home (23). This architecture allows us to sample many instances of folding from the extended state, and hence gather robust statistics regarding the folding process. For details regarding the preparation and execution of these simulations, see the supporting information (SI).
Whereas specialized hardware is typically used to generate one or several ultralong MD simulations, simulations performed on distributed computing platforms such as Folding@home produce datasets consisting of many short trajectories. The use of MSMs was a crucial advance in the analysis of such datasets (24–26). To construct a MSM, each frame of each trajectory is assigned to a discrete state. The model comprises the populations of and conditional pairwise transition probabilities between states, which provide thermodynamic and kinetic information, respectively. Since separate trajectories will feature common states, the trajectories can be threaded together through this framework and pathways between states can be determined even if the pathway is not contained in a single trajectory.
The choice of which collective variables to use when describing MD datasets is an area of active research (27). Describing the trajectories using time-structure based independent component analysis (tICA) allows us to analyze the MD dataset in terms of its slow dynamical processes (28, 29). Each component of the tICA transformation, or “tIC”, serves as a reaction coordinate for the system (30). For a protein folding dataset, the first tIC is expected to correspond to the folding process and can thus be used as a reaction coordinate for folding. The MSM is then created from trajectories that are represented by their progress along the tICs by creating microstates that group kinetically similar conformations. The thermodynamics and kinetics of different systems can be directly compared when the same representation (i.e. tICA model) and microstates are used to build a MSM for each dataset.
Results
First, we describe folding from a global perspective and compare the thermodynamics and kinetics for each force field. Then, we inspect the mechanism of beta-hairpin formation for each dataset in the context of a set of influential theoretical and experimental studies. Last, we examine the rate-determining step of the folding process.
Thermodynamics and kinetics of folding
In order to analyze folding of CLN025, we first constructed an optimized MSM for the CHARMM22* dataset (see the SI for optimization protocol and model validation). The same features, tICA model, and states were then used to derive a unique MSM transition matrix for the AMBER ff99SB-ILDN and AMBERFB15 datasets. By using a consistent model basis, we are able to directly compare folding of CLN025 as a function of force field. This approach allows us to summarize folding along a kinetically motivated 1-dimensional reaction coordinate (see Models for more detail). We note that our conclusions are independent of the basis chosen, and results from AMBER ff99SB-ILDN and AMBER-FB15 bases are shown in the SI.
This data is illustrated in Fig. 1 (top), where folding from the denatured state is represented by the movement from the right free-energy basins, labeled “denatured extended” and “denatured collapsed”, to the left basin, labeled “folded”. Additionally, in order to quantify the kinetics of folding, we computed the mean first passage time (MFPT) both to and from the folded and denatured states for each model. This is depicted in Fig. 1 (bottom) and the method is described in the SI.

Thermodynamics and kinetics of CLN025 folding reveal differences across force fields. Top: The free energy landscape shows overstabilization of the native state for the CHARMM22* and AMBER ff99SB-ILDN force fields at the experimental melting temperature. The black dashed line indicates the location of the crystal structure (PDB ID: 5AWL) on the folding reaction coordinate (see Models). All models populate a folded state and a denatured extended state, but only the AMBER models populate a denatured collapsed state at this temperature. Shading represents the range of free energies based on 100 bootstrapped samples between the 5th and 95th percentiles. Bottom: The mean first passage time for folding is approximately similar for all force fields; however, the mean first passage time for unfolding is slower than folding for the CHARMM22* and AMBER ff99SB-ILDN models at the experimental melting temperature. Only the AMBER-FB15 model shows approximately equal folding and unfolding MFPTs, which is consistent with melting behavior.
We found that all models share several notable characteristics. First, all models show that the folding process is rate limited by a small global barrier (Fig. 1, top). This demonstrates that the potential energy surfaces for CLN025 described by each of these force fields are qualitatively similar. Second, all were able to fold the extended, denatured protein into a native conformation similar to the experimental crystal structure (dashed line, Fig. 1, top). The minimum of the folded basin is found at a similar location on the reaction coordinate for all models. This implies that the most stable folded conformations are also very similar. Third, the mean first passage time (MFPT) for folding was found to be on the order of 10 ns. This is evidenced by the short and comparable folding MFPTs for all models (Fig. 1, bottom).
The examined models also differ in several ways. First, their description of dynamics at the experimental melting temperature differ. During melting, the native and denatured states should be equally populated, and the folding and unfolding rates should be the same. We found that the AMBER-FB15 model displays equally deep folded and unfolded basins, well as approximately equal folding and unfolding MFPTs. This is aligned with experiment at the same temperature. In contrast, the CHARMM22* and AMBER ff99SB-ILDN models exhibit disproportionately high unfolding barriers, and unfolding MFPTs much slower than their corresponding folding MFPT. This represents overstabilization of the native state at the experimental melting temperature. Such a phenomenon is a common limitation of protein force fields (31, 32).
We expect that melting behavior would be achieved for the CHARMM22* and AMBER ff99SB-ILDN models at temperatures higher than the experimental melting temperature.* Our comparison of the dynamics at the experimental melting temperature shows the discrepancies of the CHARMM22* and AMBER ff99SB-ILDN dynamics with experiment at the same temperature. Since the CHARMM22* and AMBER ff99SB-ILDN models do not show melting, these models differ considerably in the structure of the denatured state ensemble. At 340 K, the AMBER ff99SB-ILDN and AMBER-FB15 models populate both an extended denatured state as well as a compact denatured state. This compact denatured state describes a hydrophobically collapsed structure. In contrast, the CHARMM22* model populates only the denatured extended state. Detailed analysis of these states can be found in the following sections.
Mechanism of beta-hairpin formation
The theory of beta-hairpin formation has converged on two leading mechanisms that were developed to explain the folding of the C-terminal fragment of Protein G (residues GLY-41 to GLU-56). The first, suggested by (9, 33) to explain relaxation kinetics observed from T-jump experiments, proposes that the hairpin turn forms first from the extended state. The beta-sheet then “zips” from the turn to the terminus via the formation of a series of cross-strand hydrogen bonds; in so doing, the structure becomes collapsed. The second mechanism, formulated by (10) and by (11), proposes that hydrophobic collapse occurs first and the turn is formed from the collapsed structure. The mechanism of (10) includes the same “zipping” of hydrogen bonds from the turn to the terminus, whereas the mechanism of (11) proposes formation of hydrogen bonds starting near the middle of the beta sheets and propagating outward in both directions. Many subsequent studies of this fragment also produced irreconcilable results (see, e.g., works cited in (34) and (35)).
Upon the design of CLN025 and its predecessor chignolin (36), these 10-residue systems were used to study beta-hairpin formation and continued to produce contradictory results (35, 37). In 2012, (8) used T-jump experiments combined with infrared and fluorescence spectroscopy to empirically measure the relaxation kinetics of the turn and terminal regions of CLN025. It was found that above 308 K, folding cannot be described using a two-state model. Above this temperature, (8) showed that the turn, beta sheet, and hydrophobic collapse processes occur on significantly different timescales, with a faster rate observed for beta sheet and hydrophobic cluster formation. Additionally, as temperature increases toward the melting temperature, the timescale separation increases. These results suggest a mechanism in which interactions of the terminal hydrophobic residues first cause the extended structure to collapse into a native-like topology, after which small local rearrangements occur, forming the turn and the remaining native state contacts. While this experimental characterization describes the ordering of major conformational changes, it does not resolve the relative order of specific hydrogen bond formation in the beta sheet.
We analyzed the sequence of events in our simulation datasets by using the models created above. To assess whether the turn had formed, we tracked the existence of three hydrogen bonds characterizing the turn (purple distances, Fig. 2). To determine whether the structure had collapsed, we used a binary metric based on the radii of gyration of the two hydrophobic terminal residues (TYR-1 and TYR-10; dark gray residues, Fig. 2).† Finally, to monitor the completion of beta sheet formation, the three terminal hydrogen bonds were monitored (dark gray distances, Fig. 2). We elaborate on these feature sets in the SI.

Subprocess-specific features are used to monitor the CLN025 folding mechanism. Hydrogen bonds of the native state turn, for which distances were calculated, are shown in purple. Hydrophobic residues, for which the radii of gyration were measured, are highlighted in dark gray. Distances of the native state beta sheet hydrogen bonds are also highlighted in dark gray.
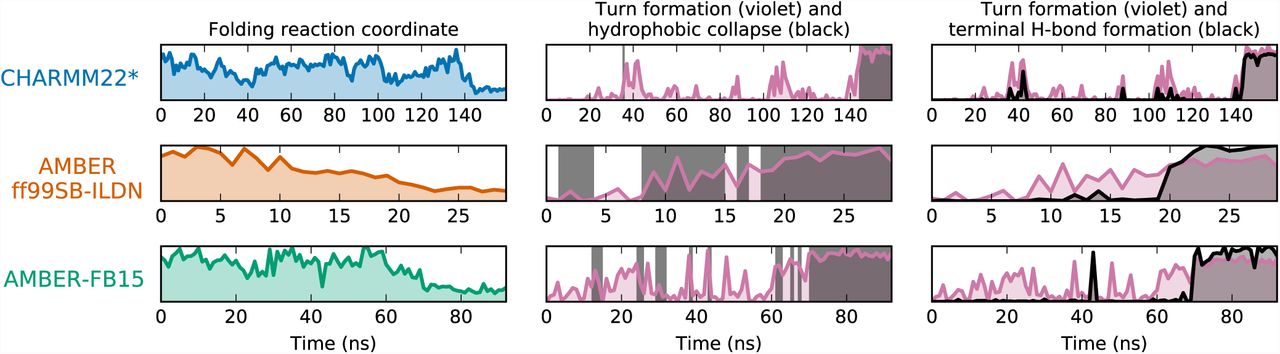
Fig. 3 shows a representative trajectory for each of the three MD datasets. First, it is interesting to note that in the CHARMM22* dataset, folding occurs as a concerted mechanism: the protein is either denatured extended or folded, and the turn formation and collapse occur simultaneously and quickly from the extended state. The model of CHARMM22* at this temperature does not resolve the mechanism enough to compare or contrast it with the existing theories of beta-hairpin formation. In the AMBER datasets, however, the turn and hydrophobic collapse occur gradually with instances of collapse (formation of hydrophobically collapsed structures) preceding the completed turn. Furthermore, in the AMBER trajectories the hydrogen bonds at the terminus form after the hydrogen bonds at the turn, providing evidence for the hydrogen bond “zipping” process proposed by (9, 33) and corroborated by (10). Visualization of additional pathways for each force field are provided in the SI, and a example movie of each force field is provided as a supplementary file.

Representative folding trajectories reveal the mechanism of CLN025 folding. Each row represents a single trajectory that starts in an extended state and ends in the folded state. The left column shows the folding reaction coordinate (see Fig. 1), where low y-axis values correspond to the folded state. In the center and right columns, the formation of the three hydrogen bonds characterizing turn formation are shown in purple, where high values on the y-axis indicate the turn is formed. In the center panel, dark gray shading is used to show when the structure is collapsed. In the right panel, the formation of the three terminal hydrogen bonds is shown in dark gray, where high y values indicate that all three bonds are formed. In the CHARMM22* model, the collapse and formation of both turn and terminal hydrogen bonds occur nearly simultaneously. In the AMBER models, the appearance of collapsed structure precede the turn, and the turn hydrogen bonds form before the terminal hydrogen bonds.
Rate-determining process
The original beta-hairpin formation theories also disagree on the rate-limiting step of the folding process. (9, 33) hypothesized that the formation of the turn from the extended state determined the rate of beta-hairpin formation. (10) agreed that the first step of the mechanism determined the rate, but in their mechanism the hydrophobic collapse preceded the turn and thus the collapse from the extended state characterized the rate-limiting step. The mechanism of (11) identifies the rate-limiting step as the inter-conversion between collapsed conformations; i.e. the formation of the turn and native hydrogen bonds from a compact state.
In order to analyze the separate processes involved in beta-hairpin formation in our datasets, we constructed MSMs over two specific feature sets designed to characterize either hydrophobic collapse or turn formation. These sparse feature sets isolate the process of interest so that structures in the MD dataset are differentiated only by characteristics relevant to the appropriate process. Because the MSM timescales describe the timescales of conformational change, the longest timescale of each MSM corresponds to the relaxation time of each process of interest (19, 38–40). These values can be directly compared with process-specific experimental relaxation timescales (8). In order to estimate the rate of hydrophobic collapse, the features selected were the radius of gyration of the two terminal hydrophobic residues (TYR-1 and TYR-10). To estimate the rate of turn formation, we calculated distances between the hydrogen bonded contacts in the turn region of the CLN025 crystal structure depicted in Fig. 2. The dihedral angles associated with these hydrogen bonds were also included, since it has been shown that only certain turn dihedrals can lead to the correct secondary structure (41). We then constructed an optimized MSM for each feature set (see the SI for feature descriptions, optimization protocol, and model validation). The per-model reaction coordinate and slowest relaxation timescale for these two processes are depicted in Fig. 4.

Isolating the processes involved in CLN025 folding reveals differences in their timescales and kinetics. The timescale plots show that all models calculate the turn formation to occur on a slower timescale than the hydrophobic collapse. The experimental values from (8) are represented by the gray shading. The reaction coordinate free energy plots above the timescale plots show that for all models hydrophobic collapse occurs downhill while turn formation occurs over a barrier. The rate-limiting step is the slower formation of the turn from the collapsed state. The ruggedness of the hydrophobic collapse landscapes indicates the existence of collapsed conformations in the denatured ensemble. Uncertainty in MSM timescale and experimental values are indicated by the thickness of the line. Shading represents the range of free energies based on 100 bootstrapped samples between the 5th and 95th percentiles.
First, we note that the relative ordering of timescales agrees with the experiments of (8). For all force field datasets, we observe a separation between the timescales corresponding to slower turn formation and faster hydrophobic collapse. Next, we note that the reaction coordinates corresponding to the hydrophobic collapse describe downhill pathways. In contrast, the reaction coordinates corresponding to turn formation feature a barrier between the turned and not turned conformations. From the relative ordering of timescales and the shape of the collapse and turn pathways, we agree with the conclusions of (8) and find that the rate-determining process for beta-hairpin formation is the formation of the turn from a pre-collapsed structure. This is also consistent with the rate-limiting step for beta-hairpin folding proposed by (11) and supported by the experiments of (8).
Discussion
In summary, our aggregated MD analysis suggests a beta-hairpin folding mechanism in which the extended state collapses into a hydrophobic cluster, followed by a slower process in which the hairpin turn forms over a barrier within the denatured collapsed state. The order of these conformational changes agree with the experimental conclusions reported by (8) for CLN025. Additionally, the resolution of MD simulations has allowed us to also model the formation of specific native state hydrogen bonds. We observe that the hydrogen bonds are formed by a “zipping” mechanism from the turn toward the terminus. Our findings demonstrate mixed agreement with the early theories of beta-hairpin formation; namely, our results support the “turn zipper” process of hydrogen bond formation (9, 33), the collapse-then-turn mechanism (10), and the rate-determining process comprising rearrangement within a collapsed state (11).
Performing this analysis simultaneously with datasets built from three different protein/water force field combinations demonstrates the force field dependence of CLN025 simulations at the experimental melting temperature. We find that simulations performed with CHARMM22* and AMBER ff99SB-ILDN yield an overstabilized native state and unequal folding and unfolding rates, which indicates that a higher simulation temperature would be necessary to obtain melting behavior. In contrast, AMBER-FB15 simulations show behavior consistent with melting. Furthermore, while the folding mechanism can be determined using either AMBER dataset, the CHARMM22* dataset does not contain a compact denatured state at the simulated temperature, nor does it resolve the ordering of hydrogen bond formation via the “zipper” mechanism. We recommend that modelers who wish to use MD simulation to interrogate the denatured state ensemble of a protein and/or its role in the protein folding process choose a force field that accurately represents denatured state properties at the temperature of interest, and highlight that the AMBER-FB15 model yields behavior consistent with experiment at the simulated temperature. We anticipate that protein force fields that are accurate beyond the native state and sensitive to temperature dependence will enable further insight into more complex protein systems.
Materials and Methods
Complete descriptions of all methods used in this work are available in the SI.
Simulations and MSMs
The SI describes preparation of AMBER simulations for the ff99SB-ILDN and FB15 parameter sets and provides the script used to generate initial states. All parameters of the MSMs created are enumerated in the SI along with descriptions of the analyses presented in the main text.
Data
MSM objects compatible with the MSMBuilder software have been provided for all MSMs discussed in the main text. Details of these files and instructions for loading them can be found in the SI.
Movies
Example movies for CLN025 folding in each force field are provided as supplementary files.
Software
Free, open source software implementing the methods used in this work is available in the OpenMM (42), MDTraj (43), MSMBuilder (44), and Osprey (45) packages available from http://openmm.org, http://mdtraj.org, and http://msmbuilder.org.
ACKNOWLEDGMENTS
The authors thank Lee-Ping Wang and Muneeb Sultan for helpful discussions and Jerelle Joseph for invaluable manuscript feedback. We are also grateful to Caitlin Davis and Brian Dyer for discussions about their experimental results. We acknowledge the National Institutes of Health under No. NIH R01-GM62868 for funding. We graciously acknowledge Folding@home donors who contributed to the AMBER force field simulations and D. E. Shaw Research for providing CHARMM22* simulation data.
Footnotes
This manuscript was compiled on June 21, 2017
K.A.M. designed the experiment and performed the AMBER simulations. B.E.H. constructed statistical models. K.A.M. and B.E.H. visualized results, formulated conclusions, and wrote the manuscript. V.S.P. supervised the experiment.
V.S.P. is a consultant & SAB member of Schrodinger, LLC and Globavir, sits on the Board of Directors of Apeel Inc, Freenome Inc, Omada Health, Patient Ping, Rigetti Computing, and is a General Partner at Andreessen Horowitz.
↵* The folding temperature for CLN025 with CHARMM22* is reported to be 370 K (22). The analysis of CLN025 for the AMBER ff99SB-ILDN with the TIP3P water model in (4) shows that about 85% of CLN025 is folded at 370 K; thus the folding temperature for this force field is expected to be higher.
↵† The hydrophobic collapse metric follows from the first tICA solution (i.e. the slowest process found) when only the radii of gyration of the two hydrophobic terminal residues are input into the tICA model. This is the same feature set used for the analysis of the hydrophobic collapse process in the context of the rate-determining step.





{kind=link}
{kind=link}
{kind=link}
{kind=link}