

Abstract
Most current methods to identify cell-specific RNA binding protein (RBP) targets require analyzing an extract, a strategy that is problematic with small amounts of material. To address this issue, we developed TRIBE, a genetic method that expresses an RBP of interest fused to the catalytic domain of the RNA editing enzyme ADAR. TRIBE therefore performs Adenosine-to-Inosine editing on candidate RNA targets of the RBP. However, editing is limited by the efficiency of the ADARcd and may fail to identify some RNA targets. Here we characterize HyperTRIBE, which carries a hyperactive mutation (E488Q) of ADAR. HyperTRIBE identifies dramatically more editing sites than TRIBE, many of which were also identified by TRIBE but at a low editing frequency. The HyperTRIBE data also overlap more successfully with CLIP data, further indicating that HyperTRIBE has a reduced false negative rate and more faithfully recapitulates the known binding specificity of its RBP than TRIBE.
Introduction
Pre-mRNAs and mRNAs are subjected to many post-transcriptional regulatory events, which are mediated by RNA-binding proteins (RBPs). RBPs have been found to be essential for pre-mRNA splicing, 3’ end formation, mRNA translocation from the nucleus to the cytoplasm, and translation among other processes (Gerstberger et al., 2014; Jansen, 2001; Szostak and Gebauer, 2013; Witten and Ule, 2011; Zhao et al., 1999). Numerous human diseases have been linked to RBPs, e.g., ALS (Amyotrophic Lateral Sclerosis) (Nussbacher et al., 2015). Identifying the RNA targets of a RBP is not only crucial for deciphering its function but also an important aspect of deciphering RBP-related human diseases.
The current standard technique for identifying RBP targets in vivo is CLIP (Cross-Linking and ImmunoPrecipitation) and its variants (Hafner et al., 2010; Ule et al., 2005). These methods use UV light to covalently link the RBP to its targets, immunoprecipitate the RNA-protein complex, and then purify and identify the covalently bound RNA. CLIP has definite advantages, for example the ability to identify the exact RBP binding sites on RNA, but there is also a disadvantage: its requirement for large amounts of material to make an extract and do immunoprecipitations (Darnell, 2010). It is therefore virtually impossible to perform CLIP in a cell-specific manner.
We recently developed TRIBE (Targets of RNA-binding proteins Identified By Editing) to study RBP targets in specific cells, especially in small numbers of circadian neurons of the Drosophila adult brain. This method expresses in specific cells a fusion protein of an RBP and the catalytic domain of Drosophila ADAR (McMahon et al., 2016). ADAR is a well-conserved RNA editing enzyme. It deaminates adenosine to inosine, which is read by ribosomes and reverse transcriptase as guanosine (Keegan et al., 2004). ADAR consists of two modular parts, double-strand RNA-binding motifs (dsRBMs) and its catalytic domain (Keegan et al., 2004). TRIBE replaces the dsRBMs of the Drosophila ADAR enzyme with the RBP of interest (McMahon et al., 2016). Fusion protein binding and editing specificity is therefore driven by the RBP due to the absence of endogenous dsRBMs. TRIBE-dependent editing sites are identified by deep sequencing of RNA extracted from cells of interest. The editing percentage of any mRNA nucleotide is the number of adenosines edited to inosines divided by total number of reads at that site. In the initial TRIBE paper (McMahon et al., 2016), we used a conservative criterion common in the editing field, namely, a site is only scored as edited if it has an editing percentage greater than 10%, and a read coverage greater than 20, i.e., at least 2 editing events/20 reads.
Although we were successful in identifying RBP targets in tissue culture as well as in specific Drosophila brain cells, the number of targets identified by TRIBE in tissue culture was substantially reduced compared to CLIP data with the same RBP (McMahon et al., 2016). Even assuming a rather high false-positive rate of CLIP (Darnell, 2010; Lambert et al., 2014), TRIBE may still be experiencing a high false-negative problem. It could be due to the innate editing preference of the ADARcd. It preferentially edits adenosines bordered by 5’ uridines and 3’ guanosines, i.e., a UAG sequence (Eggington et al., 2011), which is also observed in TRIBE (McMahon et al., 2016). The ADARcd also prefers to edit adenosines surrounded by a double-stranded region (Eggington et al., 2011; Matthews et al., 2016). Consequently, a substantial fraction of RBP-target mRNAs may go unidentified.
To enhance the efficiency and/or reduce the specificity of the ADARcd, we turned our attention to a mutational screen directed at the catalytic domain of human ADAR2 (Kuttan and Bass, 2012). It identified a “hyperactive” E488Q mutation, which was reported to have these precise characteristics. Ideally, incorporating this hyperactive E488Q mutation into TRIBE (HyperTRIBE) could reduce its false negative rate.
As predicted, Hrp48 HyperTRIBE, which carries this E488Q mutation within the Drosophila ADARcd (dADARcd), identifies dramatically more editing sites than Hrp48 TRIBE and shows a reduced nearest-neighbor preference. Many of these sites correspond to below-threshold TRIBE editing targets. This indicates that they are bona fide targets, which are edited more efficiently by HyperTRIBE. These targets overlap much more successfully with CLIP, indicating that HyperTRIBE has a much reduced false-negative problem and suggesting that it more faithfully recapitulates the known binding specificity of its RBP than TRIBE.
Results
The original Hrp48 TRIBE construct was made by fusing coding DNA for the Hrp48 protein followed by a short linker and then dADARcd (henceforth called TRIBE). To make the HyperTRIBE construct, we first identified the dADARcd glutamate corresponding to hADAR2 E488. That residue as well as surrounding sequence is highly conserved between hADAR2 and the dADARcd (data not shown). We then introduced the E488Q mutation into the original Hrp48 TRIBE construct via QuikChange® site-directed mutagenesis. We had difficulty making a stable S2 cell line expressing Hrp48 HyperTRIBE (henceforth called just HyperTRIBE), perhaps because of the greatly enhanced editing frequency (see below). Most experiments were therefore performed by transiently expressing fusion proteins in Drosophila S2 cells together with GFP and sorting GFP-positive cells by FACS. Despite the many fewer targets identified by transient TRIBE expression than the targets from cell lines with stable TRIBE expression (due to much shallower sequencing coverage), more than 60% of the transient targets overlap with the stable targets (Fig. S1). Editing sites were always defined as the common edited sites between two or more replicate experiments.
Expression of HyperTRIBE results in approximately 20X the number of editing events compared to TRIBE; this is normalized to the respective sequencing read coverage (Fig. 1A). Expression of the ADARcd with E488Q mutation alone does not increase the number of editing events above the endogenous level of S2 cells (Fig. 1A). This is despite the fact that the HyperADARcd is stable and expressed at comparable levels to those of the other TRIBE constructs (data not shown), so editing by HyperTRIBE like editing by regular TRIBE requires the RNA binding ability of the fused RBP (McMahon et al., 2016).

(A) Transient expression of HyperTRIBE and TRIBE in S2 cells can induce increases in both the number of genes edited and the overall number of editing events. However, these increases are much more dramatic in HyperTRIBE expressing cells. Numerically, HyperTRIBE identifies 10689 common edited sites in two replicates, whereas the corresponding number for Hrp48 TRIBE is 291. Both TRIBE constructs have a reproducibility of around 60%. No increase in editing sites or genes is observed when Hyper-ADARcd is expressed alone (16 sites identified). Only editing events reproducible in both biological replicates are included. The number of genes and sites identified are normalized to the sequencing depth of each sample and are measured by relative fold change comparative to TRIBE (see also Methods).
(B) A much larger fraction of target genes are edited at multiple sites by HyperTRIBE than by TRIBE. The median number of editing sites per gene is 3 for HyperTRIBE and 1 for TRIBE. The histogram indicates the percentage of target genes containing 1 to more than 10 editing sites. These genes with multiple sites may be transcripts bound more stably by Hrp48.
(C) Three examples of commonly identified genes by HyperTRIBE, TRIBE and CLIP shown in IGV genome browser. Hrp48-ADARcd CLIP data used is from previous publication (McMahon et al., 2016). Typically, the multiple editing sites in HyperTRIBE cluster nearby the original sites identified by TRIBE. The height of bars indicate editing frequency in TRIBE data and CLIP signal strength in CLIP data.
(D) Expression of HyperTRIBE in all neurons (with elav-gsg-Gal4 driver) in fly brains can also increase the number of editing sites and genes compared to TRIBE. Editing events are identified and normalized as indicated in (A).
The ratio of HyperTRIBE-edited genes compared to TRIBE-edited genes is only 8 (Fig. 1A), 3-fold lower than the editing site ratio, indicating a substantial increase in the number of edited sites per gene in HyperTRIBE. Indeed, HyperTRIBE generates many more multiple-edited genes with a median of 3 edited sites per gene comparing to a median of 1 for TRIBE (Fig. 1B). Some HyperTRIBE editing sites are near the original editing sites identified by TRIBE (Fig. 1C), suggesting that the higher editing rate of HyperTRIBE is due in part to its ability to edit multiple adenosines near the original Hrp48 binding site. Moreover, the data show that unique editing sites that are on the same molecule of common sites – within a single RNA-seq read – have a higher editing percentage than all unique editing sites (Fig. 2). This indicates that multiple events on the same RNA molecule are positively correlated, i.e., once a target mRNA is bound well by HyperTRIBE, it is more likely to be edited on additional adenosines.

(A) Venn diagram of the editing sites shows that almost all of the Hrp48 TRIBE (blue) are also detected by HyperTRIBE (pink).
(B) Both TRIBE and HyperTRIBE identified genes overlap with CLIP identified genes well. Venn diagram shows the overlap of all genes identified by TRIBE (224 in total, blue), HyperTRIBE (3085 in total, pink) and Hrp48-ADARcd CLIP (4327 in total, green) (McMahon et al., 2016). The level of overlap between TRIBE and CLIP is not significantly different from overlap between HyperTRIBE and CLIP (Z-test performed, p=0.09).
(C) Many of the editing sites identified by either TRIBE method are close to CLIP peaks, but HyperTRIBE sites show a broader distribution relative to the CLIP peaks. Editing sites are categorized into three classes according to their distance from Hrp48-ADARcd CLIP peaks (McMahon et al., 2016), within 100 bp of CLIP peaks, between 100-500 bp away from CLIP peaks and beyond 500 bp from CLIP peaks. A smaller fraction of editing sites in HyperTRIBE are within 100 bp of CLIP peaks (40% versus 59%). (Z-test performed, *p=0.01, **p <0.0001, HyperTRIBE compared against TRIBE)
(D) Both TRIBE methods show 3’UTR editing preference, although the preference is slightly lower in HyperTRIBE. Editing preference is calculated as the distribution of editing sites in each mRNA region normalized to the distribution of sequencing reads in each mRNA region. (n.s.=p>0.05, **p<0.001, HyperTRIBE compared against TRIBE)
To validate this enhanced editing result in vivo, we performed cell-sorting experiments on flies expressing HyperTRIBE in all adult fly brain neurons using the elav-gsg-Gal4 driver (Abruzzi et al., 2015; McMahon et al., 2016). Similar to the tissue culture result, HyperTRIBE exhibits a 10-fold increase in the number of edited sites and a 5-fold increase in the number of edited genes compared to TRIBE (Fig. 1D).
We next compared the HyperTRIBE editing data with our previous CLIP data as well as with regular TRIBE editing data in Drosophila S2 cells. HyperTRIBE not only identifies 282 (97%) of the edited sites and 220 (98%) of the edited genes identified by TRIBE (Fig. 2A, 2B) but also correlates well with CLIP results; 73% of the sites are in common. (The number is 78% for TRIBE; Fig. 2B). However, there is a striking difference of 66% vs 4% in the ability of HyperTRIBE and TRIBE to recognize the CLIP-identified genes, respectively (Fig. 2B, Fig. S3). This distinction suggests that HyperTRIBE significantly lowers the false negative rate of TRIBE and thereby provides a much more complete binding signature of the RBP. We obtained similar results with FMRP HyperTRIBE (Fig. S4), another RBP assayed in the original TRIBE paper, indicating that the higher efficiency of HyperTRIBE is not limited to Hrp48.
We next analyzed the distance of the edited sites from the CLIP peaks. They presumably locate the positions of RBP binding. The HyperTRIBE edited sites are located further away from the CLIP peaks comparing to the TRIBE sites (Fig. 2C). For example, HyperTRIBE has only 40% of its sites located within 100 bp of the CLIP peaks versus 60% for TRIBE, whereas the fraction of edited sites further than 500 bp from the CLIP peaks is 29% and 15%, respectively (Fig. 2C). Presumably, the more distant sites are so poorly edited by TRIBE that they fall below the required 10% threshold (see below).
Distribution of the edited sites within each mRNA region, e.g., coding sequence, 5’UTR and 3’UTR, is a good indicator not only for the distance of the editing events from the RBP binding sites but also for RBP binding specificity. Hrp48 has been shown to preferentially bind to mRNA 3’UTR regions with both CLIP and TRIBE assays (McMahon et al., 2016). When normalized for read coverage in the different mRNA regions, HyperTRIBE still preferentially edits 3’UTR sites but with a 3.5-fold preference, somewhat less than the 5-fold preference of Hrp48 TRIBE (Fig. 2D). And coding sequence (CDS) editing preference has increased in HyperTRIBE compared to TRIBE, from 0.4 to 0.7. This is probably due to the ability of HyperTRIBE to edit adenosines distant from the RBP binding sites with higher efficiency.
The ADARcd with the hyperactive mutation (E488Q) has been shown to have less neighboring sequence preference surrounding the edited adenosine (Kuttan and Bass, 2012). Not surprisingly, this preference for 5’ uridines and 3’ guanosines is significantly reduced for the sites identified by HyperTRIBE compared to TRIBE (Fig. 3A). We also tested whether the preference for double strand structure surrounding the editing sites is diminished in HyperTRIBE, but there was no obvious difference between the two TRIBE methods (Fig. 3B). This result is surprising and of mechanistic interest and so is discussed below.
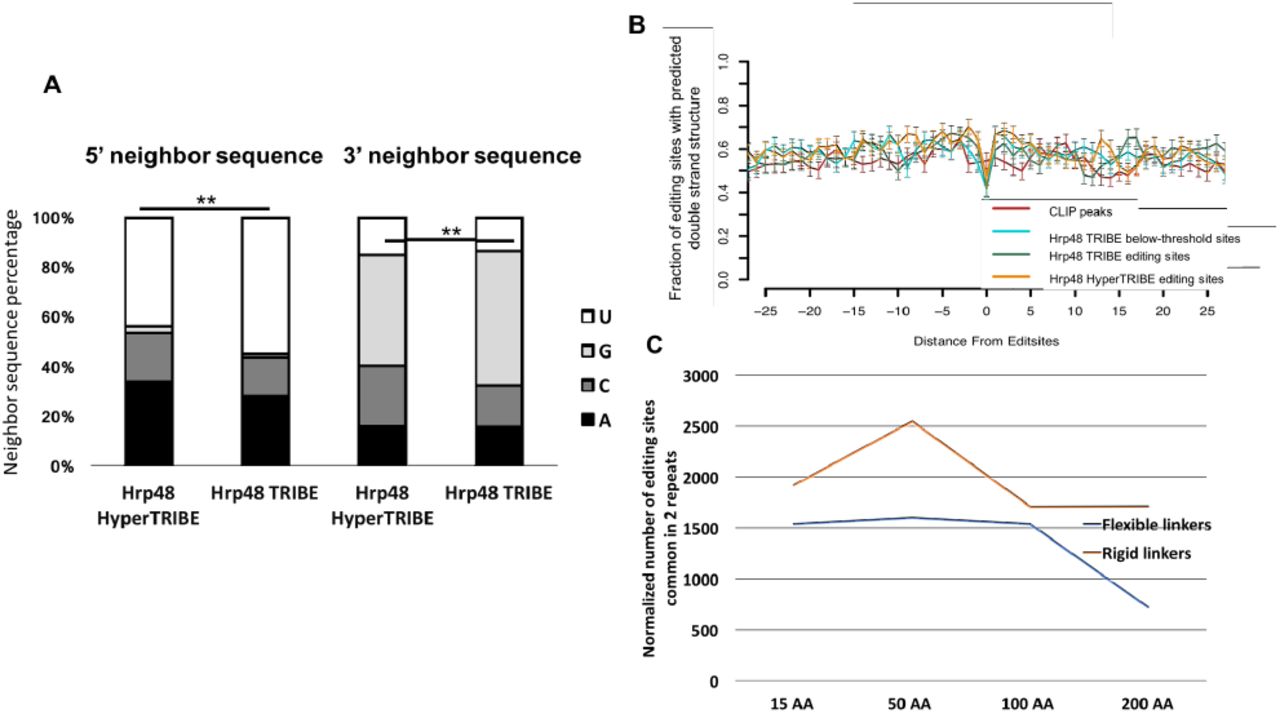
(A) 5’ and 3’ immediate neighbor sequence preference is reduced in HyperTRIBE. ADAR shows neighbor preference of uridine at 5’ and guanosine at 3’. This preference is lower in HyperTRIBE than in TRIBE (Z-test performed, **p<0.0001, percentage of uridine in 5’ neighbor and percentage of guanosine in 3’ neighbor tested, HyperTRIBE against TRIBE).
(B) Predicted of double-strandedness around the TRIBE, HyperTRIBE editing sites and CLIP peaks. Double strandedness was examined with a custom pipeline that uses UNAFold to fold RNA sequence in silico, in regions surrounding Hrp48 CLIP peaks (red), TRIBE below-threshold sites (blue, see also Fig. 4), TRIBE editing sites (green) and HyperTRIBE editing sites (yellow) and plotted as fraction of sites with predicted (A) double strandedness (see also Methods). 100 sites were blindly selected from each pool as the input for this analysis. X-axis represents the relative distance from the editing sites, and Y-axis indicates the average predicated double strandedness for each site. Error bars shows the standard error.
(C) Editing efficiency of HyperTRIBE slightly decreases as the linker length increases. Flexible linkers are repeats of (GGGGS)n of 15 AA, 50 AA, 100 AA and 200 AA length (blue), and rigid linkers are repeats of (EAAAK)n of 15 AA, 50 AA, 81 AA and 162 AA length (yellow). Minor modifications are made to the peptide sequence with amino acids of similar property for the convenience of cloning. Editing sites are identified and normalized as indicated in Fig. 1A.
Because only a minimal seven amino acid linker was used between Hrp48 and the ADARcd in the original TRIBE paper (McMahon et al., 2016) and this one, we investigated whether expanding the linker and altering its character would impact HyperTRIBE editing. However, editing efficiency is only about two-fold decreased even with a 200aa flexible linker (Fig. 3C). This suggests that RNA flexibility is the dominant feature for promoting interactions between the edited subregions and the ADARcd. It also suggests that other means of delivering the ADARcd to RNA should be possible, for example via protein dimerization schemes (Stankunas et al., 2003).
To further address possible reasons for the additional HyperTRIBE editing, we compared the editing frequencies of the sites identified by HyperTRIBE and TRIBE. We divided the HyperTRIBE editing sites into two categories, unique sites and common sites; the latter sites were also identified by TRIBE. Although the unique sites are edited at similar frequencies to the common sites in TRIBE, the common sites have a much higher HyperTRIBE editing percentage (Fig. 4A). This is not only true on average but also when the sites are examined individually (Fig. 4B). This indicates that common sites are also favored by HyperTRIBE.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Bar graph shows the editing percentage of the editing sites uniquely identified in HyperTRIBE and editing percentage of the common editing sites in HyperTRIBE and in TRIBE respectively. Editing percentage was shown as the weighted average. Editing percentage of the sites commonly identified by TRIBE and HyperTRIBE is increased in HyperTRIBE (Student t-test performed, **p<0.0001).
(B) An increase in editing percentage in HyperTRIBE is observed for most sites. Scatter plot shows editing percentage of HyperTRIBE editing sites (Y-axis) and TRIBE editing sites (A) (X-axis). Editing sites that are commonly identified by TRIBE and HyperTRIBE are shown. Line represents y=x is shown for reference (pink).
(C) About 30% of the 4017 below-threshold sites in TRIBE show elevated editing percentage in HyperTRIBE allowing them to be identified in the analysis pipeline (Z-test performed to test enrichment, P< E-100). HyperTRIBE editing sites that are present in TRIBE but below the 10% threshold set in the analysis pipeline are shown. Some of the editing sites in TRIBE have more than 10% editing in one replicates but less than 10% in the other. These sites are below-threshold by definition, but their average editing percentage could be over 10%. A red line indicating 10% threshold is shown for reference.
(D) 67% and 71% of TRIBE below-threshold target genes overlap with HyperTRIBE and CLIP respectively. Venn diagram shows the overlap of genes edited below-threshold by TRIBE (3473 in total, blue), genes identified by HyperTRIBE (3085 in total, pink) and Hrp48-ADARcd CLIP (4327 in total, green) (McMahon et al., 2016). The difference of overlap between below-threshold genes by TRIBE and CLIP (71%) versus overlap between HyperTRIBE and CLIP (73%) is weakly significant (Z-test performed, p=0.015).
Is it possible that the unique sites are also quantitatively different between HyperTRIBE and TRIBE? This would mean that they may also be edited by TRIBE but below the 10% threshold required to meet the editing criterion. Indeed, there are 4017 adenosines edited at a below-threshold level in TRIBE, i.e., at least one editing event for each specific adenosine in both replicates but with less than 10% editing percentage. The correspondence between replicates for these adenosines and HyperTRIBE editing sites is highly statistically significant (P< E-100). More than 30% of these adenosines correspond to HyperTRIBE editing sites (Fig. 4C). We conclude that below-threshold TRIBE-edited adenosines in TRIBE make a significant contribution to the extra editing sites in HyperTRIBE (see Discussion) and that much of the distinction between HyperTRIBE and TRIBE is likely quantitative and not qualitative.
Not surprisingly perhaps, the below-threshold sites are located further away from the CLIP peaks than the above-threshold sites, which probably contributes to their lower editing percentage (Fig. S5). Moreover, the 4017 below-threshold editing sites identify 3473 different genes, which now overlap well with genes identified by Hrp48 HyperTRIBE and Hrp48-ADARcd CLIP (Fig. 4D). These data taken together indicate that HyperTRIBE substantially reduces the false negative problem of TRIBE and is a superior method for identifying RBP targets in vivo.
Discussion
Our recently developed method called TRIBE (McMahon et al., 2016) expresses in vivo a chimeric protein, which is a fusion of an RBP of interest to the catalytic domain of ADAR (ADARcd). Like the normal ADAR enzyme, the TRIBE protein performs Adenosine-to-Inosine editing on RNAs recognized by the TRIBE fusion protein. However, editing by TRIBE is probably inefficient, giving rise to a high false negative rate for identification of RBP target RNAs. We present here the in vivo characterization of HyperTRIBE, which carries the mutation E488Q within the ADARcd. This mutant ADAR protein was identified in a yeast screen by Bass and colleagues (Kuttan and Bass, 2012). It has a reduced nearest neighbor sequence preference and is a more efficient enzyme. Indeed, HyperTRIBE with the Drosophila RBP Hrp48 identifies dramatically more editing sites and genes than TRIBE. The data taken together indicate that HyperTRIBE much more faithfully recapitulates the known binding specificity of its RBP than TRIBE.
One positive indication is that HyperTRIBE results overlap much more successfully than TRIBE results with our previously published Hrp48 CLIP experiments. These data were very similar whether from endogenous Hrp48 or from overexpressed TRIBE fusion protein (McMahon et al., 2016), indicating that the TRIBE protein interacts with RNA similarly to endogenous Hrp48. The much better overlap of HyperTRIBE and CLIP data suggests that HyperTRIBE has a much reduced false-negative problem. A different HyperTRIBE fusion protein, containing the RBP FRMP, also has many more editing sites and genes compared to regular TRIBE with this RBP [(McMahon et al., 2016), Fig. S4]. As this is also the case for a HyperTRIBE fusion protein containing the translation regulator eIF4E-BP (Hua Jin, personal communication), a more faithful identification of RBP targets by HyperTRIBE may be generally the case. Importantly, perfect overlap with HyperTRIBE data is not necessarily expected, i.e., there is no evidence that iCLIP data do not contain false positives. Indeed, very weak or ephemeral interactions with transcripts that cross-link very efficiently to the RBP might constitute CLIP false positives.
Despite these positive indications, might there be a substantial source of HyperTRIBE false positives? Activity by the ADARcd alone is a likely candidate. However, we never obtained any substantial numbers of editing events (McMahon et al., 2016), even with the HyperADARcd (Fig. 1A); this is despite comparable expression levels to the HyperTRIBE fusion protein (data not shown), i.e., the vast majority of editing events require an RBP when the ADARcd its neighboring dsRNA binding domains. As tethering dramatically increases encounters between the ADARcd and RNA sequences near the RBP binding sites, our interpretation is that this also increases the editing rate for some adenosines above the 10% threshold required to score positively. As previously discussed (McMahon et al., 2016), we suspect that the RBP moiety allows the enzyme to sample at high local concentration transient double-stranded intramolecular structures; they are presumably in dynamic equilibrium with single-stranded RNA.
Nonetheless, the fraction of edited adenosines by TRIBE was still low, indicating that a low fraction of enzyme-RNA encounters result in successful editing and suggesting that false negatives rather than false positives were the major issue with TRIBE. This presumably reflects the sequence and structural requirements of the ADARcd. The argument above about transient structures notwithstanding, they are the double-stranded character surrounding a bulged adenosine and nearest neighbor sequence preferences. These two sequence/structural features either enhance flipping of the substrate adenosine out of the helix and/or successful A to I conversion by the ADARcd, both of which should enhance editing efficiency (Kuttan and Bass, 2012; Matthews et al., 2016). As the HyperTRIBE mutation is unlikely to influence some global feature of RNA structure, we assumed that it reduces the RNA sequence/structural requirements for successful editing. Indeed, a reduced sequence requirement is indicated by Fig. 3A.
However, the dramatically increased editing frequencies of the common sites indicates that the same structure and sequence experiences substantially enhanced editing frequencies with HyperTRIBE compared to TRIBE (Fig. 4A). This argues against a reduced structural requirement and suggests either enhanced base flipping, perhaps by enhanced amino acid insertion by the HyperTRIBE ADARcd into the RNA A helix, or a higher rate of successful editing/flipping event. Even more surprising was the realization that many of the new HyperTRIBE editing sites correspond to very weak but proper TRIBE editing sites. Their TRIBE editing frequencies are well below an arbitrary threshold, in our case set at 10% editing, but the correspondence of these sites between replicate TRIBE experiments indicates that they are reproducible and therefore genuine editing sites. Two implications of these data are that 1) even very low editing frequencies are real editing sites if they are identified in replicate experiments and 2) there are many more bona fide editing events with regular TRIBE and even normal in vivo editing than are recognized with normal thresholds. All of these considerations taken together indicate that HyperTRIBE is a superior approach and will facilitate the identification of RBP targets in mammalian as well as fly neurons.
Method
Molecular Biology
RBP-ADARcd with E488Q mutation was created by performing Quikchange® Site-directed Mutagenesis on pMT-RBP-ADARcd-V5 plasmid (McMahon et al., 2016). Primers 5’-TCGAGTCCGGTCAGGGGACGATTCC and 5’-GGAATCGTCCCCTGACCGGACTCGA were used to induce point mutation to the underlined nucleotide. 15 AA, 50 AA and 100 AA flexible linkers and 15 AA, 50 AA rigid linkers (Amet et al., 2009) were chemically synthesized by Integrated DNA Technologies, Inc. and cloned into pMT-Hrp48-ADARcd-E488Q-V5 plasmid using Gibson Assembly® from NEB. The other linkers were created by PCR duplicating the fragment and cloning. Transient expression of TRIBE constructs was performed by co-transfecting pMT TRIBE plasmids with pActin-EGFP to Drosophila S2 cells using Cellfectin® II from Thermo Fisher Scientific. Cells were allowed 48 hours after transfection for adequate expression of GFP before sorting with BD FACSAria™ II machine for GFP positive cells. Total RNA was extracted from the sorted cells with TRIzol™ LS reagent. TRIBE protein expression was induced with copper sulfate 24 hours before FACS sorting. Expression of all fusion proteins was assayed by transient expression in S2 cells and western blot against V5 tag (Invitrogen, 46-1157). TRIBE stable cell lines used in this paper are the same as the original TRIBE paper (McMahon et al., 2016). GFP labeled neurons are manually selected by using a glass micro-pipette from dissected, digested and triturated fly brains as described in (McMahon et al., 2016) and (Abruzzi et al., 2015; McMahon et al., 2016)
Standard Illumina TruSeq® RNA library Kit was used to construct RNA-seq library from S2 cells. Manually-sorted cells are subjected to RNA-seq library protocol as decribed in (Abruzzi et al., 2015; McMahon et al., 2016). All libraries were sequencing by Illumina NextSeq® 500 sequencing system using NextSeq® High Output Kit v2 (75 cycles). Each sample were covered by ~20 million raw reads.
RNA-editing Analysis
The criteria for RNA editing events were: 1) The nucleotide is covered by a minimum of 20 reads in the sequencing run; 2) More than 80% of genomic DNA are A at this site (use the reverse complement if annotated in the reverse strand); 3) A minimum of 10% G is observed at this site in mRNA (or C for the reverse strand). Genomic DNA of S2 cell and background fly strain are sequenced to identify and exclude possible polymorphism on DNA level. RNA sequencing data were analyzed as described in (McMahon et al., 2016; Rodriguez et al., 2012), with minor modifications. The scripts used in the analysis are available on GitHub (https://github.com/rosbashlab/TRIBE). Background editing sites found in samples expressing Hyper-ADARcd alone were subtracted from the TRIBE identified editing sites both in S2 cells and in fly neurons. Overlap of editing sites from two datasets were identified using bedtools with parameters “-f 0.9 -r”.
Quantification of RNA sequencing reads distribution was performed with read_distribution algorithm in RSeQC v2.3.7 (Wang et al., 2012).
RNA structure folding was carried out with UNAFold (Markham and Zuker, 2008) on flanking sequences of TRIBE editing sites, Hyper TRIBE editing sites, TRIBE below-threshold editing sites and CLIP binding sites without nearby editing sites. One hundred sites were selected at random from each set, where a flanking region of 250nt both 5’ and 3’ of the editing site or CLIP peak was folded with UNAFold parameters (hybrid-ss-min –suffix DAT –mfold=5,8,200 –noisolate). Base pairing was counted in the predicted minimum free energy (MFE) and predicted suboptimal structures. A profile of double strandedness is created for each sequence, which is then averaged over all the 100 sequences and plotted.
Fly lines
HyperTRIBE injection plasmid were generated by site-directed mutagenesisof pJFRC7-20xUAS-Hrp48-ADARcd-V5 plasmid to introduce the E488Q mutation. The Hyper-ADARcd only construct is generated in the same way. The transgenes were injected by Rainbow Transgenic Flies, Inc. (Camarillo, CA). UAS-RBP-ADARcd-V5; UAS-eGFP flies (Bloomington stock center #1522) were crossed to elav-gsg-Gal4 driver line to allow adult-only expression of the fusion proteins in all neurons due to the lethality of constitutive pan-neuronal expression (McMahon et al., 2016; Osterwalder et al., 2001). Prior to manual cell sorting, food containing RU486 (0.2 μg/ml, Sigma) was used to induce transgene expression in young flies (~3 days old) for 3 days.
Reference




