

Abstract
The multi-scale organization of eukaryotic genomes defines and regulates cellular identity and tissue-specific functions1–3. At the kilo-megabase scales, genomes are partitioned into self-interacting modules or topologically associated domains (TADs) 4–6. TADs formation seems to require specific looping interactions between TAD borders 7,8, while association of TADs can lead to the formation of active/repressed compartments 9. These structural levels are often seen as highly stable over time, however, recent studies have reported different degrees of heterogeneity 10,11. Access to single-cell absolute probability contact measurements between loci and efficient detection of low-frequency, long-range interactions is thus essential to unveil the stochastic behaviour of chromatin at different scales. Here, we combined super-resolution microscopy with state-of-the-art DNA labeling methods to reveal the variability in the multiscale organization of chromosomes in different cell-types and developmental stages in Drosophila. Remarkably, we found that stochasticity is present at all levels of chromosome architecture, but is locally modulated by sequence and epigenetic state. Contacts between consecutive TAD borders were infrequent, independently of TAD size, epigenetic state, or cell type. Moreover, long-range contact probabilities between non-consecutive borders, the overall folding of chromosomes, and the clustering of epigenetic domains into active/repressed compartments displayed different degrees of stochasticity that globally depended on cell-type. Overall, our results show that stochasticity can be specifically modulated to give rise to different levels of genome organization. We anticipate that our results will guide new statistical models of genome architecture and will be a starting point for more sophisticated studies to understand how a highly variable, multi-scale organization can ensure the maintenance of stable transcriptional programs through cell division and during development.
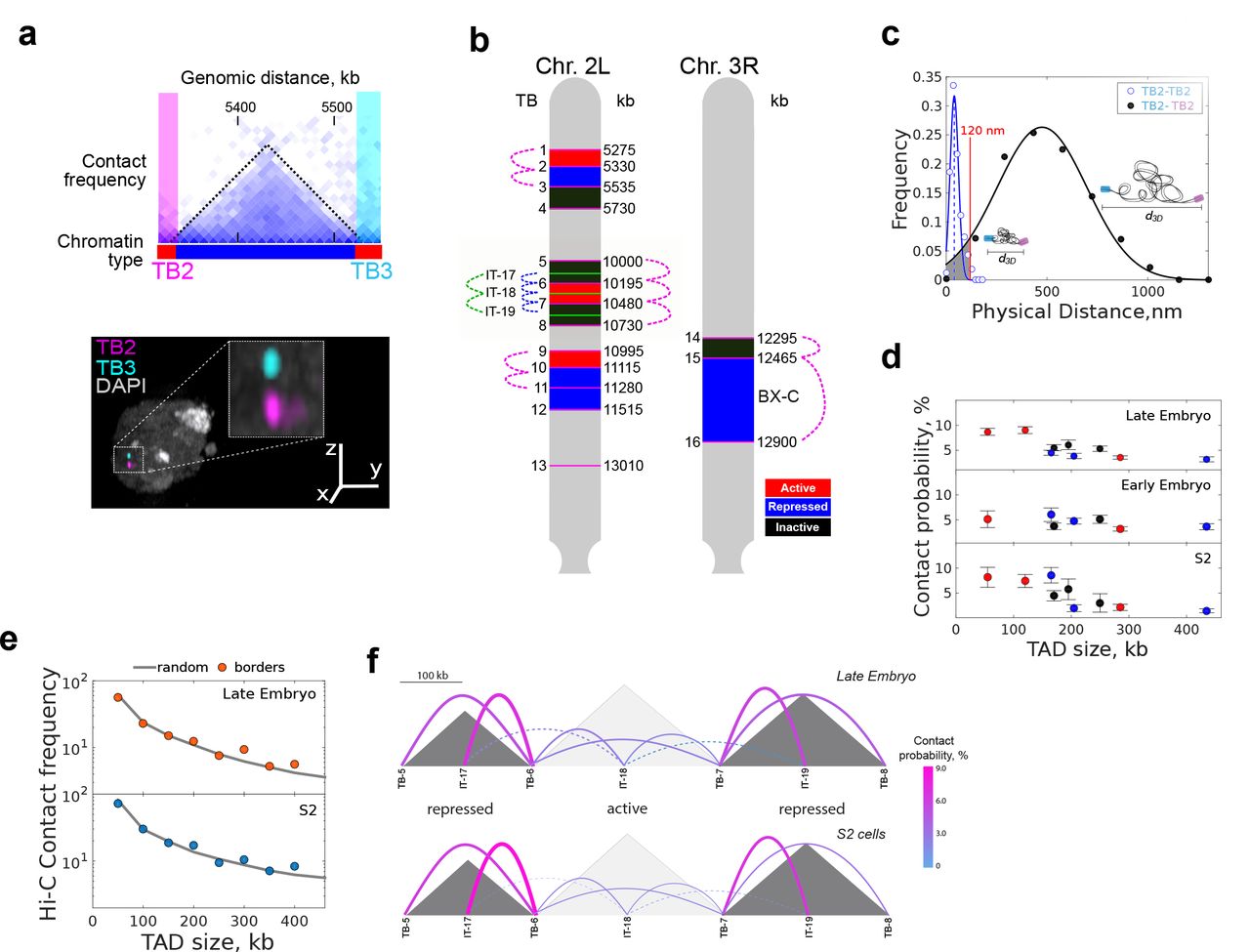
A major mechanism for TAD formation in mammals involves the stable looping of TAD borders 8 Stable looping between TAD borders was also recently proposed to be relevant for the maintenance of transcriptional programs during Drosophila development 7. However, long-lived stable interactions are unlikely to allow for rapid responses in gene regulation. To study this apparent contradiction, we developed a method to dissect the changes in TADs organization at the single-cell level in three transcriptionally distinct Drosophila cell types: early (stage 5) and late (stage 16) embryos; and an immortalized cell line (S2). Pairs of TAD borders were labeled with Oligopaints libraries 12 and imaged using multi-color three-dimensional structured illumination microscopy (3D-SIM) (Fig. 1a). TAD chromatin types were defined as active, repressed or inactive following the distribution of epigenetic marks (Fig. S1a). Borders flanking TADs with different chromatin states were imaged in chromosomes 2L and 3R (Figs. 1b and S1b), and appeared in microscopy as well defined foci (Fig. 1a). A large proportion of cells (60-70%) displayed a single foci (Fig. S1c), whose size increased proportionally with the genomic length of the library (Fig. S1d), consistent with a high degree of homologous pairing independently of the ploidy of each cell type (Fig. S1d) 13,14. Distances between TAD borders were Gaussian distributed for all cell types (Figs. 1c and S1f-h). Remarkably, the width of these distributions was comparable to the mean distance between TAD borders, revealing a high degree of structural variability, independently of TAD size or epigenetic state (Figs. 1c and S1i). Further, the linear relation between dispersion and physical distance (Fig. S1i) suggests that this variability is regulated by the polymer properties of the chromatin fiber.

(a) Top, region of Hi-C contact matrix of chromosome 2L. Black dotted line demarcates a single TAD and pink and cyan boxes represent the TAD borders (TB) labelled by Oligopaint. Chromatin epigenetic state is indicated at the bottom using the colorcode of panel b. Bottom, representative three-color 3D-SIM image. DAPI is shown in white, TB2 in pink and TB3 in cyan. Scalebar = 1 μm.
(b) Oligopaint libraries used in this study (TB1-16 were at TAD borders and IT17-19 within TADs). All libraries were in chromosomes 2L or 3R, as indicated in the sketch. Colored boxes display the chromatin type of TADs flanked by each library. Red: active, blue: repressed, black: inactive. Dotted colored lines indicate the combinations of distances between libraries used for the other panels of this figure.
(c) 3D distance distributions between TB2-TB2 and TB2-TB3. Mean colocalization resolution, estimated from two-color labelling of a single border, was 40 nm (vertical blue dashed line). Blue and black solid lines represent Gaussian fittings. Absolute contact probability between libraries was obtained from the integral of the area of the Gaussian fitting (shaded gray) below 120 nm (see Fig. S1e). N = 161 and 556 for TB2-TB2 and TB2-TB3 respectively, from more than three biological replicates.
(d) Absolute contact probability between consecutive borders as a function of genomic distance. Chromatin state of TADs being flanked is color-coded as defined in panel 1b. Error bars represent SEM.
(e) Hi-C normalized contact frequency between consecutive TAD borders (circles) and random loci (solid gray line) as a function of genomic distance for S2 and late embryonic cells. Matrix resolution = 10kb. Two biological replicates for each cell-type were performed.
(e) Schematic representation of contact probability between and within TADs (solid colored lines) for late embryo and S2 cells at the chromosomal region shaded in panel b. Sizes of TADs (grey shaded triangles) is proportional to genomic length (scale bar on top). Chromatin type is indicated at the bottom of each TAD. Thickness of the lines and color indicate absolute contact probability (see color code in scalebar on the right). Dotted lines indicate inter-TADs contacts. Early embryo measurements are depicted in Fig. S1j. Numbers of cells for each pair of libraries is provided in Fig. S1f-h.
Next, we quantified the absolute contact probability between consecutive borders by integrating the probability distance distributions below 120 nm (99% confidence interval obtained from single library two-color control experiments, Figs. 1c and S1e). Notably, the contact probability between consecutive TAD borders was below 10%, independently of cell type or of the epigenetic state of the TAD being flanked (Fig. 1d). Consistently, Hi-C contact frequencies between consecutive TAD borders vs. random genomic loci were indistinguishable (Fig. 1e). These results, combined with the lack of enrichment of CTCF and cohesin at TAD borders in Drosophila15, suggest that TAD assembly does not involve stable loops in flies, but rather can be explained by an ‘insulation-attraction’ mechanism 16 This model may provide an alternative explanation for the formation and maintenance of more than 50% of metazoan TADs whose boundaries are not formed by looping interactions as defined by Hi-C experiments 8
In agreement with this model, absolute contact probabilities within TADs and between their borders were similar (Fig. 1f and S1j), with inactive/repressed TADs displaying higher contact probabilities than active TADs (7 ± 1% vs. 2.7 ± 1%). Contact probabilities within TADs were in all cases considerably higher that with neighboring TADs (Fig. 1f), indicating that stochasticity is locally modulated at the TAD level. Of note, contacts across TAD borders were not uncommon (∼3%, Fig. 1f), implying frequent violations of boundary insulation at TAD borders. These results indicate that confinement of chromatin into TADs may require only small differences in absolute contact probabilities (∼2 fold). Thus, condensation of chromatin into TADs may arise from a multitude of low-frequency, yet specific, intra-TAD contacts.
Recent Hi-C studies suggested that stable clustering between neighboring active TAD borders regulates transcriptional programmes that persist during development 7 We directly tested this hypothesis by measuring the contact probabilities between non-consecutive TAD borders (Fig. 2a). Hi-C contact frequencies among TAD borders increased exponentially with absolute contact probabilities (Fig. 2a and S2a). Our results highlight the ability of Hi-C to enhance the detection of high probability contacts and also suggest the need to relate Hi-C data to physical distances with a nonlinear relationship. This would allow a better discrimination of low-frequency contacts (1-3%, Fig. 2a) such as those observed within and between TADs (Fig. 1f) and a more realistic conversion of Hi-C maps into 3D folded structures.
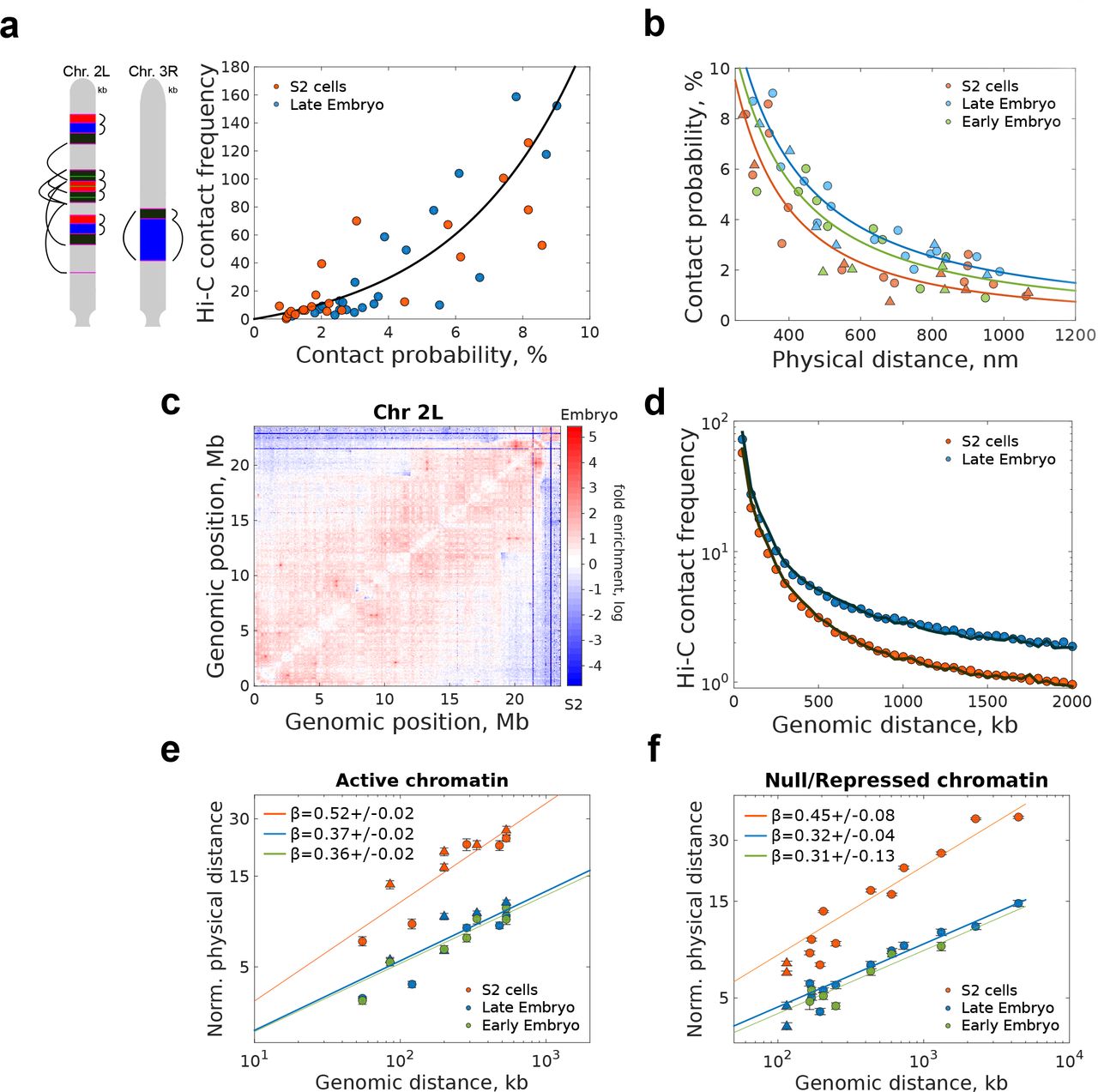
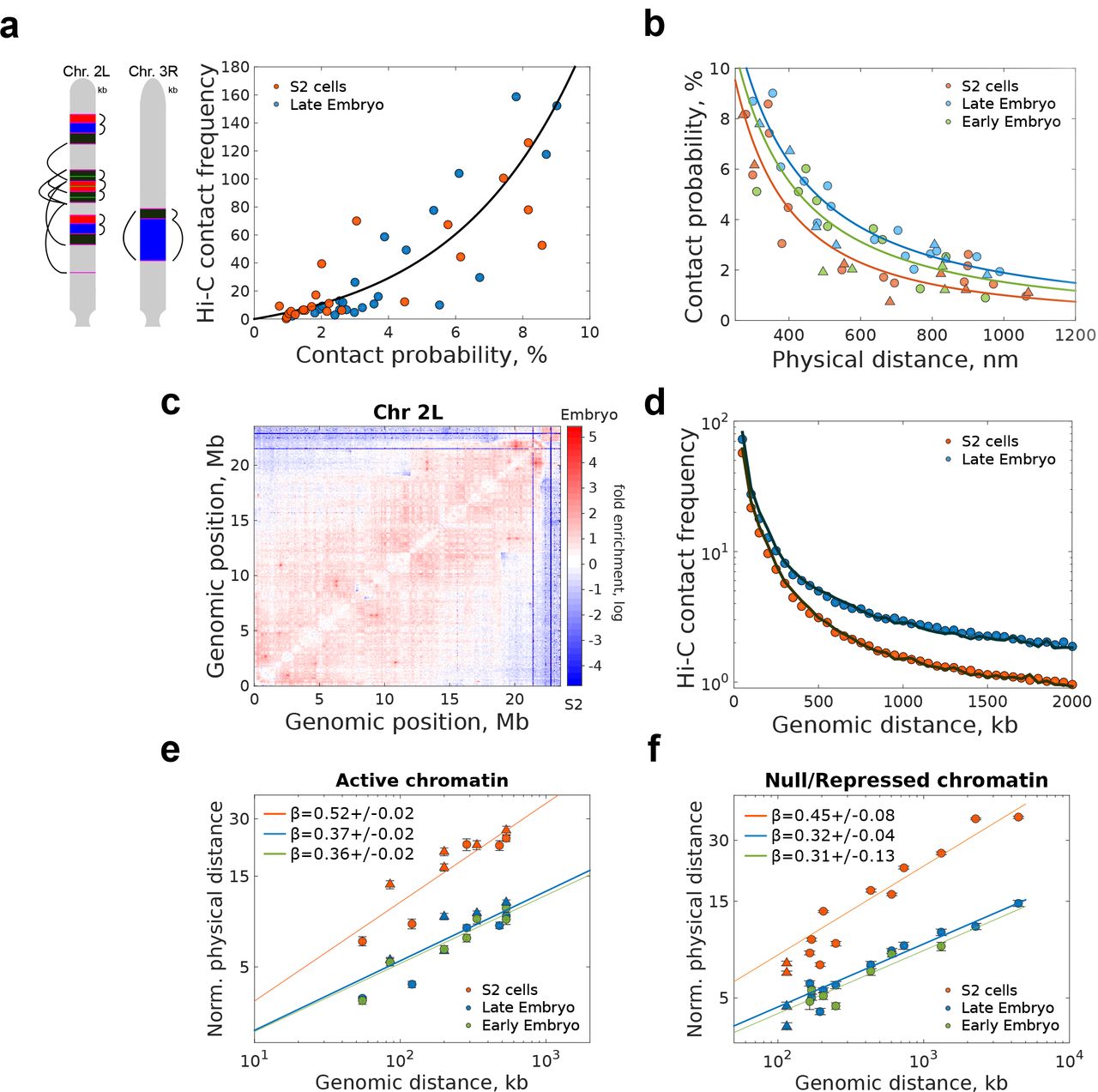
(a) Left, schematic representation of pairwise distance measurements between consecutive and non-consecutive borders, with color code and positions as in Fig. 1b. Right, Hi-C contact frequency vs. microscopy absolute contact probability for consecutive and non-consecutive domain borders for embryo and S2 cells. Solid black lines represent an exponential fitting. Independent fits for each cell-type are provided in Fig. S2a. Matrix resolution = 10kb. N for microscopy pairwise measuremnts is provided in Fig. S1 f-h. N = 2 for Hi-C data, from at least three and two biological replicates, respectively.
(b) Absolute contact probability vs. physical distance for consecutive and non-consecutive TAD borders (filled circles). Solid lines represent power-law fittings with scaling exponent values described in Fig. S2b. Triangles represent measurements within TADs.
(c) Matrix of relative frequency of Hi-C contacts for late embryo vs. S2 cells for chromosome 2L. Contact frequency ratio is color coded according to scale bar. Matrix resolution = 50kb. N=4, biological replicates.
(d) H i-C contact frequencies between TADs borders vs. genomic distance for embryo and S2 cells. Solid lines represent the average contact frequency for randomly chosen positions in the genome. Matrix resolution = 10kb. N=2, biological replicates.
(e-f) Log-log plot of the end-to-end physical distance vs. genomic length of chromatin domains, normalized by the power-law fit pre-exponential factor for active (e) or inactive/repressed chromatin (f) for different cell types. Solid lines indicate the power-law fits, with the scaling exponent β Circles and triangles are depicted as described in panel 2b. Error bars represent SEM. Non-normalized data and fits can be found in Figs. S2d-e. N > 140 for each end-to-end measurement, from more than three biological replicates (see Fig. S1 for actual number of cells per library combination).
Contact probabilities between non-consecutive TAD borders were in all cases low (<9 %, Fig. 2b) and decreased monotonically with physical and genomic distance following a power law behaviour (Figs. 2b and S2b-c). Notably, the decay exponents were different between cell types (Figs. 2b and S2b), indicating that levels of stochasticity are globally modulated between cell-types, possibly reflecting cell-type specific transcriptional programs. To test whether this tendency held genome-wide, we calculated the ratio between normalized Hi-C contact maps of embryos and S2 cells. For all chromosomes, embryos displayed a higher relative contact frequency than S2 cells below a few Mb (Fig. 2c and Fig. S2d), in accordance with our microscopy results. Furthermore, the frequency of contacts between non-consecutive TAD borders genome-wide was similar to that of random genomic loci for both cell types (Fig. 2d). As the large majority of TAD borders in Drosophila contain active chromatin 17,18, our results are inconsistent with stable preferential looping of active borders 7,19, and rather indicate that these contacts are rare or short-lived. This interpretation is consistent with the transient assembly and disassembly of transcription clusters in human cells 20
Next, we sought to determine if this modulation in contact probabilities resulted from cell-type specific changes in the local folding properties of the chromatin fiber. To this end, we measured the end-to-end distance (d3D) for active or inactive/repressed chromatin domains of varying genomic (dkb) lengths. For all cell and chromatin types, we observed a power-law scaling behavior (d3Dα  ) (Fig. 2e-f and S2e-f) with scaling exponents being higher for active than for repressed domains, consistent with previous measurements in Kc167 cells 21. Remarkably, scaling exponents were considerably lower in embryos than in S2 cells, for both types of chromatin. TAD border localization is conserved between cell types 18,22, however, our results show that TAD conformation and structural heterogeneity strongly depend on cell type. This cell-type specificity in TAD organization results from the interplay between the degree of chromatin compaction and the frequency of stochastic long-range interactions.
) (Fig. 2e-f and S2e-f) with scaling exponents being higher for active than for repressed domains, consistent with previous measurements in Kc167 cells 21. Remarkably, scaling exponents were considerably lower in embryos than in S2 cells, for both types of chromatin. TAD border localization is conserved between cell types 18,22, however, our results show that TAD conformation and structural heterogeneity strongly depend on cell type. This cell-type specificity in TAD organization results from the interplay between the degree of chromatin compaction and the frequency of stochastic long-range interactions.
To quantitatively dissect stochasticity at larger genomic scales, we labeled 69 quasi-equidistant TAD borders encompassing 90% of chromosome 3R (Fig. 3a and S3a-b). Tens of foci were resolved in embryonic and S2 cells by 3D-SIM (Fig. 3a). The probability distance distribution p(r) between any two foci within each cell exhibited moderate single-cell variations (Fig. 3b), but were considerably different between cell types (Figs. 3b and S3c). The chromosome elongation and volume, obtained from the maximum pairwise distance (Dmax) and the radius of gyration (Rg, Fig. 3c), decreased to almost half between S2 and late embryonic cells, with early embryonic cells adopting intermediate values (Fig. 3b). The number of foci imaged in S2 cells was consistent with a very low frequency of physical interactions between domain borders (see discussion in Fig. S3). Interestingly, in early and late embryos the number of observed foci was considerably reduced (Fig. 3b, right panel), consistent with higher probabilities of long-range interactions for these cell-types. The lower number of foci detected was not associated with the smaller volume of embryonic cell nuclei causing the probes to be closer than the resolution limit of 3D-SIM microscopy (Fig. S3e). Finally, changes in Hi-C contact frequency of S2 vs. late embryo for the 69 TAD borders were notable in the sub-Mb scale (200-600 kb), and they extended to genomic distances as high as ∼10 Mb (Fig. 3d), suggesting that changes in chromosome compaction between cell types arises from an increased frequency of interactions affecting all genomic scales. All in all, these data indicate that chromosome folding is highly variable, with mild, cell-type specific increases in the probability of long-range contacts being sufficient to produce large changes in the manner in which chromosomes occupy the nuclear space (Fig. 3c).
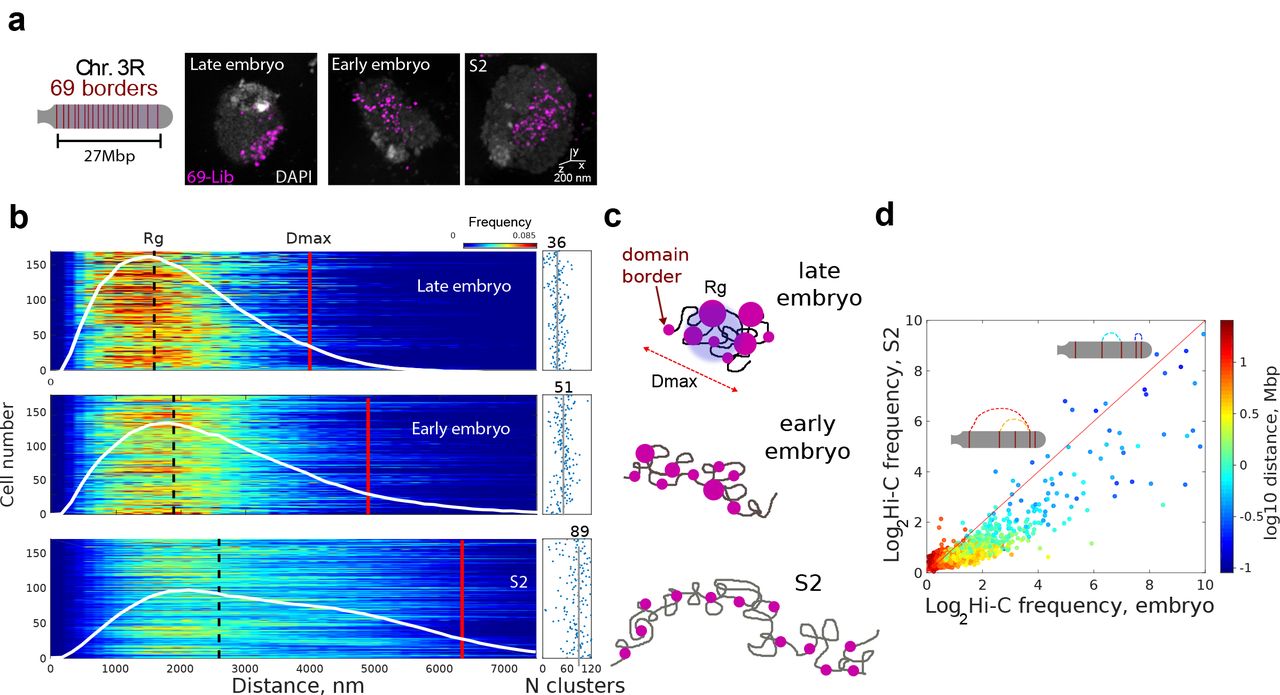
(a) Left, schematic representation of 69 domain borders labelled by a single Oligopaint library (Lib-69) in Chr. 3R. Each probe spanned ∼20 kb, probes were separated by 320 kb in average (see Fig. S3a-b). Right, representative two-color 3D-SIM images for all studied cell types. DAPI signal (white) and Lib-69 (pink) are shown.
(b) Left panel, single-cell probability distance distribution p(r) between all pairs of foci imaged by 3D-SIM. White line represents the population averaged p(r) frequency. Detailed Rg and Dmax values are shown in Fig. S3. Dmax is defined as the distance that comprises <97% of the area under the p(r) function. Right panel, number of foci per cell for each condition with mean population values shown as solid vertical lines and indicated above. N = 180, from more than three biological replicates.
(c) Schematic representation of the chromosome structure for each cell type. Solid grey line represent the DNA fiber and pink circles represent domain borders with sizes proportional to the number of regrouped borders.
(d) H i-C contact frequencies of S2 vs. late embryo cells for all the pairwise combinations of the 69 borders. Solid red line represents the relation expected if frequencies of interactions between the 69 borders were equal between cell-types. Insets depicts chromosome 3R and different combinations of genomic distances and frequencies of interaction between borders. Matrix resolution = 50kb. N=4, from at least three biological replicates.
Inter− and intra-chromosomal Hi-C maps have revealed that active and repressed TADs may associate to form two types of compartments (namely A and B). 9,23. To study this higher-order level of organization in single cells and at the single-molecule level, we immunolabeled active and repressive epigenetic marks (histones H3K4me3 and H3K27me3, respectively) and performed multicolor direct stochastic optical reconstruction microscopy (dSTORM) 24–26, a method that provides a higher spatial resolution than 3D-SIM. dSTORM imaging revealed that active and repressive histone marks distributed non-homogeneously across the cell nucleus, forming discrete nanometer-sized compartments for all cell types (Figs. 4a and S4a). Repressed and active chromatin marks were strictly segregated at the nanoscale for all cell types, as revealed by independent co-localization quantification methods and additional non-colocalizing epigenetic marks (Figs. 4b and S4b-c) 27. Interestingly, active marks were often observed at borders of/or demarcating large repressed compartments, mirroring their alternating one-dimensional genomic distributions (Fig. 4c).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) Two-color dSTORM image of active (H3K4me3, blue) and repressive (H3K27me3, red) chromatin marks in a representative S2 cell. Images of early and late embryo are displayed in Fig. S4a and panel c.
(b) Violin distributions of Manders colocalization coefficient between active and repressive chromatin marks for all cell types.
(c) Representative zoomed images of two-color dSTORM for the three cell-types investigated. Black arrows indicate the localization of small active chromatin domains in the periphery of large repressive domains. Lower panel display active and repressive marks Chip-Seq enrichment profiles for late embryo. Note that active marks locate at the borders of large repressive domains.
(d-e) dSTORM rendered images of Alexa-647-labelled (d) H3K27me3 and (e) H3K4me3. Images show density maps computed from the area of the polygons obtained from the Voronoï diagram with scale defined on top. Zoomed regions display detected compartments (highlighted with different colors). Additional, images for all cell types and chromatin marks are displayed in Fig. S5a-b.
(f-g) Population based distribution of epigenetic domains sizes as obtained from dSTORM and predicted from ChiP-seq data for H3K27me3 (f) and H3K4me3 (g). PDF is probability density function. Single cell distributions of physical sizes and Chip-Seq data are shown in Figs. S5c-d and S6b, respectively. N=60, from two to three biological replicates in microscopy imaging.
(h) Percentage of clustering for active and inactive chromatin marks for each cell type. Error bars = SD. One-sample t-test p-values: *p<0.01; **p<0.001.
To investigate if active and repressed compartments also varied among cell types and development, we resorted to one-color dSTORM using Alexa 647 as the fluorophore of choice (results were similar when using other fluorophores, Fig. S4d). Compartments were detected using a Voronoi diagram-based algorithm (Figs. 4d-e) 28 In all cases, active compartments were smaller than repressive compartments in agreement with two-color dSTORM observations (Figs. 4c-e and S5a-b). Interestingly, for both marks the number of compartments and their sizes showed variations between single cells of the same type (Figs. S5c-d). To further evaluate if changes in compartment sizes correlated with changes in local chromatin folding, we quantified the density of single molecule detections in active and repressed compartments. Notably, the local density of compartments was higher for both types of marks in embryonic cells than for S2 cells (Fig. S6a), consistent with our previous findings (Figs. 2e-f).
To study whether the nanoscale organization of repressive and active marks reflected the epigenomic domain organization from ensemble genome-wide methods, we predicted the physical sizes of epigenomic domains (Fig. S6b) and compared them with those obtained by direct observation. The predicted size distributions failed to recover the largest compartments observed by microscopy (Figs. 4f-g and S6b). We reasoned that large compartments are likely to arise from clustering of smaller epigenetic domains (‘clustered compartments’). To quantify this phenomenon, we calculated the percentage of compartments not accounted for by the distribution of epigenetic domains. This percentage of clustered compartments was below <10% for embryonic cells and almost absent in S2 cells (Fig. 4h). Repressive and active compartments showed different degrees of clustering (Figs. 4f-h), indicating that stochasticity can be specifically modulated by transcriptional/epigenetic states. This is likely due to the different mechanisms of clustering formation at play, such as Polycomb regrouping of repressed genes 29vs. transient interactions of active genes 30,31. It is important to note, however, that the large majority of compartments (∼90%) could be accounted for by the predicted distributions of epigenomic domains, consistent with the majority of the epigenetic domains described by genome-wide methods existing at the single-cell level. In addition, these results are consistent with the cell-type specific higher-order organization of chromatin arising from stochastic contacts between chromosomal regions harboring similar epigenetic marks, likely reflecting cell-type specific developmental and transcriptional programs.
In this work, we showed that genome in Drosophila is not driven by stable or long-lived interactions but rather relies on the formation of transient, low-frequency contacts whose frequencies are modulated at different levels. Stochasticity is modulated locally at the TAD level by specific intra-TAD interactions, and globally at the nuclear level by interactions of TADs of the same epigenetic type. Furthermore, stochasticity is also regulated between cell-types. These modulated stochasticities reveal a novel mechanism for the spatial organization of genomes. These evidences could be critical for a more accurate understanding of how different cell types interpret genomic and epigenomic states to produce different phenotypes. Most current spatial models of genome architecture rely on interpreting interaction maps from Chromosome Conformation Capture based experiments, which capture relative frequencies of interactions between loci at close spatial proximity. Translation of relative contact frequencies into spatial distances is challenging. Our direct single-cell measurements of absolute contact probabilities, full distance distributions, and dissection of low frequency events for different chromatin and cell types will complement existing methods to refine the next generation of statistical models of genome architecture. Our results call for more sophisticated studies to reveal how a highly stochastic genome organization can ensure the maintenance of stable transcriptional programs through cell division and during development.
Author contribution
D.I.C, A.M.C.G. M.G. and M.N. designed experiments and conducted research. A.V. J.B.F., M.G. and M.N. developed software for image analysis. D.I.C, A.M.C.G., A.V. and F.B. designed Oligopaint probes. F.B. performed fly handling. M.D. and M.A.M-R. performed Hi-C bioinformatics analysis. D.C., C.H. and S.D. synthesized and purified oligopaints libraries and performed S2 cells handling. D.I.C, A.M.C.G. M.G., G.C. and M.N. wrote the manuscript. All the authors reviewed and commented the data.
Competing financial interests
The authors declare no competing financial interests.
Methods
Cell culture, embryonic tissue preparation and sample fixation
Drosophila S2 cells were obtained from the Drosophila Genomics Resource Center. S2 cells were grown in serum-supplemented (10%) Schneider’s S2 medium at 25 °C. Fly stocks were maintained at room temperature with natural light/dark cycle and raised in standard cornmeal yeast medium. Following a pre-collection period of at least 1 hour, fly embryos were collected on yeasted 0.4% acetic acid agar plates at and incubated at 25 °C until they reached the desired developmental stage: 2-3 h or 12-14 h (total developmental time) for early for late embryos, respectively. Embryos were mechanically broken and immediately fixed as described32 by using 4% PFA in PBS for 10 min at room temperature (RT). S2 cells were allowed to adhere to a poly-l-lysine coverslip for 30 min in a covered 35-mm cell culture dish before 4% PFA fixation.
Immunostaining
Cells were permeabilized with 0.5% Triton X-100 for 10 min and blocked with 5% of bovine serum albumin (BSA) for 15 min at RT. Primary antibodies anti-H3K27me3 (pAb-195-050, Diagenode and ab6002, Abcam), anti-H3K4me3 (cat#04-745, Millipore and ab1012, Abcam), anti-Polycomb33 and anti-Beaf-3234 (made from rabbit by Eurogentec) were coupled to Alexa Fluor 647 or Cy3b as described elsewhere27. Antibodies were used at a final concentration of 10 μg/ml in PBS and 1% BSA. Coverslips were incubated overnight at 4 °C in a humidified chamber and washed 3 times with PBS before introducing fiducial markers diluted 1/4000 (Tetraspeck, #10195142, FisherScientific). Coverslips were mounted on slides with 100 μl wells (#2410, Glaswarenfabrik Karl Hecht GmbH & Co KG) in dSTORM buffer composed of PBS, glucose oxidase (G7141-50KU, Sigma) at 2.5 mg/ml, catalase at 0.2 mg/ml (#C3155-50MG, Sigma), 10% glucose and 50 mM of β-mercaptoethylamine (MEA, #M9768-5G, Sigma). Coverslips were sealed with duplicating silicone (Twinsil, Rotec).
Oligopaint libraries
Oligopaint libraries were constructed from the Oligopaint public database (http://genetics.med.harvard.edu/oligopaints). All libraries consisted of 42mer sequences discovered by OligoArray2.1 run with the following settings: −n 30 −l 42 −L 42 −D 1000 −t 80 −T99 −s 70 −× 70 −p 35 −P 80 −m ‘GGGG;CCCC;TTTTT;AAAAA’ −g 44. Oligonucleotide for libraries 1-18 and BX-C were ordered from CustomArray (Bothell, WA). The procedure used to synthesize Oligopaint probes is described below. Chr3R-69 borders oligonucleotides were purchased from MYcroarray (Ann Arbour, MI). Oligopaint probes for this library were synthesized using the same procedure as for the other libraries except for the initial emulsion PCR step. Secondary, fluorescently-labeled oligonucleotides were synthesized by Integrated DNA Technologies (IDT; Coralville, IA for Alexa488) and by Eurogentec (Angers, France for Cy3b). See Supplementary Table 1 for a list of Oligopaint probe sets used for libraries 1-18. Sequences for secondary oligonucleotides and PCR primers are described below (Supplementary Tables 2-4). Details for the methods used for probe synthesis are provided in Online Methods.
Fluorescence in situ hybridization (FISH)
To prepare sample slides containing fixed S2 cells for FISH, S2 cells were allowed to adhere to a poly-l-lysine coverslip for 1h in a covered 35-mm cell culture dish at 25C. Slides were then washed in PBS, fixed 4% paraformaldehyde (PFA) for 10 min, rinsed 3 times for 5 min in PBS, permeabilized 10min with 0.5% Triton, rinsed in PBS, incubated with 0.1M HCl for 10min, washed in 3 times for 1 min with 2X saline-sodium citrate − 0.1% Tween-20 (2XSSCT) and incubated in 2XSSCT/50% formamide (v/v) for at least 30min. Then, probes were prepared by mixing 20μl of hybridization buffer FHB (50% Formamide, 10% Dextransulfat, 2X SSC, Salmon Sperm DNA 0.5 mg/ml), 0.8 μL of RNAse A, 30 pmol of primary probe and 30 pmol of secondary oligo. 12μl of this mix were added to a slide before adding and sealing with rubber cement the coverslips with cells onto the slide. Probes and cells are finally co-denaturated 3 min at 78 °C before hybridization overnight at 37 °C. The next day, the slides were washed for 3 times for 5 min in 2X SSC at 37 °c, then for 3 times for 5 min in 0.1X SSC at 45 °c. Finally, they were stained with 0.5μg/ml of DAPI for 10 min, washed with PBS, mounted in Vectashield and sealed with nail polish. For a more detailed protocol, see 35
Image acquisition and post-processing of 3D-SIM data
Samples were prepared as described above and mounted on an OMX V3 microscope (Applied Precision Inc.) equipped with a 100X/1.4 oil PlanSApo objective (Olympus) and three emCCD cameras. 405 nm, 488 nm and 561 nm excitation lasers lines were used to excite DAPI, Alexa488 and Cy3, respectively. Each channel was acquired sequentially. A transmission image was also acquired to control for cell morphology. For each channel, a total of 1455 images made of 97 different Z-planes separated by 125 nm were acquired, in order to acquire a stack of 12 μm. Three different angles (60°, 0° and +60°) as well as five phase steps were used to reconstruct 3D-SIM images using softWoRx v5.0 (Applied Precision Inc.). Final voxel size was 39.5 nm in the lateral (xy) and 125 nm in the axial (z) directions, respectively, for a final 3D stack volume of ∼40 × 40 × 12 μm. Multicolor TetraSpeck beads (100 nm in diameter, Invitrogen) were used to measure x, y and z offsets, rotation about the z-axis and magnification differences between fluorescence channels. These corrections were applied to the reconstructed images. The same beads were used to validate the reconstruction process ensuring a final resolution of ∼120 nm in xy and ∼300 nm in z at 525 nm of emission wavelength. 3D-SIM raw and reconstructed images were analyzed with SlMCheck ImageJ Plug-in (University of Oxford, http://www.micron.ox.ac.uk/software/SlMCheck.php). Acquisition parameters were optimized to obtain the best signal-to-noise ratio avoiding photobleaching between the different angular, phase, and axial acquisitions.
3D nuclei segmentation from 3D-SIM data
3D-SIM images were analyzed employing homemade software written in Matlab. In order to identify nuclear shells, nuclei are first segmented by manually selecting rectangular ROIs of the DAPI signal in the XY-plane and keeping all the Z-planes and then a low-pass filter is applied to the DAPI intensities so that only the large-scale information (i.e. nuclear shape) is kept. For each plane of the 3D ROIs, an intensity threshold is computed as described by Snell et al. 36 in order to distinguish voxels inside or outside the nucleus. The average intensity threshold calculated from the threshold of the single planes is used to identify the complete nuclear shell. After nuclei segmentation, foci were identified by calculating, for each channel separately, the maximum entropy threshold of the fluorescence intensities in the 3D ROIs. By using the intensity thresholds the 3D ROIs are finally binarized (voxels above threshold are set to 1 while the others to 0) and the different foci identified as groups of connected voxels. From the group of connected voxels the center of mass was estimated with subpixel resolution. Distance between TBs was estimated as the linear distance between the closest foci imaged in two different emission channels.
Image acquisition and post-processing of two-color dSTORM data
Super-resolution experiments were carried out in a custom-made inverted microscope using objective-type total internal reflection fluorescence (TIRF) configuration employing an oil-immersion objective (Plan-Apocromat, 100x, 1.4NA oil DIC, Zeiss) mounted on a z-axis piezoelectric stage (P-721.CDQ, PICOF, PI). For 2D imaging, a 1.5x telescope was used to obtain a final imaging magnification of 150 fold corresponding to a pixel size of 105 nm. Three lasers were used for excitation/photo-activation: 405 nm (OBIS, LX 405-50, Coherent Inc.), 488 nm (OBIS, LX 488-50, Coherent Inc.), 561 nm (OBIS, LX 561-50, Coherent Inc.), and 640 nm (OBIS, LX 640-100, Coherent Inc.). Laser lines were expanded, and coupled into a single beam using dichroic mirrors (427, 552 and 613 nm LaserMUXTM, Semrock). An acousto-optic tunable filter (AOTFnc-400.650-TN, AA opto-electronics) was used as to modulate laser intensity. Light was circularly polarized using an achromatic quarter wave plate (QWP). Two achromatic lenses were used to expanded the excitation laser and an additional dichroic mirror (zt405/488/561/638rpc, Chroma) to direct it towards the back focal plane of the objective. Fluorescence light was spectrally filtered with emission filters (ET525/50m, ET600/50m and ET700/75m, Chroma Technology) and imaged on an EMCCD camera (iXon X3 DU-897, Andor Technologies). The microscope was equipped with a motorized stage (MS-2000, ASI) to translate the sample perpendicularly to the optical axis. To ensure the stability of the focus during the acquisition, a home-made autofocus system was built. An 785 nm laser beam (OBIS, LX 785-50, Coherent Inc.) was expanded twice and directed towards the objective lens by a dichroic mirror (z1064rdc-sp, Chroma). The reflected IR beam was redirected following the same path than the incident beam and guided to a CCD detector (Pixelfly, Cooke) by a polarized beam splitter cube (PBS). Camera, lasers and filter wheel were controlled with software written in Labview 37.
Analysis of two-color dSTORM data
Unless stated otherwise, all homemade software and routines were developed in Matlab. Single-molecule localizations were obtained by using Multiple Target Tracing (MTT) 38 Localization coordinates were further processed using SMLM_2C, a custom software written in Matlab 27 Fluorescent beads were used to correct for drift and chromatic aberrations. Lateral drift was corrected with 5±3 nm precision as previously described 37. Chromatic aberration correction was performed as described in 27,39. Samples with abnormal drift or lesser precision of drift or chromatic aberration correction were discarded. Clustering of localizations was performed as described in Cattoni et al. 40. Four methods were used to estimate colocalization from two-color dSTORM datasets. In the first method, we estimated the colocalization of single-molecule localizations using a custom implementation of the Coordinate-based colocalization analysis 41 adapted for whole-cell automated analysis 27. The other methods to estimate co-localization, pixel, Pearson, and Manders correlation, relied on the use of digital images. These were obtained from the list of localizations using standard procedures described elsewhere 37. Two-color digital images were then used to plot the correlation between pixel intensities (pixel correlation analysis), or to calculate the Pearson or Manders correlation coefficients 42,43
Analysis of one-color dSTORM data
Single-molecule localizations are converted into a Voronoi diagram using a modified version of the Voronoi tesselation algorithm of Levet et al. 28 Compartment segmentation is directly calculated from the Voronoi diagram using three steps. First, densities of each polygon are calculated as the inverse of their area. Densities are then thresholded using the general criteria of Levet et al. 28 Using this criterion, in which the threshold is determined by the average localization density, a random distribution of localizations did not provide any segmented polygon. Finally, polygons that have a density higher than the threshold and that are touching each other are merged to define the compartment outline. Compartment sizes are obtained by interpolating each segmented compartment on a grid of 5 nm size and calculating their equivalent diameter using standard morphological operations. Probability density functions in compartment size histograms are calculated such that the area of each bar is the relative number of observations and that the sum of the bar areas is equal to 1.
Analysis of genome-wide data
Calculations of the genomic size distributions of H3K27me3 and H3K4me3 domains (Fig. S6) were performed as follows. ChIP-chip/seq computed peaks were downloaded from ModEncode (ftp://data.modencode.org/D.melanogaster/)44 Datasets used are described in the Online Methods (Supplementary Table 5). Peaks positions and intensities were used to interpolate the signal into one-dimensional matrix with 1bp resolution and the length of each chromosome. One matrix was produced for each chromosome. These matrices were binarized using a threshold that corresponded to 0.1 of the log of the maximum intensity signal, ensuring that even peaks with very low intensity were retained. Domains were defined as continuous segments of bins with non-zero intensity. Domains with less than 2 bins (i.e. 2bp) were discarded. Domains that appeared closer than 1kb were fused together. This procedure was robust to calculate domain size distributions above 3kb (Fig. S6). Genomic distributions of domain sizes were converted to physical size distributions (Figs. 4f-g) by applying the power law dependence between genomic and physical distance used to fit experimental data in Figs. 2e-f. The exponents used depended on cell type and chromatin domain type, and are displayed as insets in Figs. 2e-f. Amplitudes were chosen to minimize the residuals between dSTORM compartment size distributions and predicted genome-wide distributions.
Chromatin states were defined according to the enrichment in the percentages of H3K4me3 and H3K27me3/PC as described in Fig. S1.
Clustering of domains of different epigenetic marks was defined as the ratio between the number of clusters of sizes larger than 150 nm obtained from Chip-seq vs. microscopy imaging. Changes in this threshold did not affect the results.
In-situ Hi-C data processing and normalization
Hi-C data was processed using an in-house pipeline based on TADbit 45 First, quality of the reads was checked using the quality_plot() function in TADbit, which is similar to the tests performed by the FastQC program with adaptations for Hi-C datasets. Next, the reads are mapped following a fragment-based strategy as implemented in TADbit where each side of the sequenced read was mapped in full length to the reference genome (dm3). After this step, if a read was not uniquely mapped, we assumed the read was chimeric due to ligation of several DNA fragments. We next searched for ligation sites, discarding those reads in which no ligation site was found. Remaining reads were split as often as ligation sites were found. Individual split read fragments were then mapped independently. Next, we used the TADbit filtering module to remove non-informative contacts and to create contact matrices. From the resulting contact matrices, low quality bins (those presenting low contacts numbers) were removed as implemented in TADbit’s filter_columns() function. Next, the matrices were normalized using the ICE algorithm 46 The normalization iterations stopped when the biases were diverting less than 10% of the previous values or a max of 10 iterations. Finally, all matrices were corrected to achieve an average content of one interaction per cell. All parameters in TADbit were kept at default values.
The resulting late-embryo and S2 Hi-C interaction maps (at 10kb resolution) of the different replicates for each experiment were highly correlated (correlation coefficient from genomic distances ranging from 10Kb to 20Mb were 0.99 to 0.75 and 0.95 to 0.45, respectively) and thus were further merged into the final datasets with more than 282 and 210 million valid pairs each (Supplementary Table 6).
Acknowledgements
We thank Brian Beliveau, Hien Hoang and Ting Wu for help with oligopaints design. This research was supported by funding from the European Research Council under the 7th Framework Program (FP7/2010-2015, ERC grant agreement 260787 to M.N. and FP7/2007-2013, ERC grant agreement 609989 to M.A.M-R.). M.A.M-R and G.C. acknowledge support from the European Union’s Horizon 2020 research and innovation programme under grant agreement 676556. This work has benefited also from support by the Labex EpiGenMed, an «investments for the future» program, reference ANR-10-LABX-12-01, the Spanish Ministry of Economy and Competitiveness (BFU2013-47736-P to M.A.M-R), and from ‘Centro de Excelencia Severo Ochoa 2013-2017’, SEV-2012-0208 to the CRG. 3D-SIM experiments were performed at Montpellier Resource Imaging. We acknowledge the France-BioImaging infrastructure supported infrastructure supported by the French National Research Agency (ANR-10-INBS-04, «Investments for the future»).
Footnotes
↵* Co-authors
References




