

ABSTRACT
AKT is a kinase that regulates numerous cellular processes in the brain and mutations in AKT are known to affect brain function. AKT is indirectly implicated in synaptic plasticity, but its direct role has not been studied. Moreover, three highly related AKT isoforms are expressed in the brain, but their individual roles are poorly understood. We find that each AKT isoform has a unique expression pattern in the hippocampus, with AKT1 and AKT3 primarily in neurons but displaying local differences, while AKT2 is in astrocytes. We also find isoform-specific roles for AKT in multiple paradigms of hippocampal synaptic plasticity. AKT1, but not AKT2 or AKT3, is required for L-LTP through regulating activity-induced protein synthesis. Interestingly, AKT activity inhibits mGluR-LTD, with overlapping functions for AKT1 and AKT3. In summary, our studies identify distinct expression patterns and roles in synaptic plasticity for AKT isoforms in the hippocampus.
INTRODUCTION
Dysregulation of synaptic plasticity is implicated in cognitive and memory impairments associated with many neurological diseases and psychiatric disorders, such as Alzheimer's disease, schizophrenia, and intellectual disability. The AKT (Protein kinase B, PKB) signaling pathway is thought to play a pivotal role in synaptic plasticity 1. AKT is a central serine/threonine kinase expressed in almost all cell types throughout the body and regulates cell growth, proliferation and metabolism. In post-mitotic neurons, the AKT pathway has significant functional impact on stress responses, neurotransmission and synaptic plasticity 2.
Synaptic plasticity describes the specific modification of neuronal connections in response to neural activity. A persistent increase in synaptic strength after stimulation is known as long-term potentiation (LTP) while a decrease in synaptic strength is known as long-term depression (LTD). Late-phase LTP (L-LTP) is a long-lasting form of LTP that requires changes in gene expression and the synthesis of new proteins 3. A form of LTD mediated by group 1 metabotropic glutamate receptors (mGluR-LTD) also requires protein synthesis 4. Interestingly, AKT plays an important role in the mammalian target of rapamycin (mTOR) pathway controlling protein synthesis, and previous studies found that induction of L-LTP or mGluR-LTD in the hippocampus leads to AKT activation measured by increased phosphorylation of serine 473 5,6.
AKT is known to include a family of three closely related isoforms, named AKT1, AKT2 and AKT3, which share a high degree of structural homology 7. Despite their homology, accumulating evidence suggests that each isoform is involved in distinct neurological disorders. Mutations in AKT1 have been associated with schizophrenia, AKT2 with gliomas and AKT3 with brain growth 8–10. Supporting these observations in humans, single-isoform Akt knockout (KO) mice also show distinct phenotypes. Akt1 KO mice have growth retardation and increased neonatal death 11,12, Akt2 KO mice suffer from a type 2 diabetes-like syndrome and Akt3 KO mice show decreased brain size 11, 13, 14. These data suggest that each isoform subserves different cellular functions to give rise to distinct phenotypes. Whether each AKT isoform plays different roles in synaptic plasticity has not been examined.
Activation of AKT has been correlated with LTP and LTD induction, implicating a role for AKT 5,6,15. However, whether AKT activity is necessary in synaptic plasticity has not been directly tested. Moreover, all three AKT isoforms are present in the brain 11. Given the numerous forms of synaptic plasticity, isoform-specific functions of AKT may provide an important mechanism for control and precision of the cellular processes supporting synaptic plasticity. Here, we show for the first time that each AKT isoform has a distinct expression pattern in the hippocampus. We then examined the role of each isoform in several hippocampal synaptic plasticity paradigms known to involve different molecular and cellular processes: early-phase LTP (E-LTP), L-LTP, low frequency stimulation (LFS)-LTD, and mGluR-LTD. Our studies provide evidence that AKT isoforms play differential roles in synaptic plasticity due to cell type-specific expression of Akt genes in the hippocampus and isoform-specific functions in protein synthesis.
RESULTS
AKT isoforms show differential expression in the hippocampus
AKT is a well-studied kinase that may play a central role in brain disorders 1. However, most studies examining AKT activity made no distinction between the activities of each isoform. We hypothesized that AKT isoforms play distinct roles in synaptic plasticity. To test this, we first examined the expression pattern of AKT1, AKT2 and AKT3 in the mouse hippocampus. We found that AKT1 and AKT3 were distributed throughout somatic layers of the hippocampus, with local differences in expression (Fig. 1). AKT1 showed more intense immunoreactivity in the cell body layer of area CA1 whereas AKT3 showed more intense staining in cell bodies within area CA3 and the hilus of the dentate gyrus. Interestingly, AKT2 showed a very different staining pattern. AKT2 was mostly expressed in cells of the molecular layer of the dentate gyrus and stratum radiatum of CA1 (Fig. 1). Isoform-specific KO tissues confirmed specificity of the staining (Fig. 1). Because the brain consists of neurons and glia cells, we next determined the cell types in which each isoform is expressed by co-staining brain slices for the neuronal marker neuron-specific nuclear protein (NeuN), the astrocytic marker glial fibrillary acidic protein (GFAP) and each AKT isoform. This triple stain approach revealed that within CA1, AKT1 was mainly expressed in pyramidal layer neurons, with no detectable staining in astrocytes (Fig. 2a,b). Interestingly, certain neurons seemed to express more AKT1 compared with neighboring neurons (Fig. 2a,b). In the CA1, AKT3 was also mainly expressed in neurons, including in the processes extending into stratum radiatum, with some weak staining in astrocytes (Fig. 2a,b). AKT2 showed no detectable co-localization with NeuN but co-localized with GFAP (Fig. 2a,b). To confirm that AKT2 is not expressed in neurons, we employed a Cre-mediated strategy of selective Akt2 gene disruption by crossing mice with floxed alleles of Akt2 to two different mouse lines with neuron-specific Cre recombinase expression (Camk2α-Cre-T29 and NSE39-Cre). Using this strategy, we confirmed that AKT2 levels were not reduced in the hippocampus with either Cre line, showing that Akt2 is not expressed in hippocampal neurons (Supplementary Fig. 1). To verify disruption of the floxed Akt2 alleles, hippocampi of Akt2loxP/loxP mice were infected with adeno-associated virus (AAV) constructs expressing EGFP-tagged Cre under the CMV promoter (CMV_Cre-EGFP), a promoter active in both neurons and astrocytes. Three weeks post-infection, we found that AKT2 levels were reduced in the hippocampus with CMV-driven Cre expression (Supplementary Fig. 1), providing further support that AKT2 is expressed in astrocytes and not in neurons. Combined, our results demonstrate differential AKT isoform expression in the hippocampus. This isoform-specific expression may lead to unique functions of each isoform in synaptic plasticity processes.
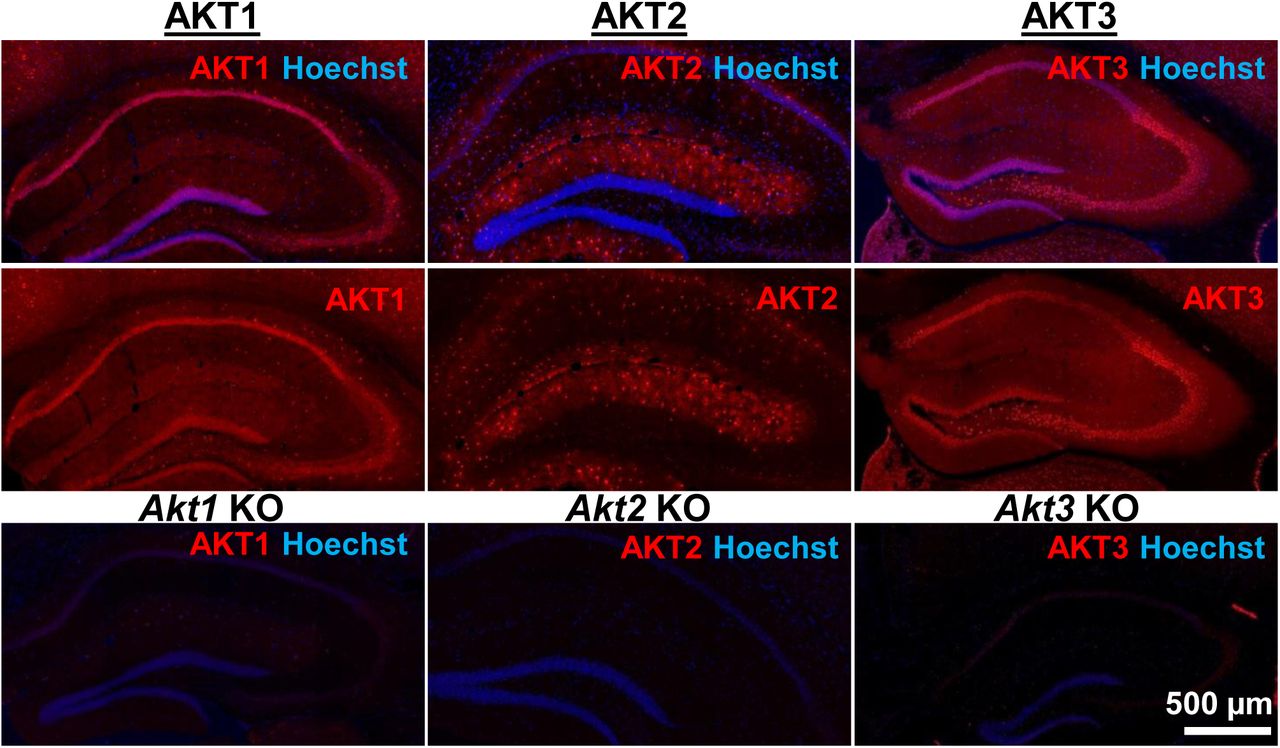
AKT isoform-specific expression in the hippocampus. Immunohistology using isoform-specific antibodies revealed distinct expression patterns for each AKT isoform in the hippocampus. AKT1 was mainly expressed in the cell body layers, with the greatest levels in stratum pyramidale of CA1. AKT2 was mostly expressed in specific cells in the molecular layer of the dentate gyrus, CA3 and CA1. AKT3 was also mainly expressed in the cell body layers of the hippocampus and showed strong expression in the hilus and CA3. Bottom panels show single Akt knockout (KO) tissue to validate the specificity of the antibodies.

Cell type-specific expression of AKT isoforms in hippocampal area CA1. (a). AKT1 was mainly expressed in neuronal cell bodies, indicated by co-localization with NeuN. Certain neurons in the pyramidal layer, stratum oriens and molecular layer showed greater expression levels of AKT1 (yellow arrows). AKT2 was specifically expressed in astrocytes, shown by colocalization with the astrocyte marker GFAP. AKT3 co-localized with NeuN like AKT1 but was also expressed in the stratum radiatum, most likely within dendrites. (b) Higher magnification showed no specific co-localization of AKT1 with GFAP and that certain neurons in the stratum oriens expressed high levels of AKT1 (yellow arrows). AKT2 was mainly expressed in the cell bodies of astrocytes. AKT3 showed high expression levels in neuronal cell bodies and dendrites and some expression in astrocytes.
AKT inhibition leads to reduced substrate activity and impaired L-LTP
AKT has been implicated in L-LTP16, but directly targeting AKT in L-LTP had not been studied. We chose to test the effects of two AKT inhibitors, AZD5363 (AZD) and MK2206-HCl (MK), on synaptic plasticity. AZD and MK have different mechanisms of action; AZD is an ATP-competitive AKT inhibitor 17 and MK is an allosteric AKT inhibitor 18. To examine AZD and MK action in the brain, we first assessed their effects on the phosphorylation of AKT and a well-characterized downstream target of AKT, glycogen synthase kinase 3β (GSK-3β) in hippocampal slices. Incubation with 10 μM MK strongly inhibited AKT activity as measured by the reduction of AKT phosphorylated on serine 473 (pAKT S473); however, phosphorylation levels of GSK-3β on serine 9 (pGSK-3β S9) were not reduced (Fig. 3a-c). At higher doses of MK (30 μM and 100 μM), pAKT continued to be reduced at similar levels (Fig. 3a,b) while pGSK-3β levels were ultimately reduced in a dose-dependent manner (Fig. 3a,c). AZD incubation resulted in increased pAKT levels (Fig. 3a,b), consistent with previous reports that ATP-competitive inhibitors of AKT result in hyperphosphorylation of the kinase itself, which does not indicate increased AKT activation but likely reflects intrinsic responses to competitive inhibition of AKT 19. Indeed, AZD significantly decreased phosphorylation levels of the AKT target GSK-3β in a dose-dependent manner, indicating AZD inhibited AKT activity (Fig. 3a,c). Recently, the AKT1/2 inhibitor A6730 (10 mM) was found to reduce levels of the AMPA receptor subunit GluA2 20, but we found no reduction even with the highest concentration of AZD or MK (Fig. 3a,d). Based on the dose response of pGSK-3β S9 levels, we chose to test 30 μM for both AZD and MK inhibitors on synaptic plasticity in the CA3-CA1 circuit of the hippocampus. After determining that incubation of hippocampal slices from wild-type (WT) mice with either inhibitor did not affect the stability of baseline field excitatory postsynaptic potentials (fEPSP) recorded in CA1 (Supplementary Fig. 2), we assessed the effect on expression of L-LTP induced by high frequency stimulation (HFS). We found that both AKT inhibitors resulted in significantly impaired L-LTP compared with vehicle treatment (Fig. 4a,b), indicating that AKT activity is required to sustain long-lasting LTP in area CA1.

AKT inhibitors MK2206 and AZD5363 effectively inhibit AKT activity in hippocampal slices. (a) MK2206 and AZD5363 blocked AKT activity effectively, shown by reduced phosphorylation levels of the AKT substrate GSK-3α on serine 9 (pGSK-3β S9), but had different effects on phosphorylation levels of AKT itself on serine 473 (pAKT S473). (b) MK2206 significantly reduced pAKT S473 levels normalized to total AKT (totAKT) levels while AZD significantly increased pAKT S473 levels (MK2206 F(3.8)=59.99, p<.001; AZD F(3.8)= 28.51, p<.001; post hoc comparisons: veh vs. MK10 p<.001, veh vs. MK30 p<.001, veh vs. MK100 p<.001; veh vs. AZD10 p<.001, veh vs. AZD30 p=.005, veh vs. AZD100 p<.001). (c) MK2206 had a dose-dependent effect on pGSK-3β S9 levels normalized to total GSK-3β (totGSK-3β) levels, with no effect at 10 μM but significantly reduced levels at 30 and 100 μM compared with vehicle treatment (MK2206 F(3.8)=74.56, p<.001; post hoc comparisons: veh vs. MK10 p=.998; veh vs. MK30 p=.002; veh vs. MK100 p<.001, MK10 vs. MK30 p=.002, MK30 vs. MK100 p<.001). AZD also had a dose-dependent effect on pGSK3β-S9 levels, with significant reduction at 10 μM in addition to the 30 and 100 μM doses compared with vehicle (AZD F(3,8)=39.69, p<.001; post hoc comparisons: veh vs. AZD10 p=.002; veh vs. AZD30 p<.001; veh vs. AZD100 p<.001; AZD10 vs. AZD100 p=.01). (d) Levels of GluA2 normalized to GAPDH was not significantly altered after MK or AZD treatment (MK2206 F(3,8)=0.279, p=.838; AZD F(3,9)=0.378, p=.771). Veh=vehicle, MK10=10 μM, MK30=30 μM, MK100=100 μM; AZD10=10 μM, AZD30=30 μM, AZD100=100 μM, 3-4 mice/group, *p<.05
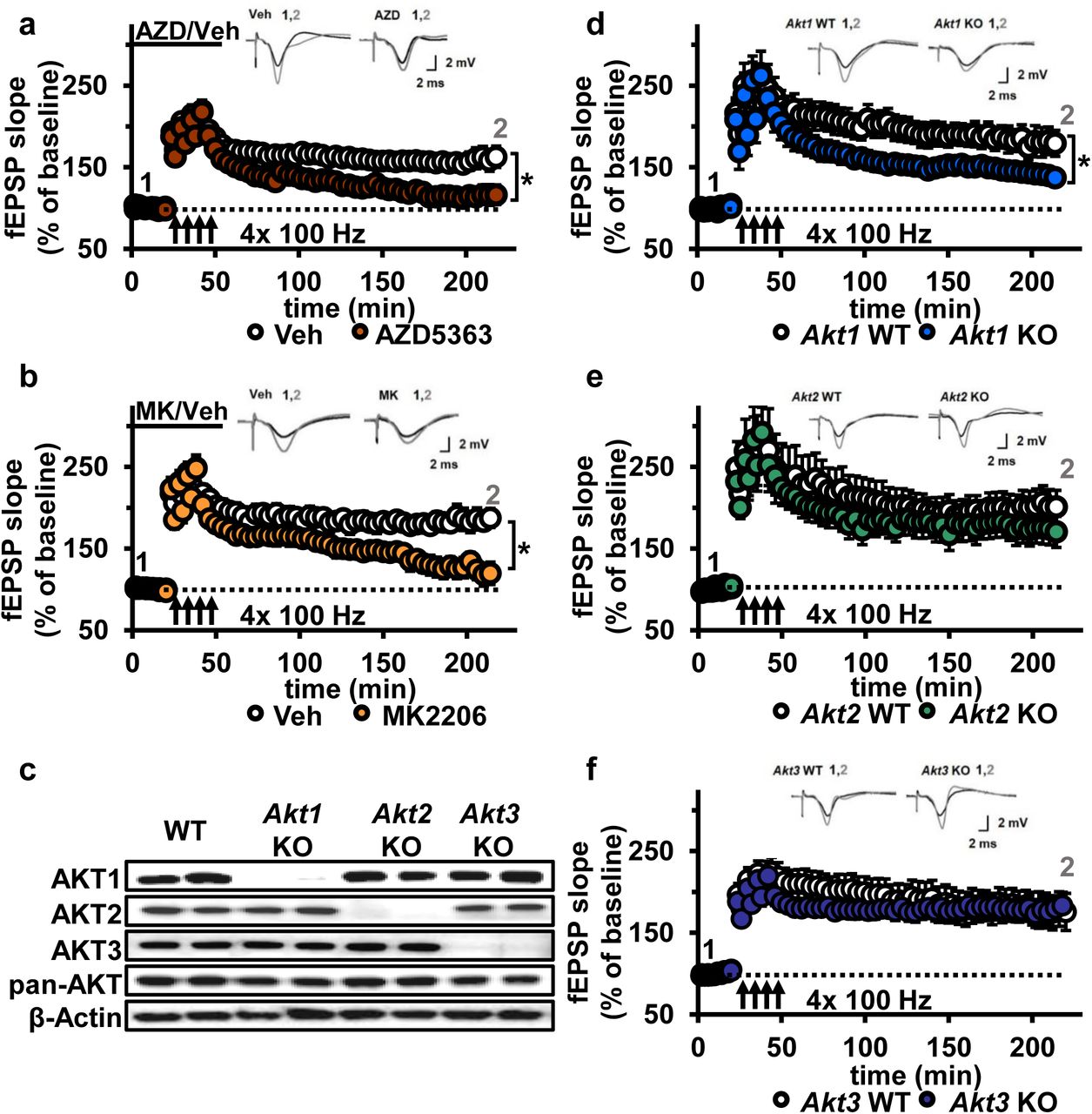
AKT inhibition and Akt1 deletion results in impaired L-LTP. (a) Inhibiting AKT activity in hippocampal slices with AZD5363 (30 JM) prior to four trains of HFS (100 Hz) resulted in significantly impaired L-LTP in hippocampal area CA1 compared with vehicle treatment (F(1,24)=6.896, p=.015), n=13 slices/group, 5 mice/group. (b) AKT inhibitor MK2206 (30 μM) also significantly impaired L-LTP compared with vehicle treatment (F(1,13)=18.639, p<.001), n=7-8 slices/group, 4 mice/group. (c) Western blot validation of AKT1, AKT2 and AKT3 removal using cortical tissue from Akt isoform-specific KO mice. β-actin, loading control. (d) Akt1 KO mice showed significantly impaired L-LTP in hippocampal area CA1 compared with WT mice (F(1,28)=5.049, p=.033), n=15 slices/group, 6 mice/group. (e) Akt2 KO mice displayed similar levels of L-LTP to WT mice (F(1,20)=1.121, p=.302), n=10-12 slices/group, 4-5 mice/group. (f) Akt3 KO mice showed similar levels of L-LTP to WT mice (F(1,20)=0.33, p=.857), n=8-14 slices/group, 5-7 mice/group. *p<.05
Loss of AKT1 leads to impaired L-LTP
To determine if a specific AKT isoform may be responsible for maintaining L-LTP in the CA3-CA1 circuit, we examined hippocampal slices from Akt1, Akt2 or Akt3 KO mice (Fig. 4c). Because AKT has been linked to AMPA and NMDA receptor function 20, we first examined the effect of deleting single Akt isoforms on basal synaptic transmission, presynaptic plasticity and short-term LTP in area CA1. We found normal synaptic input/output curves (Supplementary Fig. 3), paired-pulse facilitation (Supplementary Fig. 3) and E-LTP (Supplementary Fig. 4) in all single Akt isoform mutants, suggesting no one isoform is required for normal basal synaptic transmission, presynaptic plasticity and short-term LTP. In contrast, we found that Akt1 deletion resulted in impaired L-LTP (Fig. 4d), while slices from Akt2 or Akt3 KO mice showed normal L-LTP (Fig. 4e,f). This shows that AKT1 is the major isoform involved in the expression of L-LTP and targeted by AZD and MK inhibition to impair L-LTP in CA1.
Akt1 deletion results in impaired protein synthesis after L-LTP induction
Hippocampal L-LTP requires synthesis of proteins that are involved in modifying synaptic connections 3. Because the AKT pathway plays a well-known role in translational control, the AKT1 isoform may function in L-LTP by regulating protein synthesis. Interestingly, even though Akt3 removal had no effect on L-LTP, Akt3 KO mice have significantly smaller brains, which may indicate impaired translation. To examine protein synthesis associated with L-LTP in Akt1 and Akt3 KO hippocampal slices, we used the SuNSET method of labeling newly synthesized proteins 21. Slices from Akt1 or Akt3 KO and WT littermates received either four spaced trains of HFS to induce L-LTP or no stimulation (control). Western blotting showed that Akt1 KO slices failed to increase protein synthesis following L-LTP induction (Fig. 5a,c), consistent with the L-LTP impairment observed in these slices (Fig. 4d). In contrast, Akt3 deletion did not affect the protein synthesis response post-HFS (Fig. 5b,d), which is consistent with the normal L-LTP found in Akt3 KO slices (Fig. 4f). These results suggest an isoform-specific role for AKT1 in activity-induced protein synthesis that promotes L-LTP expression in the hippocampus.

Only Akt1 KO mice show an impaired protein synthesis response after tetanic stimulation. (a,c) Puromycin labeling of newly synthesized proteins showed that four trains of HFS in area CA1 to induce L-LTP results in increased protein synthesis levels compared with no stimulation (Ctrl) in WT hippocampal slices (t(14)= −3.52, p=.005), while stimulated Akt1 KO slices fail to increase protein synthesis from unstimulated levels (t(14)=−1.45, p=.167). GAPDH, loading control. (b,d) Akt3 KO hippocampal slices showed a normal increase in protein synthesis after four trains of HFS (Akt3 WT Ctrl vs. HFS t(12)=−2.75, p=.018; Akt3 KO Ctrl vs. HFS t=−2.20, p=.047). GAPDH, loading control. (e,g) Akt1 KO slices failed to show an increase in S6 phosphorylation (pS6 S235/236) normalized to total S6 (totS6) levels after tetanic stimulation (Akt1 WT Ctrl vs. HFS t(14)=−2.71, p=.016; Akt1 KO Ctrl vs. HFS t(14)=−1.01, p=.33). (f,h) Akt3 KO slices showed a normal increase in pS6 S235/236 levels after tetanic stimulation (Akt3 WT Ctrl vs. HFS t(14)=−2.92, p=.01, Akt3 KO Ctrl vs. HFS t(14)=−2.59, p=.02). 4-6 mice/group, *p<.05
To investigate the molecular signaling pathways linking AKT activity to translational control in long-lasting synaptic plasticity, we examined the phosphorylation levels of a well-known AKT effector, S6. S6 is a component of the 40S ribosomal subunit that mediates translation, and phosphorylation of S6 at serine 235 and 236 (pS6) is essential for S6 capbinding activity 22. We found that pS6 levels significantly increased after tetanic stimulation in WT and Akt3 KO hippocampal slices but not in Akt1 KO slices (Fig. 5e-h). This result agrees with the impaired protein synthesis after tetanic stimulation observed with AKT 1 deficiency. Therefore, AKT1 is the critical isoform supporting hippocampal L-LTP through activity-regulated signaling to protein synthesis and may compensate for AKT3 to enable normal L-LTP and associated protein synthesis in Akt3 KO conditions.
AKT activity is not required for LTD induced by low frequency stimulation
Another form of synaptic plasticity is LTD induced by low frequency stimulation (LFS-LTD), which is known to depend on NMDA receptor-mediated calcium influx and is independent of protein synthesis 4. Previously, synaptic plasticity related to brain-derived neurotrophic factor signaling was reported to involve a feedforward AKT mechanism to NMDA receptors 15. Furthermore, the AKT substrate GSK-3β was found to be important for LFS-LTD, and phosphorylation levels of GSK-3β was found to be reduced in Akt3 KO brains 23, 24. We confirmed that Akt3 deletion results in decreased hippocampal levels of pGSK-3β (Fig. 6a). In contrast, Akt1 KO mice show normal pGSK-3β levels (Fig. 6a), revealing further differential AKT isoform signaling. Therefore, AKT activity, especially AKT3, may be involved in maintaining LFS-LTD. To address this question, we examined LFS-LTD in the CA3-CA1 circuit of single Akt isoform mutant slices and found no effect of any single isoform deletion (Fig. 6b-d). To determine if these findings resulted from incomplete blockade of AKT activity or if multiple isoforms are involved, we examined LFS-LTD in WT hippocampal slices following pharmacological inhibition of AKT activity using AZD or MK. We found normal expression of LFS-LTD compared to vehicle-treated slices (Fig. 6e,f). Therefore, these experiments combined indicate that AKT is not required for LFS-LTD in CA1.

AKT is not involved in LFS-LTD. (a) pGSK-3β levels were significantly reduced in the hippocampus of Akt3 KO mice but normal in Akt1 KO mice (Akt3 KO t(20)=−4.725, p<.001; Akt1 KO t(14)=0.104, p=.918), 4-6 mice/group. (b-d) Hippocampal slices from Akt1 KO, Akt2 KO or Akt3 KO mice showed normal LFS-LTD induced by 900 stimuli of 1 Hz compared to WT slices (p>.05 for all genotypes), n=12-20 slices/group, 5-7 mice/group. (e) Blocking AKT activity in hippocampal slices with AZD5363 (30 μM) prior to 900 stimuli of 1 Hz did not affect LFS-LTD compared to vehicle-treated slices (p>.05), n=12-13 slices/group, 6 mice/group. (f) AKT activity blocked by MK2206 (30 μM) also resulted in normal LFS-LTD in hippocampal slices compared to vehicle-treated slices (p>.05), n=10-12 slices/group, 5 mice/group.
Inhibition of AKT activity leads to enhanced mGluR-LTD
Previous studies suggest AKT is involved in another form of LTD, which is mediated by mGluR (mGluR-LTD) and depends on protein synthesis 25. Upon mGluR stimulation with DHPG, pAKT S473 levels were shown to increase, suggesting increased AKT activity 5. Also, inhibiting phosphoinositide 3-kinase (PI3K) activity, a well-validated kinase upstream of AKT activation 26, results in impaired mGluR-LTD 5. However, direct interrogation of the role of AKT in mGluR-LTD expression had never been performed. Thus, we directly examined AKT function in mGluR-LTD using the pan-AKT inhibitors AZD and MK and Akt mutants.
We incubated WT hippocampal slices with AKT inhibitors prior to mGluR-LTD induction with DHPG 27. Unexpectedly, we found that inhibiting AKT activity with either AZD or MK enhanced mGluR-LTD (Fig. 7a,b). Because a previous report using LY294002 inhibition of PI3K, a major upstream activator of AKT 28, showed impaired mGluR-LTD 5, we had expected to find that direct AKT inhibition also would impair mGluR-LTD 5. We therefore repeated the experiment with LY294002. In agreement with our MK and AZD data, we found that inhibition of PI3K resulted in enhanced mGluR-LTD (Fig. 7c). These results demonstrate that AKT is indeed involved in regulating mGluR-LTD, although in a different manner than we expected.

Blocking AKT activity leads to enhanced mGluR-LTD. (a,b) Blocking AKT activity using AZD5363 or MK2206 resulted in enhanced mGluR-LTD induced with 3,5 RS-DHPG (100 μM) in hippocampal area CA1 (AZD F(1,23)=5.010, p=.035; MK F(1,40)=7.216, p=.010), n=12-20 slices/group, 5-6 mice/group. (c) Blocking PI3K using LY294002 resulted in enhanced mGluR-LTD (F(1,27)=10.809, p=.003), n=13-16 slices/group, 7-8 mice/group. (d-f) Akt1, Akt2 or Akt3 KO mice show normal mGluR-LTD compared to WT controls (p>.05), n=13-20 slices/group, 5-7 mice/group. *p<.05
To investigate isoform-specific contributions to this phenotype, we next induced mGluR-LTD in Akt1, Akt2 or Akt3 KO hippocampal slices. Interestingly, we found that all single isoform mutant slices displayed normal mGluR-LTD (Fig. 7d-f). Because pan-AKT and PI3K inhibition both resulted in enhanced mGluR-LTD (Fig. 7a-c), two or more isoforms may redundantly compensate for each other to allow normal expression of mGluR-LTD in the single Akt isoform mutants. To test this idea, we generated multiply mutant Akt mice.
Akt1/Akt3 double mutants have enhanced mGluR-LTD
Because Akt1 and Akt3 are the more similarly expressed isoforms in the hippocampus, we hypothesized that AKT1 and AKT3 may substitute for each other in regulating mGluR-LTD expression at CA3-CA1 synapses. Therefore, combined Akt1 and Akt3 deletion may result in enhanced mGluR-LTD, reproducing the effect of pan-AKT inhibition with either AZD or MK. Because Akt1/Akt3 double KO mice are embryonic lethal 12, we used a Cre-mediated strategy of selective Akt1 disruption by crossing mice with floxed alleles of Akt1 to a forebrain-specific neuronal Cre recombinase mouse line (T29) in an Akt3 KO background (cA1F/A3K mice) (Fig. 8a). Using this strategy, AKT3 is removed and AKT1 levels are significantly reduced in the hippocampus of cA1F/A3K mice compared with WT mice (Fig. 8b,c). Immunostaining of the hippocampus showed Akt1 was mostly deleted from excitatory pyramidal neurons in area CA1, while other neurons, most likely inhibitory neurons, still expressed AKT1 (Fig. 8d). Next, we examined mGluR-LTD in cA1F/A3K mice. Consistent with our results from the AKT inhibitor experiments, we found that mGluR-LTD was enhanced in cA1F/A3K mice compared with WT mice (Fig. 8e). Thus, concomitant Akt1 and Akt3 deletion results in enhanced mGluR-LTD, supporting the idea that the AKT1 and AKT3 isoforms function in hippocampal mGluR-LTD and can compensate for each other.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
AKT1 and AKT3 are involved in mGluR-LTD. (a) Double Akt1 and Akt3 mutant (cA1F/A3K) mice with Cre-mediated removal of both Akt1 alleles in the Akt3 KO background were generated with WT littermate controls by breeding Camk2α-Cre::Akt1loxP/+/Akt3+/- female mice with Akt1loxP/+/Akt3+/- males. (b,c) Western blot analysis showing significantly reduced AKT1 levels and no AKT3 expression in the hippocampus of cA1F/A3K mice compared with WT mice (AKT1 levels: t(6)=3.802, p=.008). (d) Immunostaining confirming reduced neuronal AKT1 expression by Camk2α-driven Cre removal of Akt1 in the hippocampus, especially in the pyramidal cell body layer of CA1. (e) mGluR-LTD is enhanced in cA1F/A3K hippocampal slices (F(1,30)=7.923, p=.009), n=16 slices/group, 5 mice/group. *p<.05
DISCUSSION
Our studies discovered novel distinct roles for the different Akt isoforms in the brain. We show, for the first time, that AKT2 expression is localized to astrocytes in the hippocampus, while AKT1 and AKT3 are primarily but differentially expressed in neurons. We also show for the first time that these differences in isoform expression are associated with different functions in the brain involving synaptic plasticity. We demonstrate that the AKT1 isoform is critical for the expression of L-LTP and regulating activity-induced protein synthesis that supports L-LTP. Interestingly, in contrast to the apparent singular requirement for AKT1 in L-LTP, we found that AKT1 and AKT3 serve overlapping roles to inhibit the expression of mGluR-LTD. To our knowledge, this is the first study to identify AKT isoform-specific expression in the hippocampus and AKT isoform-specific activity underlying different forms of hippocampal synaptic plasticity.
Because AKT controls numerous cellular processes, uncovering isoform differences is important to improve understanding about normal AKT function. Moreover, isoform-specific roles for AKT have been associated with different disease pathologies. The AKT2 isoform, for example, has been implicated in diabetes. A missense mutation in AKT2 that causes loss of AKT2 function leading to insulin resistance was identified in a family with diabetes 29. Similarly, AKT2-deficient mice have resistance to insulin and a subset develop diabetes mellitus-like syndrome 13. AKT2 also has been associated glioma, a type of brain cancer with malignant tumors derived from glial cells. High-grade gliomas have been shown to overexpress AKT2 9. Although this correlation suggests a role for AKT2 in astrocytic function, we were still surprised that our immunostaining in the hippocampus showed AKT2 only to be present in astrocytes. Close examination of our immunostaining revealed little or no detectable AKT1 and AKT3 expression in astrocytes (Fig. 2), although we cannot exclude that they may be present. A previous study using primary astrocytic culture derived from developing mouse brain tissue showed that all three isoforms were expressed in cortical astrocytes 30. Therefore, it may be possible that AKT2 expression is restricted to hippocampal astrocytes or becomes restricted post-developmentally or that signals from other brain cell types in vivo restrict AKT2 expression to astrocytes. Although we found no evidence of a role for AKT2 in the synaptic plasticity paradigms tested in this study, AKT2 might play a role in synaptic plasticity as it relates to astrocytic function for supporting neuronal activity 31.
The AKT1 isoform specifically has been linked to schizophrenia, a neurological disorder believed to represent brain dysconnectivity with a molecular basis in aberrant synaptic plasticity 32. AKT1 is one of the effectors in the well-established pathway from neuregulin 1 (NRG1-ERBB4-PI3K-AKT1 pathway), where each of these components have genetic polymorphisms that have been linked to schizophrenia 8, 33. These polymorphisms may lead to lower AKT1 protein levels in schizophrenia 8, 34, although increased AKT1 expression and activity also have been reported in schizophrenia 35, 36. Human olfactory neurosphere-derived cells collected from schizophrenia patients were shown to have reduced protein synthesis 37. Additionally, the antipsychotic haloperidol used to treat schizophrenia can activate the AKT pathway, thereby inducing protein synthesis 38. Together, these reports may suggest protein synthesis is reduced in schizophrenia, which may be due to reduced AKT1 signaling. Because protein synthesis is crucial to maintain L-LTP 3, our finding that Akt1 deletion results in the impairment of L-LTP (Fig. 4) and the associated protein synthesis response (Fig. 5) suggests AKT1 may be an important factor in the synaptic and cognitive deficits observed in schizophrenia 32.
The AKT3 isoform is expressed in a highly overlapping pattern with AKT1 in the hippocampus (Fig. 1), suggesting overlapping roles with AKT1, but there are also notable differences between their expression (Fig. 1, 2). AKT3 is known to control brain size 11, 14. Mutations that result in enhanced AKT3 activity lead to increased brain growth 10, while deletion of AKT3 is involved in microcephaly 39. AKT3 may control brain growth through regulation of protein synthesis. Deletion of Akt3 is known to reduce phosphorylation of the ribosomal protein S6 11, which is an effector in the AKT-mTOR pathway leading to protein synthesis. Intriguingly, our results show that only AKT1-deficient mice display impaired protein synthesis-dependent L-LTP, while deletion of Akt3 has no effect (Fig. 4). These data are consistent with the idea that while both AKT1 and AKT3 can regulate neuronal protein synthesis, AKT1 is specifically recruited to support translation involved in L-LTP.
What mechanisms might underlie the differential regulation of protein synthesis by AKT1 and AKT3? Phosphorylation of S6 on S235/236 is increased after L-LTP, which correlates with increased translation 40. Consistent with our finding that Akt1 deletion leads to an impaired protein synthesis response, Akt1 KO slices fail to increase S6 phosphorylation following stimulation, whereas Akt3 KO slices still display dynamic range in protein synthesis and S6 activation (Fig. 5). These results show distinct roles for AKT1- and AKT3-mediated protein synthesis. AKT1 is the activity-regulated isoform, converting external stimuli into protein synthesis, while AKT3 may be more important for steady-state protein synthesis. This distinction may also correlate with the regional differences we observed in hippocampal AKT1 and AKT3 expression (Fig. 1, 2). Thus, in regions where AKT3 expression predominates like area CA3, Akt3 KO slices may show altered activity-induced protein synthesis, following stimulation of the mossy fiber-CA3 circuit for instance. Additionally, future experiments aimed at finer localization of AKT1 and AKT3 may help to answer this question.
mGluR-LTD has been widely studied in relation to the neurodevelopmental disorder fragile X syndrome (FXS) and shown to be enhanced in hippocampal slices from Fmr1 KO mice, a mouse model of FXS 41. mGluR-LTD requires protein synthesis, leading to AMPA receptor internalization 25. AKT has been implicated in the cascade mediating this protein synthesis after mGluR stimulation 5. Studies inhibiting upstream and downstream signaling of AKT have suggested that mGluR stimulation activates a cascade through PI3K-AKT-TSC-mTORC1-S6K to protein synthesis. However, there are conflicting findings about the role of this signaling cascade in mGluR-LTD. In support of the idea that AKT functions in this pathway to facilitate mGluR-LTD, pharmacological studies inhibiting PI3K 5, 42 or mTORCI 42, 43 have reported mGluR-LTD impairment. On the other hand, inhibiting mTORC1 also has been reported to have no effect on mGluR-LTD 42, 44. Likewise, loss of S6K1 has been shown to have no effect on mGluR-LTD, while S6K2 loss results in enhanced mGluR-LTD 45. Some of these divergent results may be due to methodological differences or the developmental stage at which experiments were performed. However, it remains that none of these earlier studies directly examined the role of AKT activity in mGluR-LTD. To address this gap, we targeted AKT directly using multiple approaches, including pharmacological agents and genetic removal of Akt.
Our results strongly support the idea that AKT normally acts to inhibit mGluR-LTD. We found that blockade of AKT activity with multiple separate approaches, using two different AKT inhibitors, genetic deletion of both Akt1 and Akt3 in CA1 as well as PI3K inhibition, all resulted in enhanced mGluR-LTD (Figs 7 and 8). By using two AKT inhibitors that have different mechanisms of action on AKT, complemented by genetically removing AKT, we addressed potential off-target effects of the compounds and provide converging evidence that the enhanced mGluR-LTD is due to specific inhibition of AKT. Developmental effects of AKT removal also are unlikely to contribute to the observed LTD enhancement as single Akt isoform deletion did not affect mGluR-LTD (Fig. 7). Additionally, in the double Akt1/Akt3 deletion experiment, AKT1 is removed postnatally, eliminating potential confounds from the loss of multiple AKT isoforms during development. Combined, these data show that the role of AKT in mGluR-LTD may be more complex than originally thought. AKT may not simply be a facilitator of general mTORC1-mediated translation but may only be involved in regulating distinct pools of mGluR-induced protein synthesis, some of which may act to inhibit mGluR-LTD. In agreement with this idea, the AKT-TSC pathway has been suggested to act as a brake on mTOR-regulated protein synthesis, while the extracellular-regulated kinase pathway, which is also activated by mGluR stimulation, promotes protein synthesis 44. In this case, AKT1 and AKT3 inhibition may relieve the brake on protein synthesis after mGluR stimulation, leading to altered protein synthesis or AMPA receptor internalization and subsequently enhanced mGluR-LTD. Future studies examining these possibilities will be important to resolve the role of AKT in regulating these important pathways in mGluR-LTD.
In summary, this study provides valuable new insights into the role of AKT and its isoforms in the brain. The results show expression differences between AKT isoforms in the hippocampus, affecting their role in synaptic plasticity and protein synthesis. Hence, compounds targeting single AKT isoforms may be an attractive therapeutic target for treating disorders and diseases that impact synaptic plasticity.
AUTHOR CONTRIBUTIONS
J.L. and C.A.H. conceived the project. J.L. performed immunohistology, electrophysiology and western blot experiments. H.W. performed some electrophysiology experiments. R.A.M. performed some of the western blots. B.N.K. and L.E.L. managed the mouse colony. J.L., H.W. and C.A.H. wrote the manuscript.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as mean values ± SEM. Data were statistically evaluated using SPSS software. The use of parametric tests was determined with the Shapiro-Wilk test for normality and Student's t test or ANOVA were applied. Outliers were excluded using Grubb's method. Repeated measures (RM) ANOVA were used for electrophysiological experiments with time as the within-subjects factor. Significant effects were followed by Tukey's post hoc testing. All statistical tests were two-tailed with p<.05 considered as statistically significant.
ACKNOWLEDGMENTS
For technical assistance, resources, and funding, we thank Thomas F. Franke, Michael Roche, CU Boulder BioFrontiers Microscopy Core, Alzheimer's Association MNIRGDP-12-258900 (C.A.H.), NARSAD 21069 (C.A.H.), NIH R01 NS086933 (CAH), Linda Crnic Institute Seed grant (CAH), NIH F31 NS083277 (H.W.), NIH T32 MH019524 (H.W.), Simons Foundation SFARI 27444 (CAH), and Sie Foundation (J.L.).
REFERENCES




