

Abstract
Mitochondrial inheritance, genome maintenance, and metabolic adaptation all depend on organelle fission by Dynamin-Related Protein 1 (DRP1) and its mitochondrial receptors. DRP1 receptors include the paralogs Mitochondrial Dynamics 49 and 51 (MID49/MID51) and Mitochondrial Fission Factor (MFF), but the mechanisms by which these proteins recruit DRP1 and regulate its activities are unknown. Here we present a cryoEM structure of human, full-length DRP1 bound to MID49 and an analysis of structure- and disease-based mutations. We report that GTP binding allosterically induces a remarkable elongation and rotation of the G-domain, Bundle-Signaling Element (BSE) and connecting hinge loops of DRP1. In this nucleotide-bound conformation, a distributed network of multivalent interactions promotes DRP1 copolymerization into a linear filament with MID49, MID51 or both. Subsequent GTP hydrolysis and exchange within the filament leads to receptor dissociation, shortening through disassembly, and concomitant curling of DRP1 oligomers into closed rings. The dimensions of the closed DRP1 rings are consistent with DRP1-constricted mitochondrial tubules observed in human cells. These structures are the first views of full-length, receptor- and nucleotide-bound dynamin-family GTPases and—in comparison with nucleotide-free crystal structures—teach us how these molecular machines perform mechanical work through nucleotide-driven allostery.
Introduction
Pioneering work in yeast and other model systems revealed that fragmentation of the mitochondrial reticulum disperses units of the organelle during cell division1–3, coordinates morphological adaptation with metabolic demand4–7, and quarantines damaged units for turnover8–10. Recent work also led to the discovery of the role mitochondrial fission plays in regulated cell death pathways11–19, brain development and synaptic function20–23, and how certain pathogens disrupt these processes and hijack mitochondrial resources24–26. Finally, there is a growing understanding of how interorganelle contacts between the ER and mitochondria initiate mitochondrial fission27,28, and how this process regulates mitochondrial genome duplication and integrity29,30. The master regulator that unites these processes across eukaryotic evolution is the membrane-remodeling GTPase DRP14,7,31–37.
DRP1 is necessary but not sufficient for mitochondrial fission because receptor proteins must recruit the enzyme to the Outer Mitochondrial Membrane (OMM). In mammals, these include the paralogs MItochondrial Dynamics proteins MID49 and MID51 or the Mitochondrial Fission Factor, MFF 32,35,38–41. Following receptor-dependent recruitment, DRP1 assembles into polymers that encircle mitochondria and, through still poorly understood mechanisms, channels energy from GTP binding, hydrolysis, and nucleotide exchange into a mechanochemical constriction13,41–46. In addition to DRP1 and its OMM receptors, a recent study revealed that a second member of the dynamin-family of GTPases, dynamin-2, enacts the final fission event downstream of DRPI-driven constriction of a mitochondrial tubule47. Thus, mitochondrial division is a stepwise reaction regulated by DRP1 receptor binding, oligomerization and nucleotide-dependent conformational dynamics.
We and others have reported that the OMM receptors MFF or MID49/51 are independently sufficient to recruit DRP1 in order to divide mitochondria38,39,41,48. We also reported that MID49/51 can coassemble with DRP1 forming a copolymer with a dramatically different morphology than reported for dynamin-family members41. While these results suggest that an adaptor protein could alter the architecture of a dynamin polymer to facilitate a mitochondria-specific activity, the architecture and functions of this coassembly remain unclear.
Here, we report the structural basis of DRP1 coassembly with MID49/MID51. Our cryoEM structure reveals how nucleotide binding to the G-domain induces conformational changes that allosterically propagate through the BSE to open and elongate DRP1 and thereby expose multiple receptor-binding surfaces. MID49/51 binding stabilizes a specific alignment of DRP1 tetramers to nucleate polymerization of a linear co-filament. Then, in a path-dependent reaction, we show how GTP hydrolysis and nucleotide exchange lead to conformational constriction by the polymer. Specifically, when DRP1 subunits within the cofilament exchange and hydrolyze nucleotide, they dissociate from MID49/51 receptors and the entire polymer dynamically shortens and curls into a closed ring. Analysis of structure-based and disease-causing mutations indicate that allosterically driven rearrangements of the stalk helices—and the critical L1NS loop—support curling from linear strings into constricted rings following receptor dissociation.
Results and Discussion
To date, many structural studies of dynamin-family proteins have relied upon mutated or truncated constructs to facilitate crystallization. We purified wild-type, full-length human DRP1 including the N-terminal GTPase domain (G-domain), Bundle Signaling Element (BSE), and four-helix bundle known as the stalk (Fig. 1a). This construct also contained the lipid binding ~100 amino acid region referred to as the variable domain (VD) that sits between the third and fourth alpha helices of the stalk, analogous to the Pleckstrin Homology (PH) domain found in endocytic dynamin proteins. A crystal structure of a nucleotide-free and truncated DRP1 mutant revealed the organization of these domains and an overall similarity with the structure of nucleotide-free endocytic dynamin42,49,50. We also purified soluble truncations of MID49 and MID51 engineered to lack their N-terminal transmembrane anchors but include the cytoplasmic nucleotidyltransferase-like domain and the “Dynamin Recruitment Region” (DRR) required for DRP1 binding (Fig 1a)38,51–54.
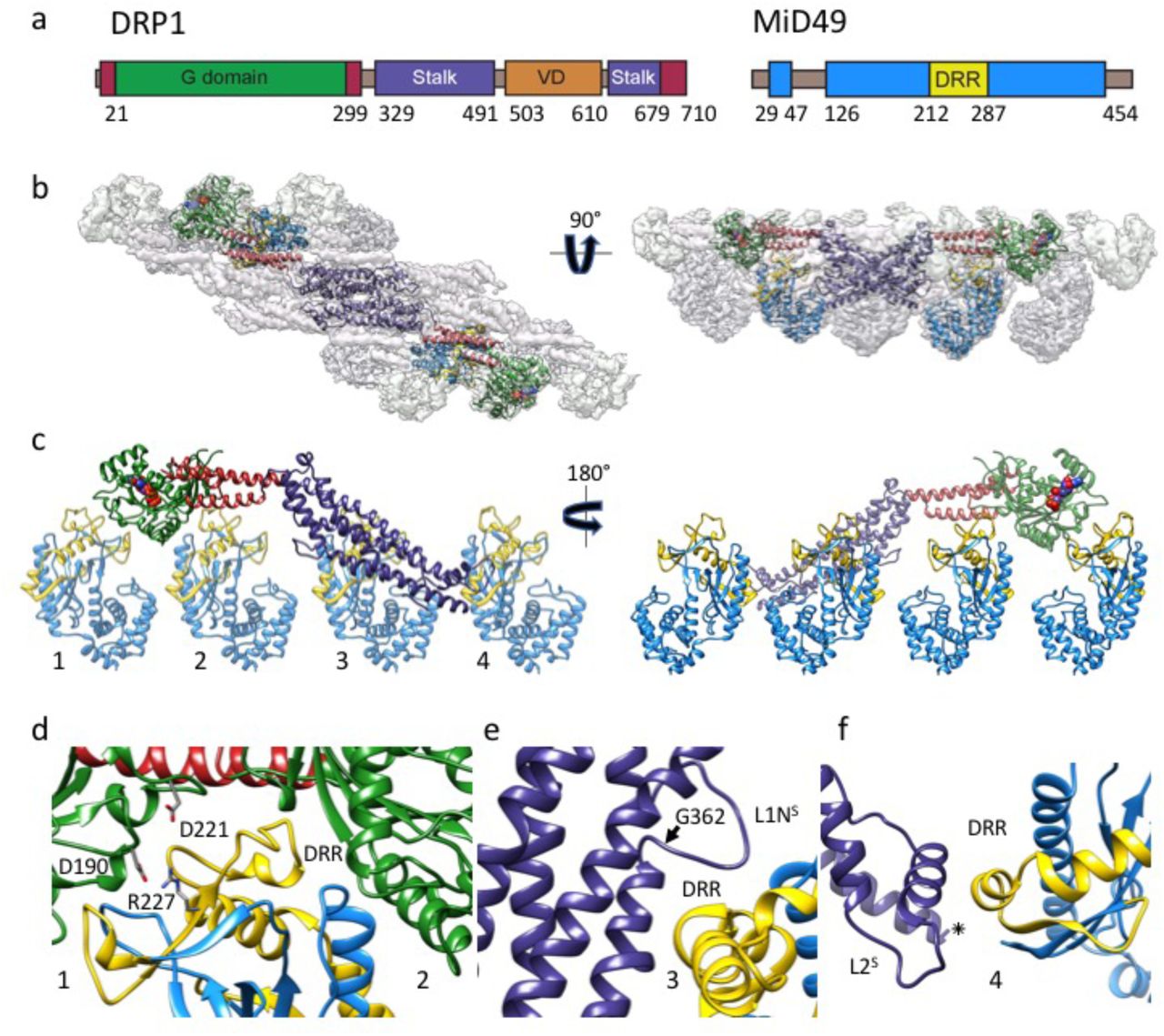
(a) DRP1 and MID49 domain arrangements. (b) Density map and atomic models for DRP1 and MID49126-454. Green: G-domain, Red: Bundle Signaling Element (BSE), Purple: Stalk, Blue: MID49, Yellow: Dynamin Recruitment Region (DRR) of MID49. (c) Each DRP1 chain contacts four different MID49 molecules, as numbered. (d) MID-interaction surfaces 1 and 2. Green ribbons on either side of the DRR come from two separate G-domains, and the residues involved in a key salt bridge for MID-interaction surface 1 are shown as sticks. (e) Rotated view of MID-interaction surface 3. Disease-associated DRP1 residue G362 (arrow) supports the conformation of the L1NS loop essential for linear copolymerization with MID receptors. (f) Rotated view of MID-interaction surface 4 and the L2S loop.
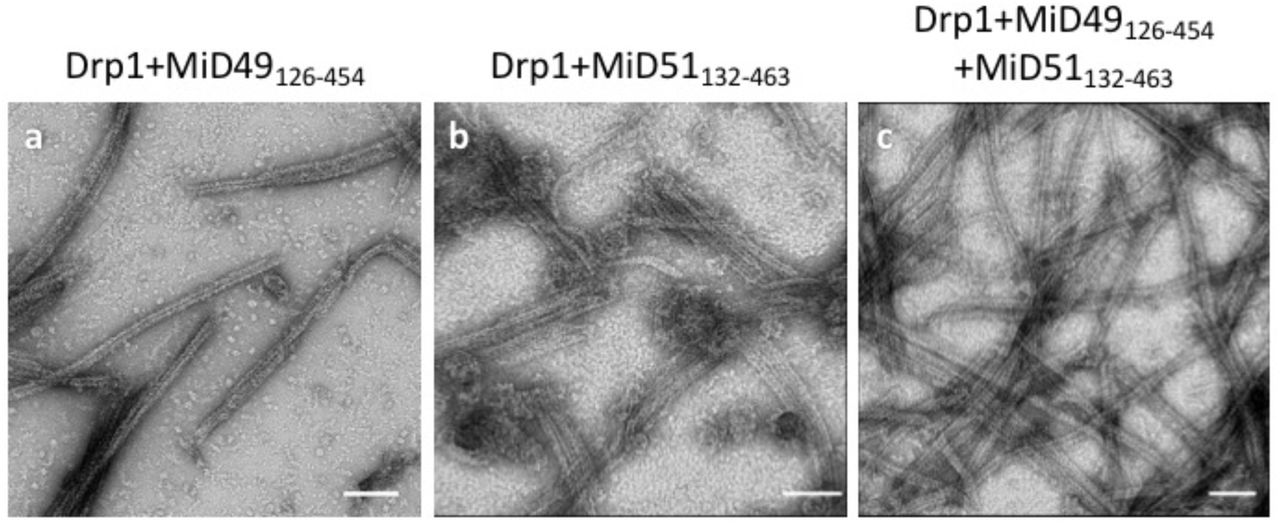
Incubating equimolar ratios of DRP1 with soluble MID49126-454, MID51132-463, or both proteins together, in the presence of GTP resulted in cofilament assembly (Extended Data Figs. 1–2). We focused on the filaments formed with MID49126-454 in the presence of the non-hydrolyzable analog GMPPCP and determined their structure from cryoEM images (Extended Data Figs. 3–9 and Table 1). 3D reconstruction resolved elongated DRP1 dimers bound stoichiometrically to MID49126-454, without assignable density for the variable domain (Fig. 1b, Extended Data Fig. 4). Surprisingly, each chain of DRP1 bound MID49 at four different sites, and each MID49 in turn bound four DRP1 molecules to yield a vast and highly avid interaction network (Figs. 1b-c, Extended Data Figs. 4 and 8). MID49 binding to four separate DRP1 molecules stabilized a linear arrangement of inter-DRP1 interfaces reminiscent of those observed for other dynamin-family proteins by X-ray crystallography (Fig. 1b, Extended Data Figs. 4 and 8)42,49,50,55–57.

DRP1 assembly states visualized with negative stain electron microscopy in the presence of different guanine nucleotides and stoichiometric MID49126-454 ([2μM] for both proteins). Bars = 100nm.

MID49126-454 and MID51132-463 form indistinguishable assemblies with DRP1 by negative stain electron microscopy. (a) DRP1 plus MID49126-454 and GMPPCP; (b) DRP1 plus MID51132-463 and GMPPCP, (c) DRP1 plus both MID49 and MID51. Bars = 100nm.

CryoEM imaging and reconstruction. (a) An electron cryo-micrograph of DRP1-MID49126-454 filaments formed with GMPPCP. Inset shows a representative 2D class average. Bar =100nm, Inset bar =10nm. (b) Oblique cross-section of the 3D reconstruction and the distribution of views determined during helical reconstruction. DRP1 density is rendered in grey, MID49 in golden yellow.

Raw particle numbers and workflow for the reconstruction protocol and imposition of symmetries. DRP1 density is rendered in grey, MID49 in golden yellow.

Local resolution estimates computed by Resmap25. (a) Histogram of voxels values, and (b) Heat map of local resolution estimates displayed for a cross-section through the reconstruction.

Fourier Shell Correlation plots for (a) the half-maps with and without imposed symmetry; and (b) model-to-map correlations for the averaged as well as each subregion of the structure using the atomic coordinates and B-factors determined using Rosetta.

Modeling of human MID49. (a) Sequence alignment between human and mouse MID49 sequences. (b) Overlay of the homology model of human MID49126-454 (blue, with yellow DRR, ribbon) modeled within the cryoEM density versus the mouse MID49 crystal structure (PDB ID: 4WOY, grey ribbon)6.

Rosetta refinement. (a) Symmetric unit of the filament with 8 chains of DRP1 and 8 chains of MID49 refined using Rosetta to enforce symmetry and account for all possible inter-molecular interfaces. (b) Atomic B-factors for the DRP1 and MID49 models (ribbon) and bound GMPPCP (space filling).
MID49’s DRR motif occupied the space between two neighboring G-domains and contacted both via MID-interaction interfaces 1 and 2 (buried surface areas of ~530Å2 and ~200 Å2, respectively, see Fig. 1b-d). The precise spacing required for this bivalent G-domain interaction explains why previous mutagenesis efforts suggested that the size and topology of the β4–α4 loop, rather than its exact sequence (which differs between MID49 and MID51) are critical determinants of binding. Single point mutations within this loop for MID49 and MID51 did not disrupt binding, while mutations that altered the length, topology or positioning of the loop did51,52,54. We found that mutating conserved DRP1 residues involved in interface 1—the energetically most significant interface—did prevent coassembly. Specifically, the D190A mutation should neutralize an interface 1 salt bridge and the D221A mutation should alter the conformation of an interface 1 loop (Fig. 1d). We observed that both mutants prevented assembly with MID49 and altered DRP1’s self-assembly properties (Extended Data Figs. 10–12).

Examples of model fitting to B-factor sharpened density for (a) a helix from the DRP1 stalk, (b) the backbone of the L1NS loop, and (c) a helix and loop from MID49.

DRP1 assembly and coassembly reactions with GMPPCP for (a) DRP1D190A alone, (b) DRP1D190A+MID49, (c) DRP1D221A alone, and (d) DRP1D221A+MID49. Bars = 100nm.
Unexpectedly, MID49 also made contact with the stalk loops of a third and fourth DRP1 molecule through MID-interaction interfaces 3 and 4 (buried surface areas of ~450Å2 and ~230Å2, respectively, Fig. 1c, e, and f). The loops involved in both interaction interfaces 3 and 4 determine the assembly properties of higher-order dynamin-family oligomers 42,49,50,55–57. MID-interaction surface 3, in particular, harbors the conserved loop L1NS and is the site of multiple disease alleles that lead to elongated mitochondrial morphology, including G362D and G363D (Fig. 1e, Extended Data Figs. 10–11 and 58–60). Prior work has also established that this loop comprises part of the intra-molecular PH domain binding site for the soluble state of endocytic dynamin tetramers (Extended Data Fig. 13)57, and is a determinant of conformational heterogeneity for these and other dynamin-family proteins55,57,61. The presence of disease alleles near this interface suggests that these mutations may compromise receptor interactions and that defects in the recruitment of DRP1 to mitochondria may contribute to pathogenesis. As discussed below, we found that the G362D mutant (Fig. 1e) failed to coassemble with MID49 and displayed altered assembly and conformational properties (Extended Data Figs. 12–15).

Multiple sequence alignment of the regions near and including the DRP1 residues mutated in this study: D190, D221 and G362. The residue numbers apply to human DRP1, isoform 2 (UNIPROT identifier: O00429-3 also known as DLP1a).

Size exclusion chromatography traces for DRP1 wild-type and mutants used in the study. (a) Comparison between wild type (WT) versus DRP1G362D, (b) WT versus DRP1D221A, and (c) WT versus DRP1D190A. (d) Standards for molecular weight comparison.
Understanding the allosteric coupling between nucleotide binding, hydrolysis or exchange and the conformational repertoire of dynamin-family GTPases remains an unmet challenge62,63. We observed that the GMPPCP-bound G-domains and the BSE of DRP1 adopt strikingly different conformations in the cryoEM density compared to the nucleotide-free crystal structure42. In addition to other nucleotide-induced conformational changes within the G-domain, the most salient are the closing of the G2/switch-1 loop to form a closed “lid” over the nucleotide (Figs. 2a-c). The closure of the switch-1 lid propagates through the adjacent beta sheet to push the α-helices of the BSE into an orthogonal position (Extended Data Movie 1). When evaluated in the context of a stalk interface-2 DRP1 dimer, this conformational change is an impressive 90° rotation of the G-domain and a 40Å translation toward the stalk (Fig. 2d, Extended Data Movies 2-3). Two of the three DRP1 surfaces that engage the DRR of MID49 (MID receptor interaction interfaces 1 and 2, Figs. 1–2) are inaccessible in the nucleotide-free state but become available for binding upon nucleotide-driven elongation (Fig. 2d, Extended Data Movies 2-3).

(a) nucleotide-free state of DRP1 G-domain and BSE as seen in a crystal structure (PDB ID:4BEJ). Arrow points to the G2/switch1 loop. (b) GMPPCP-bound G-domain-BSE conformation determined by cryoEM. (c) Overlay of A and B. Curved arrows highlight the closing of G2/switch 1 “lid” and the opening of the BSE “wrist”. (d) Global conformational change induced by nucleotide binding. Rotation and translation of the G-domain and BSE elongates the dimer and exposes MID-interaction surfaces 1 and 2 (annotated on separate monomers for clarity). The surfaces of the G-domains that engage MID receptors are rendered orange in the nucleotide-bound and elongated conformation.
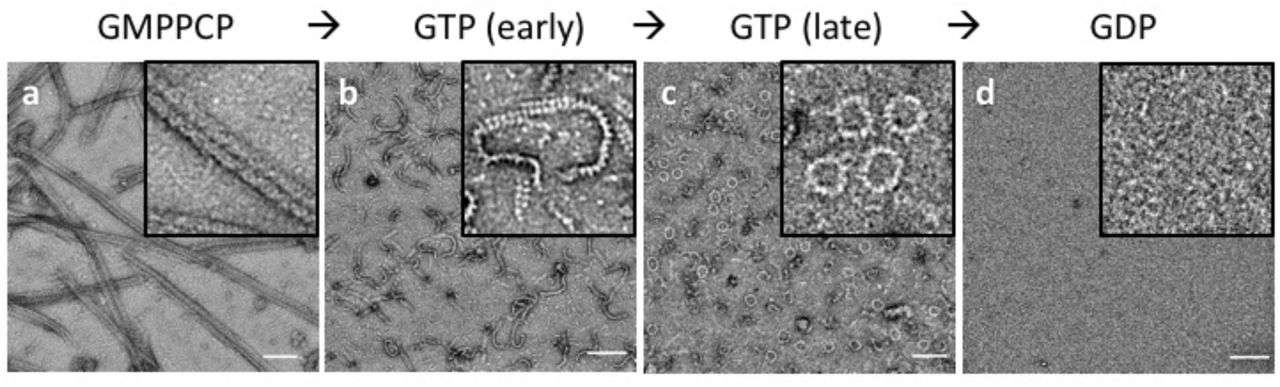
Since DRP1’s ability to polymerize into linear filaments with MID49 or MID51 depended on non-hydrolyzable GTP analogs, we next evaluated the dynamics of these filaments in the presence of hydrolyzable GTP. Following copolymerization in the presence of the non-hydrolyzable analog, we exchanged GMPPCP for GTP through dialysis and followed the reaction using negative stain transmission electron microscopy at sequential time points until the GTP was exhausted. We observed that the linear DRP1-MID49 cofilaments disassembled into small oligomers upon complete hydrolysis to GDP and exhibited a fascinating dynamic instability at intermediate time points (Fig. 3). Specifically, the long and linear filaments seen at early time points disassembled into shorter, curling oligomers that—upon reaching a reproducibly narrow range of lengths—spontaneously closed into constricted rings that were remarkably uniform in diameter (Fig. 3).
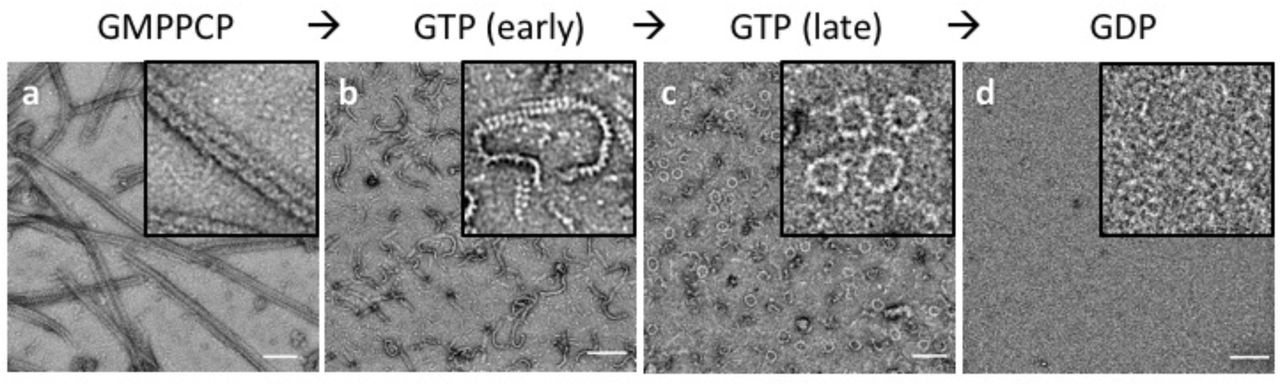
(a) DRP1-MID49126-454 linear filaments copolymerized with GMPPCP. (b-c) Subsequent exchange into GTP leads to partial disassembly and curling into closed rings. (d) GTP exhaustion leads to complete oligomer disassembly. Bars=100nm.
In a separate but related experiment, we evaluated the assembly properties of the DRP1 mutant G362D with and without MID49126-454. As described above, this disease-associated residue sits at the base of the L1NS loop that forms part of the third interface with MID49 (Figs. 1c, 1e, Extended Data Fig. 13). We found that DRP1G362D purified as a nearly monodisperse and stable dimer, rather than a mix of tetramers and higher order species observed for the wild-type, full-length protein (Extended Data Fig. 12). In addition, DRP1G362D exclusively formed rings, not filaments, with or without MID49126-454 and in the presence of GMPPCP or GTP (Fig. 4a, Extended Data Figs. 14–15). These rings resembled those observed with wild-type DRP1 in all respects except that the wild-type protein only formed closed rings from the pre-formed linear MID49 copolymers through the path-dependent reaction described above (Fig. 3 versus Fig. 4). Using the non-hydrolyzable GTP analog GMPPCP with the DRP1G362D rings also improved structural homogeneity, presumably because the wild-type rings remain dynamic and eventually disassembled in the presence of GTP upon complete hydrolysis to GDP (Fig. 3).
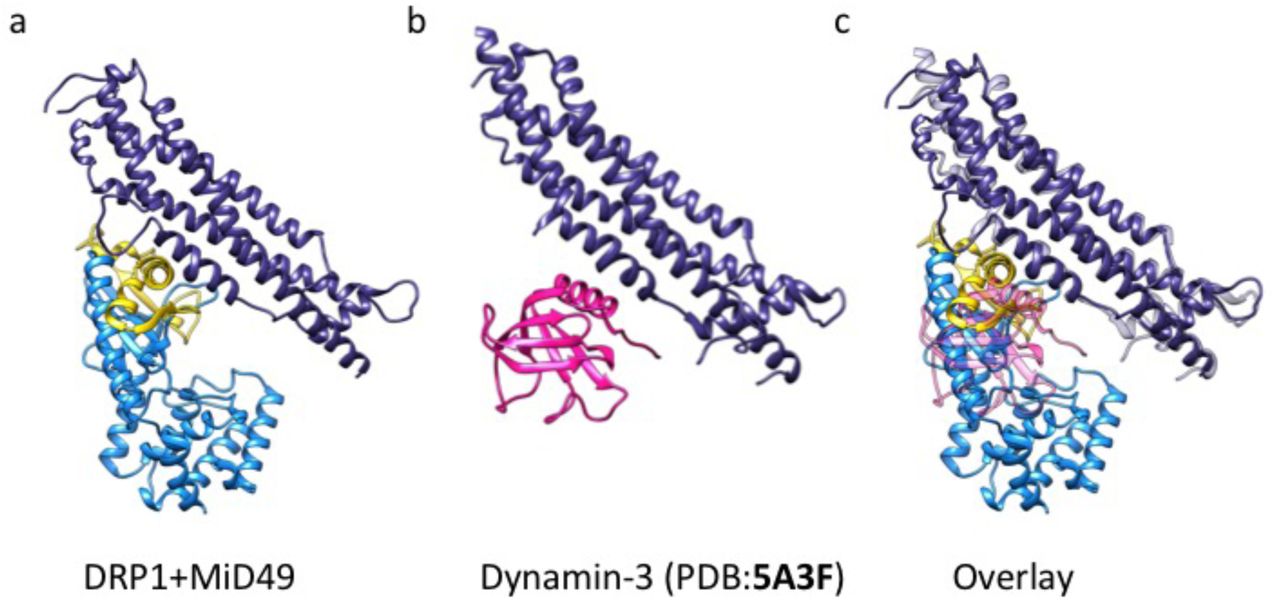
Structural similarities between the third DRP1-MID49 interaction interface that includes the L1NS loop and the interaction between the Pleckstrin Homology (PH) domain and the stalk of Dynamin-3 (PDB ID:5A3F)26. (a) DRP1-MID49 interaction at MIDinteraction interface 3, (b) the PH domain bound to the stalk of Dynamin-3, and (c) Overlay of a and b.

DRP1G362D assembly and coassembly reactions with GMPPCP or GTP. DRP1G362D forms rings but not linear filaments without MID49 (a,c); and with MID49 present (b,d); with GMPPCP (a-b); or with GTP (c-d). Bars = 100 nm.

DRP1G362D forms 12-dimer closed rings. (a) 2D class average of the rings; (b) 2D class average of infrequent, orthogonal or “side” views used as a constraint during model building; (c) “top” and (d) “side” projections of the model; (e) “top” and (f) “side” views of the final model rendered as ribbons. Bars =100Å. Green: G-domain, Red: Bundle Signaling Element (BSE), Purple: Stalk region.

(a) CryoEM micrograph of DRP1G362D rings. (b) 2D class average of the predominant closed ring that comprises 12 DRP1 dimers. (c) 2D class average of a quarter of the ring revealing the secondary structure elements of the “X”-shaped DRP1 dimer. (d) 3D model of the closed ring. (e) Comparison between DRP1 tetramers observed in the nucleotide-free state (top, PDB ID:4BEJ), the GMPPCP and MID49126-454-bound linear state (middle), and the bent conformation modeled for the rings. Bar for (a) = 30nm, for (b, c) =100Å.
We imaged the DRP1G362D rings using cryoEM and used 2D class averages of the predominant 12-dimer closed ring to model the architectural differences between the linear filaments and the closed rings (Fig. 4, Extended Data Fig. 14–15). To account for the projected ring density, the G-domain and the BSE of DRP1 must move even further down toward the stalks. Moreover, while stalk interface-2 remains constant—as revealed by the X-shaped projected dimer (Fig. 4c)—the curvature of the ring dictates that stalk interfaces 1 and 3 must be extensively remodeled to allow a ~30 degree bending per dimer in comparison with the linear DRP1-MID49 copolymer (Fig. 4d, Extended Data Fig. 15, Movie 4). We did not observe any density for MID49 in the wild-type rings that form by curling of the MID49-DRP1 cofilament in the presence of GTP, nor in our higher-resolution analysis of the DRP1G362D rings that form with or without MID49 present (Fig. 4b-c, Extended Data Fig. 14–15).
We note that even the most constricted form of the closed ring is insufficient to drive complete mitochondrial fission because the inner diameter is only ~16nm. This length suggests that a constricted membrane tubule would be stable and that the structures we have observed in vitro could correspond with the highly-constricted but pre-fission state observed in living cells when another dynamin-family protein, the primarily endocytic dynamin-2, is depleted47. Thus, initial constriction by DRP1 may stabilize the high degree of membrane curvature that is suitable, perhaps even tuned, for the recruitment and final fission event catalyzed by additional dynamin-family enzymes.
Together, these findings establish four conceptual advances. First, our cryoEM structure revealed how receptor proteins like MID49/MID51 recruit and stabilize a specific nucleotide-bound conformation of DRP1 and nucleate polymerization of a cofilament. We speculate that the nearly linear properties of this polymer have adapted to encircle low-curvature mitochondrial tubules. The selective stabilization of this open, elongated conformation of DRP1 within the MID receptor cofilament explains why overexpression of the MID receptors inhibits mitochondrial fission39. Second, analysis of the MID49-DRP1 copolymer exposed how nucleotide binding induces an impressive conformational rearrangement to expose a network of multi-valent receptor binding sites. We now understand these nucleotide-driven allosteric transformations in detail and in the context of full-length and oligomeric DRP1. Third, a path-dependent constriction reaction revealed intrinsic GTP-dependent DRP1 properties that are reminiscent of microtubule dynamic instability64. In this reaction, nucleotide exchange and hydrolysis led to MID49/51 receptor dissociation, disassembly from the ends of the linear filament, and concomitant curling of the shortened filaments into closed rings. Fourth and finally, analysis of a disease mutant in the L1NS loop, DRP1G362D, exposed this loop as a vital determinant of mitochondrial receptor binding and the dynamic inter-stalk interactions that govern oligomer architecture and the ability of dynamin proteins to perform mechanical work.
Author Contributions
R.K., J.S. and A.F. conceived of the study. R.K., R.W., A.Y. and P.T. performed all experiments. R.K., R.W. and A.F. performed the computational analyses. All authors evaluated the results and edited the manuscript. R.K. and A.F. wrote the manuscript with input from all of the authors.
Data Accessibility
All of the cryoEM density maps associated with this study have be deposited in the EMDB with accession numbers EMD-8874(PROC). The atomic coordinates have been deposited in the PDB as 5WP9(PROC).
Extended data movie Legends
Extended data movie 1: Nucleotide-induced conformational changes in the G-domain and Bundle Signaling Element (BSE).
Extended data movie 2: Nucleotide-induced conformational changes in the G-domain and Bundle Signaling Element (BSE) in the context of a full-length DRP1 dimer (“side” view).
Extended data movie 3: Nucleotide-induced conformational changes in the G-domain and Bundle Signaling Element (BSE) in the context of a full-length DRP1 dimer (“top” view).
Extended data movie 4: MID receptors engage a specific nucleotide-bound conformation of DRP1 tetramers and promote formation of a linear copolymer. Subsequent nucleotide hydrolysis and exchange promotes receptor dissociation and tetramer bending in the closed rings.
1 Overall quality at a glance ⓘ
The following experimental techniques were used to determine the structure:
ELECTRON MICROSCOPY
The reported resolution of this entry is unknown.
Percentile scores (ranging between 0-100) for global validation metrics of the entry are shown in the following graphic. The table shows the number of entries on which the scores are based.

The table below summarises the geometric issues observed across the polymeric chains. The red, orange, yellow and green segments on the bar indicate the fraction of residues that contain outliers for >=3, 2, 1 and 0 types of geometric quality criteria. A grey segment represents the fraction of residues that are not modelled. The numeric value for each fraction is indicated below the corresponding segment, with a dot representing fractions <=5%
2 Entry composition ⓘ
There are 2 unique types of molecules in this entry. The entry contains 116960 atoms, of which 59040 are hydrogens and 0 are deuteriums.
In the tables below, the AltConf column contains the number of residues with at least one atom in alternate conformation and the Trace column contains the number of residues modelled with at most 2 atoms.
3 Residue-property plots ⓘ
These plots are drawn for all protein, RNA and DNA chains in the entry. The first graphic for a chain summarises the proportions of the various outlier classes displayed in the second graphic. The second graphic shows the sequence view annotated by issues in geometry. Residues are colorcoded according to the number of geometric quality criteria for which they contain at least one outlier: green = 0, yellow = 1, orange = 2 and red = 3 or more. Stretches of 2 or more consecutive residues without any outlier are shown as a green connector. Residues present in the sample, but not in the model, are shown in grey.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 Experimental information ⓘ
5 Model quality ⓘ
5.1 Standard geometry ⓘ
Bond lengths and bond angles in the following residue types are not validated in this section: GCP, MG
The Z score for a bond length (or angle) is the number of standard deviations the observed value is removed from the expected value. A bond length (or angle) with |Z| > 5 is considered an outlier worth inspection. RMSZ is the root-mean-square of all Z scores of the bond lengths (or angles).
There are no bond length outliers.
There are no bond angle outliers.
There are no chirality outliers.
There are no planarity outliers.
5.2 Too-close contacts ⓘ
In the following table, the Non-H and H(model) columns list the number of non-hydrogen atoms and hydrogen atoms in the chain respectively. The H(added) column lists the number of hydrogen atoms added and optimized by MolProbity. The Clashes column lists the number of clashes within the asymmetric unit, whereas Symm-Clashes lists symmetry related clashes.
The all-atom clashscore is defined as the number of clashes found per 1000 atoms (including hydrogen atoms). The all-atom clashscore for this structure is 2.
All (249) close contacts within the same asymmetric unit are listed below, sorted by their clash magnitude.
There are no symmetry-related clashes.
5.3 Torsion angles ⓘ
5.3.1 Protein backbone ⓘ
In the following table, the Percentiles column shows the percent Ramachandran outliers of the chain as a percentile score with respect to all PDB entries followed by that with respect to all EM entries.
The Analysed column shows the number of residues for which the backbone conformation was analysed, and the total number of residues.
All (96) Ramaehandran outliers are listed below:
5.3.2 Protein sidechains ⓘ
In the following table, the Percentiles column shows the percent sidechain outliers of the chain as a percentile score with respect to all PDB entries followed by that with respect to all EM entries.
The Analysed column shows the number of residues for which the sidechain conformation was analysed, and the total number of residues.
All (24) residues with a non-rotamerie sideehain are listed below:
Some sidechains can be flipped to improve hydrogen bonding and reduce clashes. There are no such sidechains identified.
5.3.3 RNA ⓘ
There are no RNA molecules in this entry.
5.4 Non-standard residues in protein, DNA, RNA chains ⓘ
There are no non-standard protein/DNA/RNA residues in this entry.
5.5 Carbohydrates ⓘ
There are no carbohydrates in this entry.
5.6 Ligand geometry ⓘ
There are no ligands in this entry.
5.7 Other polymers ⓘ
There are no such residues in this entry.
5.8 Polymer linkage issues ⓘ
The following chains have linkage breaks:
All chain breaks are listed below:
Acknowledgments
We thank Michael Braunfeld, Cameron Kennedy, David Bulkley, and Alexander Myasnikov and the UCSF Center for Advanced CryoEM, which is supported in part from NIH grants S10OD020054 and 1S10OD021741 and the Howard Hughes Medical Institute. We also thank the QB3 shared cluster and NIH grant 1S10OD021596-01, Jean-Paul Armache, Nathaniel Talledge for microscopy advice, and Charles Greenberg for consulting on structural modeling. This work was further supported by a Faculty Scholar grant from the Howard Hughes Medical Institute (A.F.), the Searle Scholars Program (A.F.), NIH grant 1DP2GM110772-01 (A.F.), NIH grants GM53466 and GM84970 (J.S.), and the Howard Hughes Medical Institute (R.W., J.S., D.A.). A.F. is a Chan Zuckerberg Biohub investigator.
References
Extended data References




