

Abstract
All of the drug trials of the Alzheimer’s disease (AD) have failed to slow progression of dementia in phase III studies, and the most effective therapeutic approach still remains controversial due to our incomplete understanding of AD pathophysiology. Amyloid beta (Aβ) and its cascade have been the primary focus of drug design efforts for more than a decade. However, mounting evidence indicates that mechanisms of AD etiopathogenesis are probably more complex than the previous reductionist models.
Several genome-wide association studies (GWAS) have recently shed light on dark aspects of AD from a hypothesis-free point of view. While the newly-identified AD risk genes rather raise more questions than they answer in deciphering the amyloid cascade, as a potentially overlooked finding, many of them code for receptors and transducers of cell adhesion signaling cascades. Remarkably, the hallmark genetic factors of AD, including the amyloid precursor protein (APP), presenilins (PSEN) and APOE also take part in highly similar pathways of cell adhesion regulation and coordinate contact-guidance of neuronal growth cones in brain development, albeit these Aβ-independent roles remain highly underexplored.
Here, we have revisited function of 27 AD risk genes in pathways of normal cell physiology. Our review clearly shows that a disrupted cell adhesion signaling nexus, rather than a protein aggregation process, is the central point of convergence in the unbiased genetic risk factors of AD. To further elucidate a potential relationship between aging and pathways of cell adhesion, we have conducted an exploratory bioinformatics analysis which revealed that cell adhesion is the most representative ontology of human genes larger than 500kb (p=8.0×10‒13), and these extremely large genes are mostly expressed in brain (p=2.1×10‒17) and selectively take part in synaptic composition (p=2.4×10‒14). As possible driving forces of brain evolution, large genes may coordinate complex wiring of synaptic circuits in neurodevelopment, and we suggest that they may also be vulnerable to the impact of somatic mutations in aging due to their exceptional sizes which will be assessed by statistical models. An exemplar of this notion is the giant APOE receptor Lrplb which is one of the most frequently deleted genes in various cancers and also represents the only brain-specific lipoprotein receptor. Our model, the large gene instability hypothesis, highlights alternate strategies for AD prevention, biomarker discovery and therapeutic design based on targeting genomic instability and synaptic adhesion.
Abbreviations Alzheimer’s disease (AD), Amyloid precursor protein (APP), Genome-wide association study (GWAS), Focal adhesion kinase (FAK), Postsynaptic density (PSD), Presenilin (PSEN), Src family kinase (SFK)
Introduction
More than a century has passed since the first report of a presenile dementia case by Alois Alzheimer1, and current molecular understanding of AD mostly borrows from identification of the Aβ peptide as the main constituent of senile plaques and subsequent discovery of APP and PSEN mutations in rare cases of familial AD2,3. These observations were compiled to the amyloid cascade hypothesis in the pre-genomic era, a theory which implicates Aβ species, proteinous aggregates and neurofibrillary tangles as the driving force of dementia with broad influence as the central model of AD etiopathogenesis4.
Nevertheless, due to various methodological difficulties, Aβ species has hardly been validated as a causal force of neurodegeneration in AD patients. Despite receiving support from preclinical studies, manipulating the pathways of Aβ generation and clearance has also yielded disappointing results in several clinical trials so far5. While a handful of clinical failures do not necessarily disprove a theory per se, overemphasis on a single disease model is a dangerous gamble and could be one of the many explanations for the lack of progress in AD therapeutic design6. Accuracy of the amyloid cascade hypothesis is a topic of ongoing debate, and it goes without saying that this theory may be rejected in future7-12, a critical possibility warranting development of alternate disease models for interpreting exploratory evidence including recent high-throughput genomic findings.
In contrast to the classical hallmarks of AD including senile plaques and neurofibrillary tangles that are still of questionable etiological significance13, genetic risk factors temporally precede earliest stages of brain development, aging, and neurodegeneration, and are expected to inform on causal disease pathways. The genetic architecture of common sporadic AD is highly complex, and a number of susceptibility loci have been identified by genome-wide association studies (GWAS) in large elderly populations. These novel genetic factors provide unbiased insight into molecular and cellular mechanisms of AD14-19 but their mechanistic interpretation has been under powerful influence of the amyloid cascade theory so far.
This report servers to provide an evidence-based framework for compiling pathways of AD predisposition from an Aβ-independent point of view. The rest of this manuscript is organized as follows; in the first section, we aim to revisit the role of AD risk genes in pathways of normal cell physiology. We show that 27 disease-related genes strongly converge to common pathways of cell-extracellular adhesion signaling and brain circuit development. In the second section, we will try to explain interaction of aging as the strongest risk factor of senile dementias with the genetic landscape of AD. Finally, several testable predictions are provided for assessment of our new disease model.
1.1 The APP family proteins are highly-conserved cell adhesion molecules
Derailed catabolism of the APP protein and generation of an aggregation-prone Aβ species abstract a significant proportion of molecular investigations in AD, leading to efforts to block this cascade by means of Aβ immunotherapies or design of secretase inhibitors5. In contrast, three decades after successful cloning of the APP gene20 the physiological role of its protein product remains less investigated and may be the real key to understanding disease mechanisms.
APP codes for a transmembrane protein and is highly expressed in the developing brain at the site of neuronal growth cones, structures that form motile tips of outgrowing axons and dendrites21. The Aβ peptide enhances interaction of developing neurites with extracellular adhesion molecules and promotes outgrowth of neuronal projections22,23. The full-length and membrane-tethered form of APP also interacts with extracellular laminin, heparan sulfate, fibronectin and collagen24-26, molecules which form the backbone of extra-cellular matrix and moderate contact-guidance of growth cones in synaptic circuit formation.
Interaction of APP with heparan sulfate27 and laminin24 stimulates assembly of hippocampal connections and promote neurite outgrowth28. In the other hand, antisense-downregulation of APP inhibits extension of axons and dendrites29. APP demonstrates a dose effect in growth cone guidance30 and its increased dosage in Down syndrome results in emergence of faster advancing growth cones with promoted adhesive properties and larger sizes31. In contrast, knockdown of the APP gene in zebrafish disrupts outgrowth of developing neurites32. Intriguingly, although wild-type human APP can rescue this defective phenotype, the mutated APP of familial AD fails to substitute for normal function of animal gene32.
Several cellular pathways are speculated to mediate the neurite-promoting effects of APP in neurodevelopment. The netrin pathway of neurite guidance incorporates APP as a co-receptor33, and inactivation of APP disrupts netrin signaling and diminishes axonal outgrowth34. APP also binds reelin, which is a large extracellular adhesion molecule for guidance and migration of neurons35. In this context, interaction of reelin with APP promotes outgrowth of hippocampal neurites35. Functional interaction of APP and reelin requires presence of a third cell adhesion molecule, α3β1-integrin, as well35. Integrins are the main component of focal adhesions and known to co-localize with the APP protein36,37 at dynamic neuronal adhesion sites38. Intriguingly, integrin modulates neurite outgrowth by interacting with APP39. Similarly, integrin also acts as an accessory reelin receptor for cell adhesion regulation and neuronal migration40-42. Therefore, APP, integrin, reelin (and its counterpart APOE receptors) may take part in surface adhesion ligand/receptor complexes for transduction of coherent signals.
In addition to growth cone navigating, APP also moderates spatial migration of neurons in neurodevelopment43. Triple-knockout of the APP family genes results in neuronal migration defects similar to human lissencephaly44 and two candidate ligands for APP, including pancortin and lingo1, orchestrate migration of neural precursor cells45-47. From a cellular and molecular point of view, pathways of growth cone navigation and cell migration are highly similar, as both of these events rely on specialized membrane protrusions, namely filopodia and lamellipodia for cell reshaping and anchorage. These plastic cell organizations sense the directional gradients of extracellular contact-guidance cues by means of surface adhesion receptors including integrins. Intracellularly, filopodial adhesion receptors affect rearrangement of the actin cytoskeleton for changing cell polarity and recycling focal adhesion turnover towards the protruding end and powering cell movement48.
Mounting evidence indicates that the cytoskeletal system is an important point of convergence in signaling through the APP protein. Transmembrane APP is selectively localized to the cytoskeletal-rich regions of neuronal growth cones at dynamic adhesion sites38,49, and the APP intracellular domain (AICD) which is released after γ-secretase-mediated cleavage affects rearrangement of the cellular actin cytoskeleton50. The AICD cleavage fragment of APP interacts with a number of intracellular signal transducers, including Fe65, Tip60, KAI1, DISC1, Dab1, X11, and Grb2 that have been identified to date51-53. Intriguingly, all of these transducers influence pathways of cytoskeletal rearrangement and affect cell movement:
Fe65 and Tip60 affect the cytoskeletal system and moderate cancer cell migration54.
KAI1 suppresses cancer cell migration by affecting cytoskeletal assembly55,56.
DISC1 coordinates remodeling of the actin cytoskeleton in migrating neurons and growth cone-like protrusions57. This protein rescues neuronal migration defects caused by loss of APP51.
Dab1 is a mandatory adaptor of the APOE receptors in the reelin pathway and controls cytoskeletal remodeling in neuronal migration58.
X11 is a recently-discovered modulator of the reelin pathway and affects cell movement59.
Grb2 is an adaptor molecule that links various receptors including integrin with intracellular pathways of cytoskeletal plasticity and regulates cancer cell migration60,61.
In accordance with the common function of these putative signal transducers, The APP family genes similarly affect migration and invasion of various cancer cells by affecting the cytoskeletal pathway62,63. Interestingly, the key cytoskeletal regulator Rac1 controls expression of the APP gene by a feedback-like mechanism in primary hippocampal neurons64.
Since the early stages of nervous system evolution, the APP paralogue of Drosophila (APPL) has promoted neuronal migration65. Phylogenetic evolution of the APP family genes reveals that cell adhesion is the most consistent biological function of this family66. From an evolutionary point of view, the cytoplasmic tail of APP is probably of utmost importance, as it comprises a super-conserved NPxY amino acid motif in the form of 682YENPTY687 that has remained unchanged from roundworms to humans for more than 900 million years of evolution67. This consensus motif is known to mediate endocytic sorting of receptors and, perhaps more importantly, their interaction with tyrosine-phosphorylated intracellular signal transducers68. Two intracellular adaptors of APP with established signaling roles, including Dab1 and Fe65, interact with this APP motif in a phosphorylation-dependent manner69,70. The 682Tyr residue of this APP motif undergoes phosphorylation and is essential to induction of synaptogenesis71. Of note, the consensus NPxY motif is also present in the cytoplasmic tail of all APOE receptors and activates the mandatory Dab1 adaptor of reelin pathway72.
In addition to playing physiological roles in neurodevelopment, the APP protein is evidenced to maintain its function in mature neurons. Mouse hippocampal neurons express the APP protein under physiological conditions73, and APP is present in close association with NMDA glutamate receptors that are central to memory-formation. In this context, APP maintains NMDA receptors at postsynaptic membrane and promotes neurotransmission74,75. Through its conserved NPxY motif, APP also interacts with the postsynaptic scaffold protein AIDA-176, which is a protein for regulating hippocampal synaptic transmission and palsticity77. Loss of the APP family genes disrupts synaptic function78, memory formation79, and causes an aging-related synaptic loss in mice80,81. Further supporting physiological roles in synaptic adhesion, the three APP family members (APP, APLP1, APLP2) form trans-synaptic adhesive dimers82, and cleavage of the APP protein changes synaptic adhesion and assembly83. Lastly, APP mutations are shown to disrupt this regulatory effect84. An exhaustive review of the APP protein in pathways of normal cell physiology is beyond the scope of this manuscript and the interested reader is referred to recent publications85-87.
1.2 The γ-secretase complex is a membrane-tethered enzyme for signaling of cell adhesion receptors
PSEN1 and PSEN2 genes code for catalytic subunits of the transmembrane γ-secretase enzyme and various mutations in these genes cause autosomal-dominant AD. Since cleavage of the APP protein by γ-secretase is a mandatory step for Aβ generation, accelerated catabolism of APP in the amyloidogenic pathway is considered the mechanism of AD development in individuals with mutated PSEN genes, a hypothesis which has been extrapolated to common sporadic AD as well.
Unexpectedly, PSEN mutations of familial AD were recently found to cause an almost complete loss of γ-secretase function88 and reduce generation of the putatively-neurotoxic Aβ40, Aβ42 and Aβ43 species occasionally to undetectable levels89,90. In further contradiction, knock-in mice harboring the mutated PSEN1 gene of familial AD are phenotypically similar to knockout strains which lack γ-secretase function, and both of these strains demonstrate impaired hippocampal plasticity91. This line of evidence suggests a loss-of-function mechanism for PSEN mutations of familial AD, and potentially explain the paradoxical worsening of cognitive function and accelerated brain atrophy in γ-secretase inhibitor trials of AD92,93.
In contrast to the narrow focus on derailed pathways of APP catabolism, unbiased proteomic profiling has revealed that the γ-secretase complex has a broad spectrum of substrate specificity for cell surface receptors with signaling roles94,95. For instance, the γ-secretase enzyme cleaves the APOE/reelin receptors96, as well as DSG2, TREM2 and ephrin receptors which are all coded by AD risk genes94,97,98. A candidate gene of familial AD, Notch399, is mandatorily cleaved by γ-secretase prior to signaling100. Loss of γ-secretase results in erroneous axonal pathfinding due to derailed netrin signaling101, and has also been shown to disrupt cell adhesion force generation102. While it is difficult to pinpoint a particular signaling path that mediates detrimental effects of γ-secretase dysfunction in familial AD, it is tempting to speculate pathways of synaptic adhesion and contact-guidance in its etiopathogenesis.
Recent nanoscale microscopy reveals that expression of the γ-secretase complex is selectively enriched in postsynaptic compartments during normal synaptic maturation103. A neurophysiological role for γ-secretase is further supported by observing that this enzyme interacts with a number of synaptic adhesion molecules including δ-catenin and N-cadherin, as well as glutamate receptors103,104. Cleavage activity of γ-secretase modulates synaptic transmission and adhesive properties104 and this neuromodulatory effect is disrupted by familial AD mutations105.
1.3 The APOE/Lipoprotein receptor pathway coordinates contact-guidance of neurites
APOE4 is the strongest genetic risk factor of sporadic AD and explains ~6 percent of the risk of disease development106. In contrast, the only observed correlation of the APP locus with sporadic AD has been recently reported in a large Icelandic cohort, showing that a rare protective variant, A673T, explains less than 0.6 percent of the risk of sporadic AD107. This variant does not contribute to the risk of AD in the North American population108, and its statistical significance (p=4.8×10‒7) would not have survived a similarly-powered genome-wide scan. Curiously, mechanistic interpretation of the APOE4 risk isoform still mostly borrows from putative influences on APP catabolism and Aβ clearance, and normal function of the APOE protein has received less attention.
The APOE molecule moderates transport of lipoprotein particles in various organs by binding to the family of lipoprotein receptors. Although lipoprotein receptors aid in uptake and metabolism of lipid particles, they are not simple cargo transporters, and can activate a comprehensive nexus of intracellular second messengers with specialized signaling roles109. In this context, lipoprotein receptors including APOEr2 and VLDLr are well established regulators of brain development in reelin signaling. Activation of these two receptors by reelin triggers phosphorylation of the intracellular Dab1 adaptor. This pathway affects various aspects of cell physiology, among which cytoskeletal remodeling and neuronal migration are mainstay110. The reelin pathway guides extension of hippocampal neurites111 and coordinates outgrowth of the perforant path which represents the major input to hippocampal formation112.
The APOE molecule shares its lipoprotein receptors with reelin113, and mounting evidence indicates that APOE undertakes a similar role in guiding outgrowth of developing neurites113-117. Moreover, the neurite promoting effect of APOE is isoform-dependent, with the APOE3 isoform being a more potent inducer of neuritic outgrowth than APOE4115,117.
Intracellular transducers of the APOE molecule remain less investigated in neurons, but have been explored in other cells. In macrophages, APOE activates major transducers of the reelin pathway including Dab1 and PI3K118. In vascular pericytes, the APOE molecule affects rearrangement of the actin cytoskeleton and its knockdown deranges normal cell migration119. Similar to vascular cells, the APOE isoforms also affect the proteomic signature of cytoskeletal regulators in peripheral nerves120. Taken together, this body of evidence suggests that APOE may signal through a reelin-like pathway and influence cytoskeletal assembly, neurite outgrowth and cell movement.
In addition to the APOE risk locus and the reelin gene which is correlated with AD neuropathology in postmortem human brains121, three novel AD susceptibility loci further implicate lipoprotein receptors and a reelin-like signaling pathway in this disease; F-spondin (Spon1) is correlated with rate of cognitive decline in AD and also modulates white matter microstructure in healthy humans122,123. This gene codes for a reelin domain-containing cell adhesion molecule and its ortholog localizes to integrin adhesion sites in C. elegans124. F-spondin also binds APOEr2125 and guides extension of hippocampal neurites126. Moreover, F-spondin interacts with the APP protein127, and this interaction serves to activate signaling of the reelin adaptor Dab1 in ganglion cells128. Two other AD risk loci including Sorl1129,130 and CLU respectively code for a lipoprotein receptor and a lipoprotein receptor ligand. Sorl1 regulates cell migration130,131 and CLU activates reelin transducers including Dab1 and PI3K/Akt in neurons132.
Perhaps unrelated to their roles in lipid metabolism, lipoprotein receptors take part in the architecture of postsynaptic structures by interacting with the major synaptic scaffold protein PSD95133-135 as well as neurotransmitter receptors133,135. Expression of lipoprotein receptors affect synaptic density in hippocampal and cortical neurons136, and their activation by reelin promotes synaptic plasticity137-139. Intriguingly, lipoprotein receptors share several intercellular transducers with the APP protein, including X11, Dab1, Fe65136,140 and also control transcriptional activation of the APP gene in the cell nucleus141.
1.4 AD susceptibility loci strongly converge to cell adhesion pathways
Familial AD which is caused by APP or PSEN mutations constitutes less than one percent of AD cases. In contrast, the true polygenic landscape of common sporadic AD has been partly uncovered by recent genome-wide association studies14-19. Remarkably, the majority of novel AD risk genes engage in pathways of cell adhesion, migration and contact-guidance:
DSG2 (Desmoglein-2, rs8093731) is a component of cell adhesion complexes. DSG2 gene product serves focal adhesion roles and regulates cytoskeletal assembly by interacting with β8-integrin in endothelial cells142. DSG2 also controls cell motility and its depletion affects migration of malignant melanoma cells143.
EPHA1 (rs11771145) codes for a member of the ephrin-A receptor family that controls neurite adhesion and guidance. EPHA1 also moderates cell migration through integrin-linked kinase and the cytoskeletal remodeling pathway144,145, and affects invasion and metastasis of colorectal cancer cells146.
FRMD4A147 and FERMT2 (Kindlin-2, rs17125944) code for two members of the FERM domain family linking integrin and focal adhesion kinase (FAK) with the intracellular actin cytoskeleton148,149. FERMT2 transduces cell adhesion signals and is engaged in malignant cell invasion150.
GAB2 (rs2373115), one of the earliest AD susceptibility genes to be discovered by GWAS14,151, encodes a scaffolding protein acting downstream to the integrin signaling pathway. GAB2 regulates adhesion and migration of hematopoietic cells152 and also controls cytoskeletal remodeling for migration of malignant breast cancer cells153.
CASS4 (Hepl, rs7274581) controls focal cell adhesion154 and the CAS family members take part in axon guidance by interacting with integrin155. CASS4 also affects reorganization of the cytoskeleton and moderates cancer cell invasion154,156.
CD2AP (rs10948363) codes for an actin cytoskeleton binding protein157. CD2AP regulates focal adhesion of kidney podocytes at contact sites by linking membrane adhesion complexes with the intracellular actin cytoskeleton158.
PTK2B (Pyk2, rs28834970) is a focal adhesion signal transducer and affects the cellular cytoskeleton159,160. PTK2B controls integrin-dependent migration of T-cells161 and promotes invasion of malignant glioma cells162.
PICALM (rs10792832) is a Clathrin adaptor protein and engages in membrane receptor trafficking163. Clathrin regulates endocytosis of synaptic vesicles and moderates trafficking of the glutamate receptors164. Unbiased gene-gene interaction analysis in AD has revealed that the PICALM locus interacts with DOCK1165, which is an actin cytoskeleton regulator and affects cell movement166.
INPP5D (SHIP-1, rs35349669) is a key modulator of the PI3K pathway. This protein regulates platelet adhesion by modulating integrin signaling167 and also coordinates movement of neutrophils in response to focal contact and adhesion168.
NYAP1 (rs1476679) codes for a signal transducer of the PI3K pathway. NYAP1 acts downstream to Contactin5 synaptic adhesion molecule and controls cytoskeletal remodeling in neurite outgrowth169. Of note, Contactin5 binds amyloid precursor-like protein 1170.
Amphysin II (BIN1, rs6733839) codes for a protein that binds the cytoplasmic tail of integrin171 and neuronal focal adhesion kinase172 and is therefore probably involved in integrin-dependent cell adhesion. Moreover, Amphysin I which has a high level of sequence similarity (71%) with this gene product regulates outgrowth of hippocampal neurites173 and links endocytosis mechanisms to pathways of actin cytoskeleton remodeling174.
UNC5C175 (rs137875858) codes for a receptor of the netrin pathway of axon guidance176. In addition to the noted interaction of netrin1 with the APP protein, the netrin pathway also incorporates α3β1-integrin and Down Syndrome Cell Adhesion Molecule (DSCAM) in neuronal migration and neurite outgrowth, respectively177,178.
TPBG, a recently discovered AD risk gene19, modulates cell adhesion and movement179,180. TPBG localizes at focal adhesion sites in kidney podocytes and affects formation of actin stress fibers for cell remodeling181. Deletion of TPBG disrupts cadherin-dependent cell adhesion and suppresses cell migration182.
HBEGF19 (rs11168036) encodes a protein that promotes integrin-dependent cell adhesion183. HBEGF also regulates focal adhesion kinase and moderates cell migration by affecting the actin cytoskeleton184.
USP6NL19 (RNTRE, rs7920721) modulates integrin signaling and controls focal adhesion turnover, thereby acting as a "brake" in cell migration185.
TREM2 (rs75932628), a novel AD locus identified by next-generation sequecing186, is known to interact with the plexin-A1 adhesion molecule187 which is a receptor of axon guidance. The TREM and plexin-A1 interaction is suggested to moderate cell adhesion and movement through the cytoskeletal pathway188. The Plexin pathway also opposes integrin signal and inhibits cell movement189.
TTC3, a novel familial late-onset AD locus, maps to the Down syndrome critical region190. TTC3 modulates β1-integrin signaling in cancer cells191 and its increased level affects assembly of the actin cytoskeleton and disrupts neurite extension192.
PLCG2193 (rs72824905) encodes a phospholipase enzyme which is activated by integrin and moderates cell migration194. PLCG2 activation controls adhesion of leukocytes and takes place downstream to integrin signaling195.
ABI3193 (rs616338) affects the cytoskeletal pathway and participates in formation of membrane protrusions for cell motility196. Its binding partner, the ABI3 binding protein, interacts with integrin at focal adhesion sites and suppresses malignant cell migration197,198
Taken together, the genetic architecture of AD strongly implicates various cell adhesion receptors which coordinate pathways of cytoskeletal plasticity and cell reshaping. Further aiding in formulation of a unified disease model, many of these gene products cross-talk with the integrin pathway, and this convergence spotlights the Aβ-independent roles of APP and γ-secretase in cell adhesion signaling and synapse formation.
2 The hypothesis
Our model builds on the unbiased genetic architecture of AD and puts the cell adhesion signaling pathway at the center of disease nexus. Various cell adhesion regulators including integrins coordinate cell migration, neurite elongation, and assembly of synaptic circuits in brain development, and also undertake pivotal roles in maintaining synaptic integrity and homeostasis after completion of brain development199. Specifically, cell adhesion molecules form a dense scaffold at the postsynaptic density (PSD) sites and connect neurotransmitter receptors and synaptic ion channels with the actin cytoskeleton as well as the extracellular matrix. In addition to such mechanical support, synaptic adhesion molecules also act as biochemical sensors for modulation of postsynaptic plasticity, dendritic spine reshaping, and recycling of transmitter receptors200. For instance, it has been shown that enhancing signaling of synaptic integrin by application of its agonist peptide modulates neurotransmission201 in a dose-dependent manner202. In this context, integrin promotes budding of filopodia which serve to strengthen synaptic connections by cytoskeletal plasticity mechanisms203, i.e. the same mechanism by which integrins control axonal adhesion and pathfinding during brain development204. Remarkably, many of the intracellular transducers recruited by focal adhesion cascades also act as molecular switches of synaptic plasticity, including various tyrosine kinases (SFK, PI3K and Akt) as well as the calcium signaling pathway205.
The post-developmental roles of cell adhesion pathways in synaptic function and plasticity, which is not limited to integrins (Fig. 1), may enlighten pathways of AD from an Aβ-independent perspective. We propose that the heritable component of AD is defined by several genetic factors that coordinate robust adhesion and assembly of synaptic circuits in brain development. This genetic landscape also defines the level of synaptic adhesion, integrity and circuit resilience in the post-developmental period, and individuals with a vulnerable genetic background may suffer disassembly of neural circuits due to loss of synaptic adhesion pathways in late life.

Top: Biological adhesion pathways transfer microenvironmental signals across the cell membrane, and affect cell polarity, movement and survival. Bottom: Various pathways of extracellular adhesion coordinate rearrangement of the actin cytoskeleton and thereby control reshaping of membrane protrusions for movement. FAK: focal adhesion kinase; LRP: lipoprotein receptor; Shh: Sonic hedgehog.
3 Aging and the Alzheimer’s disease
Human aging is the strongest risk factor for dementia, but etiology of its correlation with AD remains elusive. One possibility is that AD may represent a continuation of global aging process, and cellular disruptions which happen in “normal” aging may give rise to dementia when accelerated7. An elegant work has recently revealed that frontal cortex cells of healthy humans accumulate ~37 new point mutations each year206. In line with the DNA damage hypothesis of aging, loss of genomic integrity has been previously implicated in AD predisposition12,207,208.
From a statistical point of view, even if a purely-random and stochastic process causes 37 annual mutations in aging neurons, larger genes are expected to be disproportionately affected in late life. Considering the low rate of somatic mutations estimated in aging frontal cortex cells (5.7×10‒9 mutations/bp.year206), only near one percent of the copies of a median-sized human gene will be affected by at least one somatic mutation in a 65-year individual. However, in sharp contrast, the largest known human gene, CNTNAP2, which is more than 80x larger than a median-sized protein-coding locus and codes for a synaptic adhesion molecule, is expected to be highly vulnerable to somatic mutations, and only 42 percent of its copies are estimated to remain intact at the same age (Fig. 3). This high variability is due to the characteristic distribution of human gene size parameter, which spans three-orders of magnitude with a long tail encompassing a subset of extremely large genes (Fig. 2). Intriguingly, many of the largest known human genes map to chromosomal fragile sites209.

Top: Human gene length distribution has a long tail that extends towards a group of extremely-large genes in the megabase pair range. The arrow points to the giant APOE receptor, Lrp1b. Bottom: Human gene size parameter closely follows a log-normal distribution with parameters μ=ln(26.9kbp) and σ=1.4. The outlier bin near 1kbp represents the large family of olfactory receptors which have gone through extreme evolutionary expansion. Scattered circles (top) and grey bars (bottom) show the subset of large genes used in functional enrichment analyses (>500kbp).
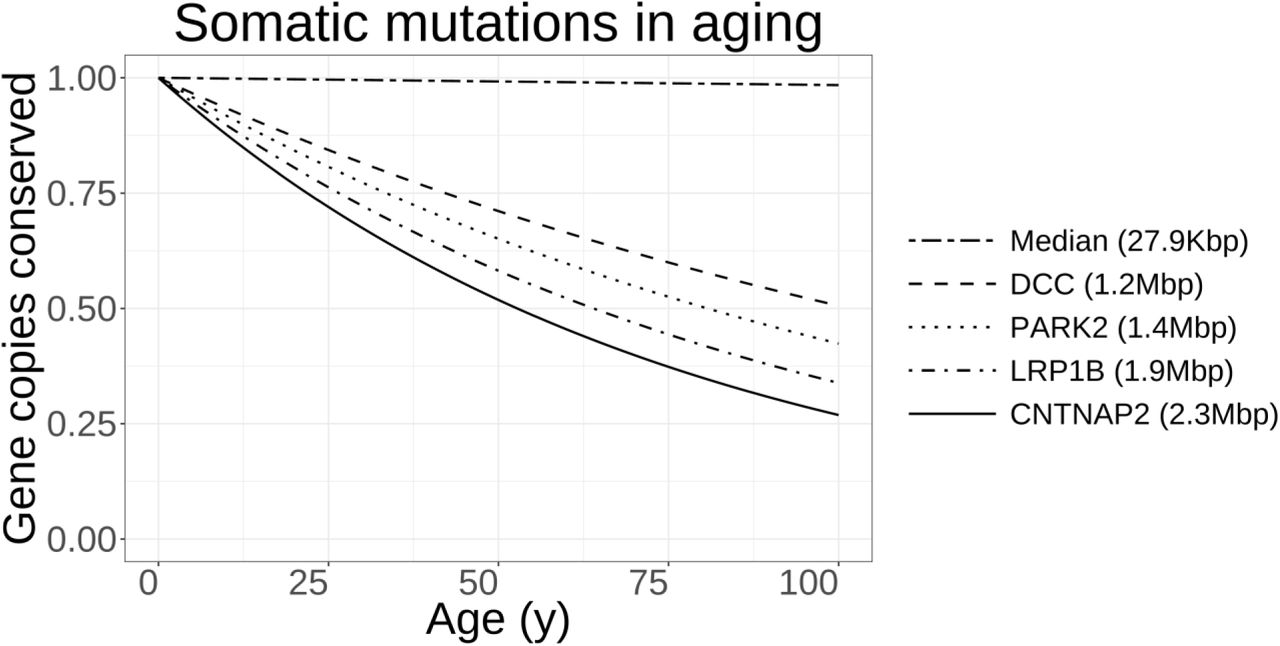
A binomial model in which somatic mutations take place at a fixed rate across the whole span of human genome has wildly different impacts on genes of various sizes. According to this model, a median-sized human gene mostly survives the mutational burden of aging, with only ~1 percent of its copies affected by any somatic mutation in late life. However, larger genes possess significantly shorter half-lives due to somatic mutations. Many of these large genes regulate synaptic function and integrity and are also known to act as tumor suppressors.
These clues compelled us to objectively investigate whether the largest human genes non-randomly take part in certain cellular mechanisms. We size-sorted all of the protein-coding human genes (n=18,181 genes with UniProt accession). Thereafter, a minimum gene length threshold of >500kb was considered, a cut-off filter that resulted in inclusion of 234 genes representing the top 1.3 percent of largest protein-coding loci. Functional annotation and tissue expression profile of this gene set of interest were investigated using Database for Annotation, Visualization and Integrated Discovery (DAVID) and standard statistical tests of gene set enrichment210,211.
The most overrepresented organ for expression of this gene set was brain (p=2.1×10‒17), followed by amygdala (p=1.9×10‒4) and hippocampus (p=8.5×10‒4). Showing strong enrichment statistics, cell adhesion (G0:0007155) was the most representative biological process related to this gene set (p=8.0×10‒13, Table 1) and the most overrepresented cellular component was synapse (G0:0045202: p=2.4×10‒14). Other significant gene ontology terms further implicated pathways of nervous system development and function, including neuron differentiation, axon morphogenesis, axon guidance, cell motion, and synaptic transmission (Table 2).
Enrichment of large human genes (>500kbp) in Gene ontology: biological process annotation. p-values are corrected for multiple comparisons.
Enrichment of large human genes (>500kbp) in Gene ontology: cellular component annotation. p-values are corrected for multiple comparisons.
The strongly nonrandom selectivity of large human genes to brain, synapse and cell adhesion is a potentially enlightening observation. We suspect that these genes may have fostered adhesion and assembly of complex synaptic circuits in cognitive evolution. As an evolutionary bottleneck, extremely large genes may also be inherently costlier to be maintained in late life, and may put individuals at a neurobiological disadvantage when the burden of somatic mutations passes a critical threshold in aging. Remarkably, potential downside of large genes in late life aging has only been weakly corrected by evolutionary forces, since average human life expectancy passed the 40-year milestone only two centuries ago212. In this regard, AD may have unmasked a DNA maintenance and repair bottleneck in the elderly brain of modern humans due to the rapid expansion of life expectancy.
While somatic mutations provide a simple explanation for synaptic adhesion failure in aging, it is worth noting that extracellular adhesion pathways also form a surveillance system for continuous checking of cell anchorage in solid organs. Abnormal loss of cell adhesion robustly activates a specialized apoptosis program known as anoikis213, with a number of AD risk loci controlling such anoikis pathways including the reelin pathway which regulates anoikis of mesodermal cells214 and the ephrin cascade of axon guidance which moderates anoikis in cancer215. Integrin is also a well-characterized regulator of anoikis cell death (e.g. see216 and reviews217,218). The interrelationship between pathways of neurite adhesion and cell survival is at a level that they occasionally rely on dual-functioning dependence receptors, as has been determined for netrin and ephrin cascades of axon guidance219,220.
4 Predictions
AD patients may suffer faster accumulation of DNA damage and somatic mutations in their aging neurons due to a combination of heritable factors and environmental exposures. This insult eventually disrupts cellular pathways controlled by unstable genes of synaptic maintenance and neuronal survival. Derailed signaling of the APOE-lipoprotein receptor axis may be a prototypic feature of cellular dysfunction in sporadic AD (Fig. 4).
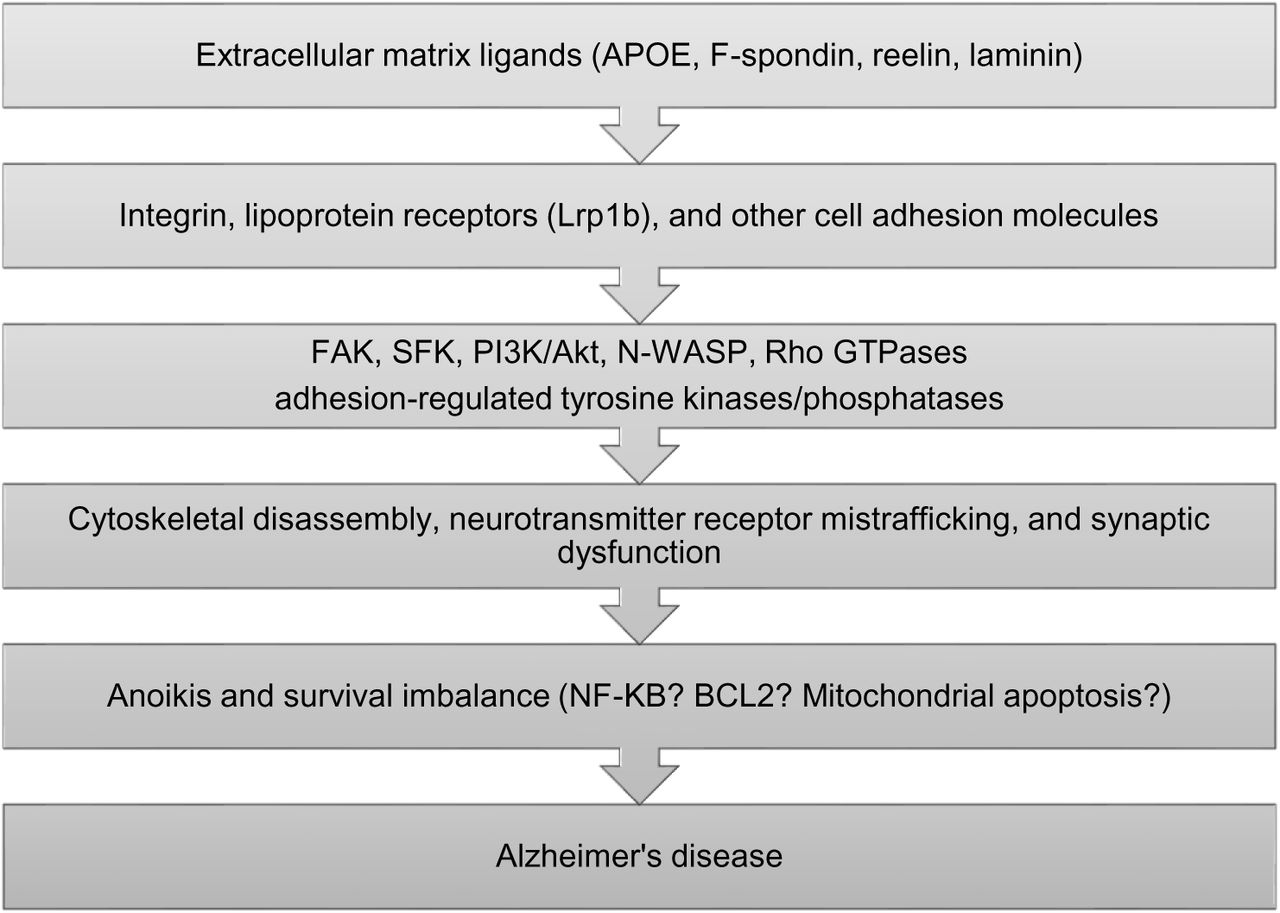
A simplified cascade of sporadic AD pathogenesis based on APOE signaling disruption. FAK: focal adhesion kinase; SFK: Src-family kinase.
We predict that mutational instability of the Lrp1b gene in human aging potentially explains the APOE4 risk factor of AD:
Lrp1b is the largest member of the lipoprotein receptor family and represents the 8th largest human gene overall. Lrp1b is also one of the most frequently deleted genes in 3,131 cancer specimens and maps to a common chromosomal fragile site223.
Coding variants within Lrp1b are correlated with cognitive stability in aging and AD224,225.
Lrp1b controls focal adhesion, cytoskeletal remodeling and cell migration226, pathways that functionally align with the genetic architecture of AD. Lrp1b has affinity to APOE particles and also interacts with APP at cell membranes, possibly acting as a co-receptor complex for cell signal transduction227,228.
Lrp1b interacts with two postsynaptic proteins including PSD95134 and PICK1229. PSD95 is the major synaptic scaffold protein and connects glutamate receptors with the synaptic actin cytoskeleton. PICK1 regulates postsynaptic function and glutamate receptor function230.
Unlike other APOE receptors that are generally expressed in various organs, Lrp1b demonstrates a restrictive pattern of brain expression227, and hippocampal CA1 neurons and amygdala show the highest Lrp1b transcription levels in the Allen human brain atlas231 (Fig. 5).

Top: Correlation of genetic polymorphisms of the Lrp1b locus with several MRI measures of brain volume in ENIGMA-2 meta-analysis232,233 (https://www.enigma-brain.org/enigmavis/visualizer/visualizer). Bottom: Spatial expression of Lrp1b in different human brain regions in six postmortem brain samples, Allen human brain atlas (http://human.brain-map.org).
We predict that AD-type cognitive decline is correlated with somatic mutations in certain large synaptic genes including Lrp1b. Although previous models have already implicated oxidative stress and DNA damage in AD12,207,208,234, high-throughput results do not support an oxidative etiology for somatic mutations in various organs, since oxidative stress typically causes G:C→T:A transversions235,236, whereas aging cells demonstrate a clock-like signature of somatic mutations with enrichment of C:G→T:A transitions237,238. Intriguingly, the C:G→T:A fingerprint was recently observed as the dominant type of somatic mutations in neurons236,239,240. We predict that the most frequent type of genomic instability in AD may also be similar to that of “normal” aging in neurons and cancer cells. The reason for this preponderance of C:G→T:A transitions is currently unknown, and might reflect transcriptional stress, spontaneous cytosine deamination, or DNA repair failure236,241.
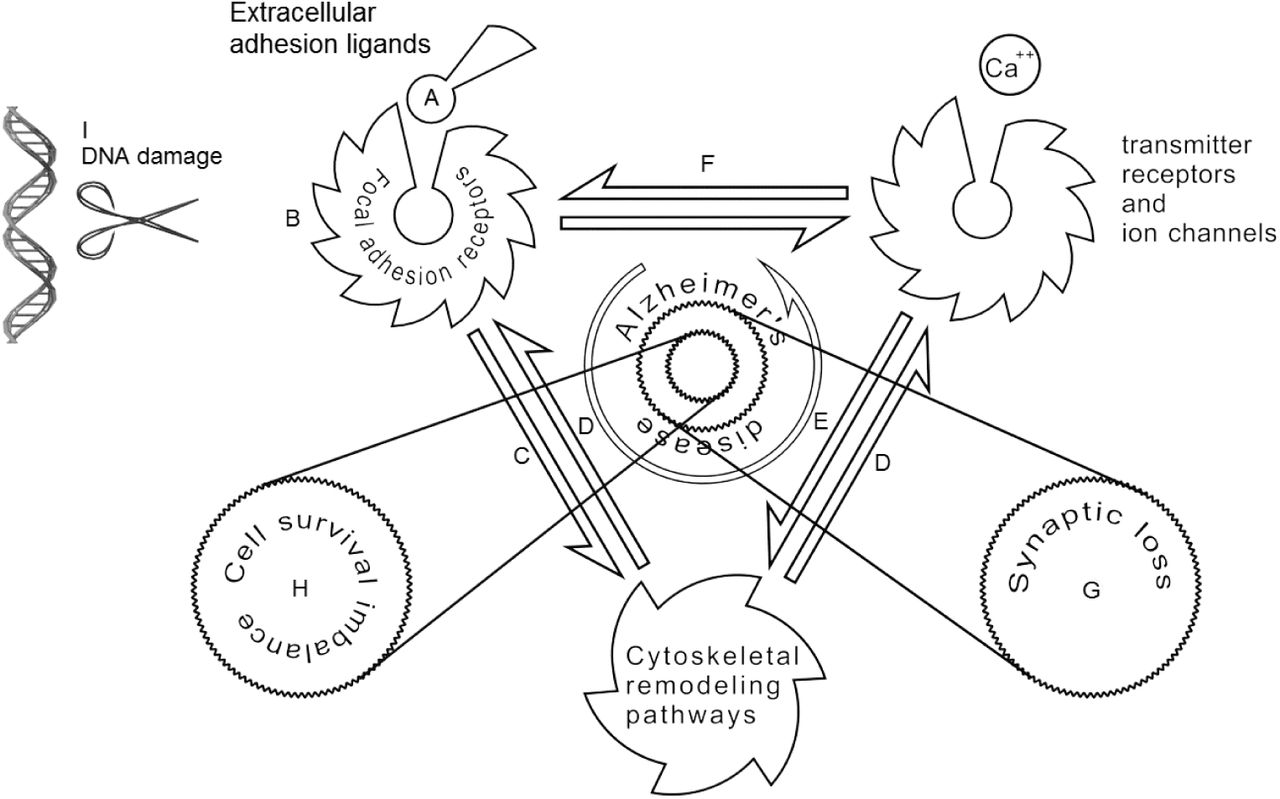
It is noteworthy that Lrp1b only serves to provide one example of vulnerable genes in brain aging, and the true genetic landscape of sporadic AD and senile neurodegenerations in general is not reducible to the APOE pathway (Fig. 6). In a manner that may be similar to engagement of various tumor suppressor genes in different cancers, brain-wide expression of several unstable synaptic genes may cause heterogeneity of dementia syndromes. For instance, the genetic architecture of Parkinson’s disease implicates many genes of the synaptic vesicular trafficking system including the extremely large tumor suppressor PARK2242, and the vulnerable dopaminergic neurons selectively express two known tumor-suppressor cell adhesion genes, including DCC243 and AJAP1244.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The proposed mechanisms of synaptic loss and neuronal death in AD. The extracellular matrix and its cell adhesion molecules (A) modulate neuronal adhesion receptors (B). Cell adhesion pathways affect remodeling of synaptic cytoskeleton as well as other modulators of plasticity and neuronal survival, e.g. various SH3 domain containing proteins (C). The postsynaptic density scaffold is anchored with the synaptic actin cytoskeleton through linking proteins, e.g. PDZ domain containing proteins (D). Function and trafficking of the neurotransmitter receptors are controlled by cytoskeletal pathways (E) as well as cell adhesion molecules (F). Disruption of cell adhesion pathways in AD impairs synaptic stability and causes dendritic spine loss (G), and may eventually lead to neuronal survival imbalance by triggering anoikis-like mechanisms (H). Selective vulnerability of large genes in aging may be the cause of cell adhesion disruption in AD (I).
5 Future perspectives
Conditional knockout of Lrp1b as well as other modulators of the APOE signaling axis (F-spondin, reelin, Dab1) after completion of brain development may aid in modeling cognitive aspects of AD in laboratory animals. Intriguingly, conditional knockout of the Lrp1 gene which is closely related to Lrp1b, but lacks its brain-specific expression profile, results in neurodegenerative changes after 12 months of animal aging245. The single study of Lrp1b knockout mice did not follow the course of abnormal phenotypes past this age134. Lrp1 and Lrp1b may also be functionally redundant and partially compensate for loss of gene function in single knock-out models.
Since even the most aggressive forms of AD remain clinically-silent for decades, the short lifespan of laboratory animals may not permit effective interaction of genetic factors and environmental exposures to take place similar to human dementias. Therefore, accelerating the aging process by crossing AD animal models with DNA-repair defective strains246 or exposure of models to genotoxic UV irradiation may prove informative, but usefulness of these gene×environment models relies on validation of DNA damage accumulation in aging human brain and its characteristics.
Our hypothesis is not based on any form of etiological relevance for Aβ species or neurofibrillary tangles in the disease cascade, and redefines these neuropathological features as bystander epiphenomena downstream to other causal factors. For AD drug design, enhancing function of the integrin-lipoprotein receptor signaling axis and their downstream second messengers including adhesion-regulated tyrosine kinases (FAK/SFK, PI3K→Akt, and Erk) or regulators of cytoskeletal actin (e.g. Rho GTPases, cofilin, and WAVE) as well as blocking anoikis pathways may prove useful. Nevertheless, even the strongest AD risk locus, APOE, fails to explain ~94 percent of the disease variance alone, and single pathway therapeutic approaches may provide limited benefit in clinical trials. As a potentially more effective method, our model warrants engineering of novel gene delivery vehicles for restoring neuronal expression of unstable genes before somatic mutations reach a level that results in clinical presentation of dementia.
In conclusion, our proposal, the large gene instability hypothesis, implicates mutational vulnerability of the complex genes of synaptic adhesion and homeostasis as the primary etiology of AD, and suggests a shift of paradigm from the protein aggregation process to DNA damage mechanisms for disease prevention and drug design.
Acknowledgements
S.S. is supported by NIH Big Data to Knowledge (BD2K) grant U54 EB20403.
Footnotes
S.S. Imaging Genetics Center USC Stevens Neuroimaging and Informatics Institute Keck School of Medicine of USC 4676 Admiralty Way Marina del Rey, CA 90292 Tel: (323) 44-BRAIN sourena.soheili-nezhad{at}loni.ini.usc.edu
M.Z. School of Cognitive Sciences Institute for Research in Fundamental Sciences PO 193955746 Tehran, Iran Tel: +98 21 mz{at}ipm.ir
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.
- 10.
- 11.
- 12.↵
- 13.↵
- 14.↵
- 15.
- 16.
- 17.
- 18.
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.
- 98.
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.
- 115.↵
- 116.
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.↵
- 181.↵
- 182.↵
- 183.↵
- 184.↵
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.↵
- 205.↵
- 206.↵
- 207.↵
- 208.↵
- 209.↵
- 210.↵
- 211.↵
- 212.↵
- 213.↵
- 214.↵
- 215.↵
- 216.↵
- 217.↵
- 218.↵
- 219.↵
- 220.↵
- 221.
- 222.
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.↵
- 230.↵
- 231.↵
- 232.↵
- 233.↵
- 234.↵
- 235.↵
- 236.↵
- 237.↵
- 238.↵
- 239.↵
- 240.↵
- 241.↵
- 242.↵
- 243.↵
- 244.↵
- 245.↵
- 246.↵




