

ABSTRACT
Due to improved instrument sensitivity and access, the use of metabolomics is gaining traction for the study of many organisms and pathogens. For the intracellular malaria parasite, Plasmodium falciparum, both targeted and untargeted metabolite detection has improved our understanding of pathogenesis, host-parasite interactions, parasite response to antimalarials, and impacts of resistance. However, protocols for purification are not optimized for investigations of intracellular pathogens and noise-limiting analysis parameters are not well defined. To explore influential parameters, we purified a diverse set of in vitro grown intra-erythrocytic P. falciparum parasites for untargeted metabolomics studies. Following metabolite identification, data processing included normalization to double stranded DNA, total protein, or parasite number to correct for different sample sizes and stage differences. We found that parasite-derived variables were most appropriate for normalization as they separate sample groups and reduce noise within the data set. However, these post-analysis steps did not remove the contribution from the host erythrocyte, in the form of membrane rich ‘ghosts’, and levels of technical sample variation persisted. In fact, we found that host contamination is as influential on the metabolome as sample treatment. This analysis also identified metabolites with potential to be used as markers to quantify host contamination levels. In conclusion, purification methods and normalization choices during the collection and analysis of untargeted metabolomics heavily affect the interpretation of results. Our findings provide a basis for development of improved experimental and analytical methods for future metabolomics studies of P. falciparum and other intracellular organisms.
Importance Molecular characterization of pathogens, such as the malaria parasite, can lead to effective treatment strategies and improved understanding of pathogen biology. However, the distinctive biology of the Plasmodium parasite, such as its repetitive genome and requirement of growth within a host cell, hinders progress towards this goal. Untargeted metabolomics is one promising approach to learn about pathogen biology and how it responds to different treatments. By measuring many small molecules in the parasite at once, we gain a better understanding of important pathways that contribute to this response. Although increasingly popular, protocols for parasite isolation from the host cell and various analysis options are not well explored. The findings presented in this study emphasize the critical need for improvements in these areas to limit misinterpretation due to host metabolites and correct for variations between samples. This will aid both basic biological investigations and clinical efforts to understand important pathogens.
Introduction
Malaria continues to be responsible for hundreds of thousands of deaths annually, most of which result from infectionwith the protozoan parasite, Plasmodium falciparum(1).Characterization of the biology of this importantpathogen can lead to improved treatment strategies. The molecular mechanisms behind interesting P. falciparum phenotypes are challenging to understand due to a lack of traditional methods of investigation in this organism,such as forward and reverse genetics. Unbiased ‘omics approaches (transcriptomics and proteomics) are widely used but the limited annotation of the parasite genome makes these data sets challenging to interpret. One way to alleviate this lack functional knowledge is to use network-based modeling to facilitate data interpretation (2). Additionally, the measurement of direct mediators of the phenotype, such as metabolite reactants and products of enzymatic reactions, can improve our ability to make predictions about cellular function under certain conditions. For this reason,metabolomics is becoming increasingly popular to study P. falciparum (3-12). These studies have allowed for a greaterunderstanding of malaria pathogenesis (13), strain-specific phenotypes (11), and host-parasite interactions (9). Although metabolomics can successfully identify metabolic signatures that correlate well with biological function, such as time and dose-dependent response to antimalarial treatment (3, 5) and resistance-conferring mutations (12) there are distinct challenges that need to be considered when performing metabolomic studies in P.falciparum.
Challenges such as host contamination, limited parasite yield, and parasite stage-specificities arise due to certain properties of this organism (see Table 1). For example, experimental samples typically have few parasites and abundant host material. One contributing factor is that parasitemias are limited during in vitro culture and clinical infections (<5% or five infected erythrocytes per 100 total (14,15). Additionally, P.falciparum is an intracellular parasite during the asexual cycle in the human blood stream; the host erythrocyte accounts for up to a 10-fold more cellular material over early state parasites (16, 17). Due to our ability to enrich for late stage parasites using magnetic purification (18), the study of the larger later stage parasite has historicallyallowed for efficient genomic, transcriptomic and proteomic analysis of parasite biology. These stages have typically been thought of as more metabolically active than the early stage parasites due to increased activity of well-studied cellular pathways, including robust hemoglobin degradation (19), nuclear genome replication, and protein synthesis (20, 21). The study of the smaller early stage of the parasite is particularly hard to achieve due to difficulty isolating adequate amounts of parasite material as a result of few effective enrichment methods (22). Thus, studies must be designed in a manner to overcome these challenges, limiting sample-to-sample variation and optimizing metabolite recovery (i.e. total number of metabolites detected).
In this study, we sought to define critical parameters that would help overcome these challenges and allow the collection of highquality metabolomics data. We show that diverse sample groups can be differentiated, but the choice of analytic parameters for data processing and host cell contamination both heavily influence the parasite metabolome. In particular, we investigated normalization approaches to assess the impact of host contamination and found that the adjustment to parasite-derived variables better remove sample noise.However, even appropriate normalization fails to remove host noise completely, as host contamination is as influential on metabolome as sample treatment. Thus, we propose that the combination of improved purification and analytic parameters will generate more accurate measures of the metabolome, increasing the utility of unbiased metabolomics to investigate intracellular parasite biology.
RESULTS
Parasite sample groups are metabolically distinct
To ensure our metabolomics approach can identify obvious differences in sample groups, we compared parasite groups that differed in stage, origin, and growth conditions (Fig. 2A). Distinct purification procedures were used for preparation of each sample group (see Materials and Methods and Fig. 1), resulting in different amounts of parasite material (Fig. 2B, Table S1). Replicates of sample group 1, which were merely lysed from host cells with a mean parasitemia of 1.14%, contained between 1.3-6.9 x 106 total parasites. Sample group 2 was enriched for late stage parasites using magnetic purification to a mean parasitemia of 53.6% (Table S1). These replicates contained between 4.7 x 107 to 6.7 x 108 total parasites (up to 100-fold more individual parasites). Despite these differences, mean protein abundance was insignificantly different across replicates of each sample group and was more variable in sample group 2 (group 1SD: 12.7, group 2 SD: 38.2, see supplementary information for code and Fig. 2B).Sample group 1 had a mean of 115.3 µg/ml of protein, and sample group 2 had a mean value of 107.6 µsg/ml. Cell number and DNA abundance are positively correlated, as expected (r = 0.8037, p-value = 0.00002, Fig. 2B); these values are not perfectly correlated because the late stage parasites in sample group 2are actively replicating DNA, and, thus, have increased and variable genome copy number per cell. Protein does not correlate with parasite number or DNA abundance (data not shown, see supplementary information for code).
We conducted metabolomics on the samples described above (Fig. 1). Cultured parasites were lysed from host erythrocytes and analyzed via UPLC-MS. In comparison 1, we detected 375 total metabolites that were annotated by Metabolon, Inc.; 143 of these were detected in every sample and represented 10 energy associated metabolites, 159 lipid species, 108 peptides and amino acids, 40 nucleotides, 28 cofactors, 20 carbohydrates, and 10 others (Fig. 2C).Samples from group 1 contained between 182-242 metabolites while those from group 2 contained between 267-368 metabolites (Fig. 2C). Fifteen metabolites are found in every group 1 sample, but not all group 2 samples, and 111 metabolites are found in everygroup 2 sample but not all group 1 samples. Thus, distinct samples, due to parasite origin, stage, growth conditions, and purification differences, have distinct metabolomes.

1) Laboratory-adapted P. falciparum clones are cultured in host erythrocytes. 2) If enrichment of late stage parasites is desired (dotted line), cultures can be passed through a magnetic column to retain paramagnetic late stage-infected erythrocytes (black dots inside red circles). Samples for parasite count determination were collected at steps 1 and 2, depending on the sample group (see Materials and Methods). 3) Erythrocytes (infected and uninfected) are lysed using saponin, but parasites remain intact (black dots). Wash steps are used to remove hemoglobin and other intracellular erythrocyte contents (red material). Samples for total protein determination were collected at this step. 4) Soluble metabolites (purple dots) are extracted from precipitated protein (grey pellet) using methanol (droplets). Samples for DNA content determination were taken at this step, prior to methanol extraction. 5) Metabolites are detected by liquid chromatography followed by mass spectroscopy. Metabolites are identified by comparison to a library of authenticated standards. 6) Abundance data for each metabolite is normalized to an appropriate parameter (i.e. DNA content or parasite number), log transformed, centered to median, and scaled to variance, prior to employing statisticalcomparisons.
Normalization parameters influence sample variation
Normalization methods can influence results (23), but have not been explored in the use of metabolomics for Plasmodium nor other intracellular pathogens. To explore the importance of various normalization approaches, we performed principal component analysis with all sample metabolomes using either unnormalized data or three normalization methods: quantification of parasite number, double stranded DNA, and total protein amount. Each normalization method yields distinct principle component structures and clearly separates sample groups (Fig. 2D). In all cases, principle component (PC) 1 primarily represents between group variation, and PC2 represents within group variation (Fig. 2D). Without normalization, PC 1 and 2 summarize 78.4% of sample variation. These principal components from parasite number and DNA normalization summarize 87.7 and 80.6% of sample variation, respectively. With protein normalization, 79.1% of variation is summarized. PC2 tends to separate sample group 1 better than those samples within group 2 (Fig. 2D).
The metabolites that most contribute to group or sample variation are not the same with each normalization approach (Table S2). Thus, metabolome differences between groups are dependent on normalization approach.Yet, there are several striking trends across analyses. For example, the PC structure following protein normalization closely mimics that of theunnormalized data and, similar metabolites contribute to PC1 and PC2 in both analyses. Sphingomyelin species contributeto within group variation (PC2), and orotidine and dipeptides contribute to between group variation (PC1; Table S2). Upon DNA or parasite number normalization, phenylalanine, tryptophan, leucine, putrescine, and sedoheptulose 7-phosphate contribute to PC2, or within group variation (Table S2).Contrary to protein amount (see Discussion), DNA and parasite number normalization are parasite-derived and, thus, these two measurements are preferable for normalization. The choice of which parasite-derived variable to use for normalization should be based on the experimental question. Accordingly, we normalize to parasite number during our subsequent comparison of sample groups 1 (early stage) and 2 (late stage; see Fig. 2); normalization to DNA amount would not be appropriate because these different stages have known genome copy number differences (late stage parasites are actively replicating their DNA, whereas ring stage parasites are haploid). Furthermore, we normalize to DNA content during our subsequent comparison within a group (i.e. replicates of samples group 1, see Fig. 3). In this case, normalization to this parasite variable is more appropriate because these measurements are collected immediately prior to mass spectrometry metabolite processing (Fig. 1) in our experimental design and are the most representative of analyzed samples.

A.Comparison made throughout Figure 2. Group 1 contains ten samples of early-stage parasites grown in AlbuMAX-based media in three blood batches, treated with antimalarial (see Materials and Methodsand Table S1). These parasites were isolated at a low parasitemia and purified from host material using saponin lysis. Group 2 contains ten samples of late-stage parasites grown in a human serum-based media in four blood batches, treated with antimalarial (see Materials and Methodsand Table S1). Group 2 parasites were purified magnetically to achieve high parasitemia and lysed from host cells with saponin.B. Sample characteristics. Samples (group 1 in circles and group 2 in triangles) were evaluated for DNA, parasite count, and protein amount prior to analysis. C. Summary of detected metabolites. Not all metabolites were detected in each sample. The majority of metabolites detected were lipid species. Sample groups are color coded with group 1 in red and group 2 in blue. A full list of identified metabolites are listed in supplemental data (see Github). D. Normalization affects measured metabolome. Principle component (PC) analysis was performed prior to normalization (left), as well as using three different normalization methods (left to right, total protein, parasite number, and DNA). Circles indicate group 1 samples and triangles indicate group 2 samples. For PC decompositions, see Table S2.

{kind=link}
{kind=link}
{kind=link}
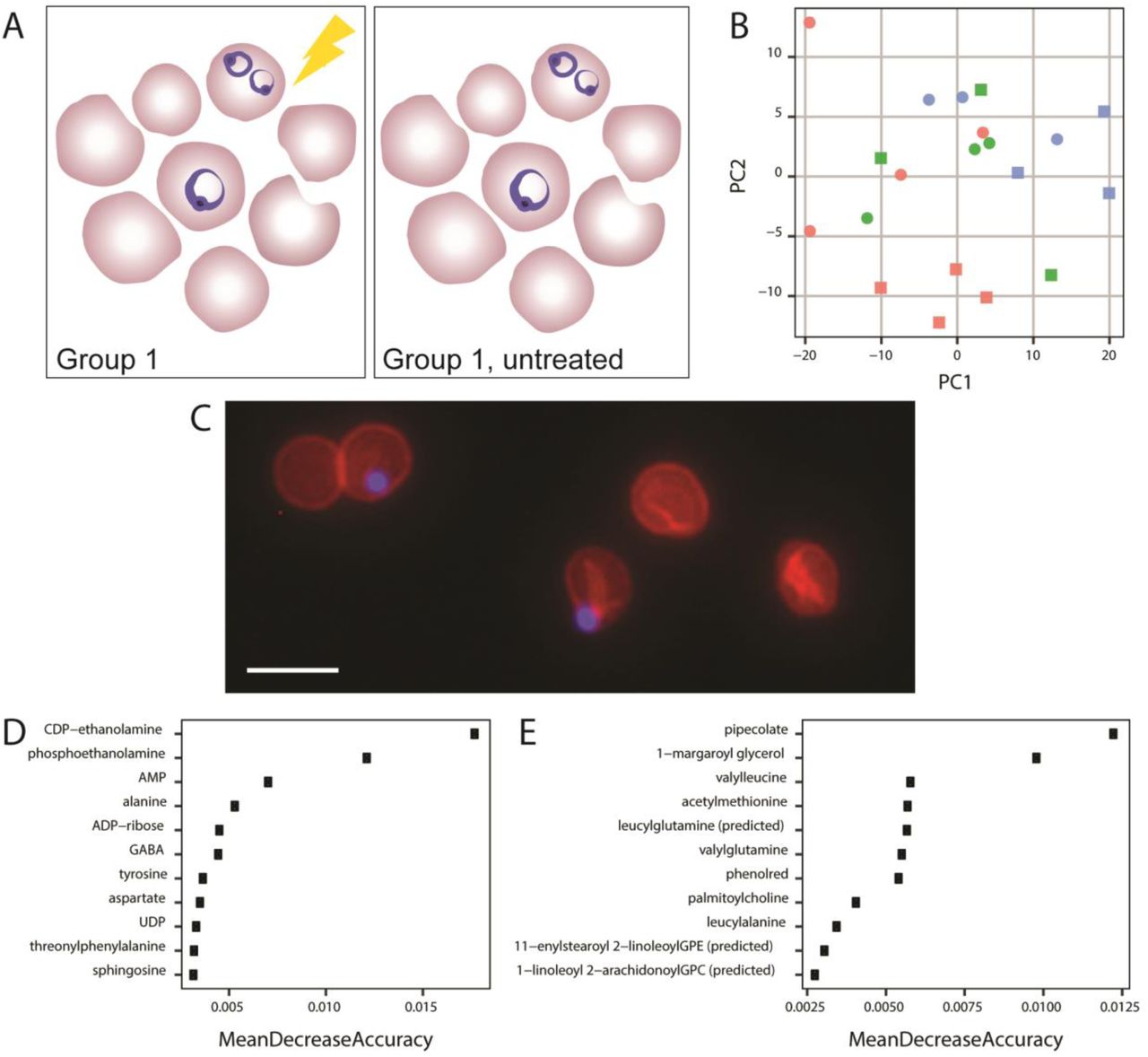
A. Comparison made throughout Figure 3. All samples were grown in AlbuMAX-based media in three blood batches and purified from host material using saponin lysisduring the early stage. Group 1 was treated with antimalarial for 6 hours and group 1, untreated, did not undergo treatment (see Table S1); samples were matched for blood batch. B.Metabolome principle component analysis. PCA of DNA normalized, median-centered metabolomes of early stage parasites from Influential parameters for P. falciparum metabolomics 31Comparison 2. Squares indicate antimalarial treated samples and triangles indicate untreated samples. Blood batches are indicated by color. C. Visualization oferythrocyte ghosts containing parasites. Fluorescent imaging (40X) reveals parasites (blue, DAPI) retained within erythrocyte ghosts (red, phycoerythrin conjugated CD235a antibody) following saponin treatment. Scale bar represents 10µm. D. Metabolites predictive of blood batch. Top ten most predictive variables in the blood batch Random Forest classifier. E. Metabolites predictive of antimalarial treatment. Top ten most predictive variables in the antimalarial treatment Random Forest classifier.
Remnants of the erythrocyte host contribute to metabolite pool
Beyond comparing the metabolomes of artificially distinct samples groups, we explored the metabolic changes induced by antimalarial treatment. We collected metabolomics from treated and untreated early stage parasites that were identical in growth conditions and purification approach, and were matched for blood batch (Fig. 3A, Table S1, see Materials and Methods for group 1). Following data processing, the metabolomes of antimalarial treated and untreated parasites fail to cluster via PCA (Fig. 3B). Accordingly, univariate statistical analysis revealed no differentially abundant metabolites between treated and untreated samples (see supplemental information for code).
When considering possible explanations for this result, microscopy revealed that parasites lysed from host cells remain embedded in erythrocyte membranes and washes fail to isolate parasite material (Fig. 3C). This result emphasized that erythrocyte ‘ghosts’ (cell membranes with associated metabolites) remain abundant in the sample and heavily contribute to the metabolome (see Discussion). In fact, univariate statistical analysis only revealed one metabolite with increased abundance in one blood batch (1-arachidonoyl-GPE; see supplemental information for code). Thus, the metabolome is likely influenced by both blood batch and antimalarial treatment, with the noise induced by each variable overshadowing group differences.
To further explore the host contribution to the metabolome, we built two Random Forest classifiers to identify metabolites that are associated with either erythrocyte ghosts or antimalarial treatment. We first built a classifier to predict blood batch in early-stage parasites (Fig. 3A). These samples likely have large host contribution due tothe inability to enrich for erythrocytes infected with early stage parasites. Ninety-five metabolites (of 298), including AMP, ADP-ribose, aspartate, and sphingosine improved classifier accuracy in predicting blood batch (most influential depicted in Fig. 3D, see supplemental information forcode); the remaining metabolites had no effect on classifier performance or worsened its predictive capabilities, indicating they are not associated with blood batch due to high variability or association with other features that differentiate samples. This classifier predicted blood batch with a 30% error rate. Thus, a subset of the measured metabolome was predictive of blood batch.
To determine if blood batch is as influential on metabolome as antimalarial treatment, we built a similar classifier to predict treatment within early stage samples (Fig. 3A). Early stage parasites were classified into two treatment conditions with a 30% class error rate. One hundred and eighteen metabolites (of 298) improved classification accuracy (see most influential in Fig. 3E, and supplemental information for code), including pipecolate and several dipeptides. Thus, sample metabolome can classify both blood batch and sample group, indicating sample treatment and blood batch influence the metabolome.
DISCUSSION
Here, we explore metabolomics methods used in in vitro study of intraerythrocytic P. falciparum. The parasite’s intracellular lifestyle introduces challenges in implementing traditional protocols,predominately due to limited amounts of parasite material and host metabolite contamination. In our study, we sought to determine critical parameters for the collection of high quality metabolomics data despite these challenges. In particular, we investigated normalization approaches and conducted a detailed assessment of the impact of host contamination. Overall, we found that only parasite-derived variables are best suited to use during normalization. Despite these analytic approaches, host noise permeates the analysis, as host contamination is as influential on metabolome as antimalarial treatment. Thus, improvements in both purification and analytic parameters must be combined to generate accurate metabolomes and increase our ability to learn more about the parasite’s biology.
Normalization of metabolite levels aims to limit technical or non-biological variation, thus enhancing interpretation of results. Normalization can be calculated by a variety of methods and is implemented either before or after analysis (Table 1 (24, 25). Often, pre-analysis normalization is conducted by isolating the same number of cells for analysis (26) but this is not typically used in the study of P. falciparum as generating adequate biomass can be challenging. Furthermore, sample adjustments following the use of inaccurate quantification methods may introduce more variability. Post-analysis normalization methods are also routinely used; these include the use of internal standards (25, 27), corrections for protein amount (often used for supernatant or cell-free metabolomics (28)), DNA content (an approach validated in mammalian cells (29)), or cell number (typically used for bacterial populations (30)).A common approach used in the study of P. falciparum involves an uninfected erythrocyte control to adjust for the presence of host metabolites (7, 10, 27, 31-33). However, use of this control without other forms of normalization led to the misattribution of host metabolites to the parasite (34). Selecting the correct method of normalization in P. falciparum metabolomics studies is essential to ensure that parasite-derived metabolites, and not host-derived metabolites, are measured and interpreted to make conclusions.
Note: most parameters do not have strict recommendations, as they are dependent on experimental design. Grey highlights indicate methods that were employed and evaluated during this study.
We explore three post-analysis normalization approaches: protein, DNA, and parasite number. We argue the host erythrocyte heavily contributes to protein abundance, and, thus, this metric is not solely parasite-derived. In our analysis, this was most clearly observed when comparing protein abundances between our sample groups (Fig. 2B). We expected a proportional increase in protein amount as parasite size increases throughout the intraerythrocytic life cycle (from sample group 1 to 2; early to late stage); however, this increase was not detected, implicating host erythrocyte contribution. Furthermore, heavy host contamination explains the observations that 1) there is an increased level of protein variability in group 2 (explained by the wider range in parasitemia level and thus host erythrocyte contribution, Table S1), 2) host/media metabolites such as kynurinine, phenol red, and HEPES were detected in this analysis (see below and supplemental data), and 3) protein normalization minimally changes the PCA data structure and top contributing metabolites (Fig. 2D and Table S2).
In sharp contrast, total DNA amount and parasite count are entirely parasite-derived; mature uninfected erythrocytes are anucleated, without detectable DNA (35), and are excluded when determining parasite count (see Materials and Methods). When metabolites were evaluated following DNA and parasite count normalization, more nuclear material and total parasites were observed in later stages (group 2, Fig. 2B). These data are not surprising, as late-stage parasites are known to amplify DNA content up to twenty times during their asexual life cycle (36). A greater cell count in late stage samples can be attributed to the higher parasitemia that is achieved through magnetic purification of late stage trophozoites and schizonts (37). To our knowledge, normalization to parasite-derived material has not been described in detail in previous metabolomics studies of P. falciparum. We propose that similar to studies in Leishmania(38-40), normalization to parasite-derived measurements should become standard during metabolomics analysis of these intraerythrocytic parasites (Table 1).
Clearly, parasite-to-parasite sample variation can influence metabolomics data, but we also found host erythrocyte material can heavily impact a sample’s metabolome. Many studies employ erythrocyte lysis prior to sample purification ((8, 32) and our current study, see Materials and Methods). However, this approach does not eliminate the potential for host contamination; host membrane fragments devoid of internal components, colloquially referred to as erythrocyte “ghosts,” remain in purified samples (Fig. 3C). Despite this concerted effort to limit host metabolites through lysis, our studies support heavy erythrocyte contribution to the P. falciparum metabolome. Several metabolites were detected in group 1 and 2 metabolomes that have not previously been measured as produced or consumed in Plasmodium. For example, kynurenine is known to be present in erythrocytes and is derived from the amino acid L-tryptophan (41, 42). Although no known production or consumption has been reported in the parasite, kynurenine was detected in 13 of our 30 samples, most frequently in the group 2 (late stage parasites, see supplemental data). This finding indicates some metabolites may be from the host, not the parasite, or the parasite has greater metabolic capabilities than previously understood. Similarly, media components such as phenol red (phenolsulfonphthalein) and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) were measured in parasite metabolomes (see supplemental data). Neither are produced or consumed by the parasite but likely remained associated with our cells following in vitro culture in media that contains these metabolites (i.e. RPMI, see Materials and Methods). The abundance of phenol red and HEPES, as well as cholesterol (a metabolite excluded from parasite membranes (43,44)) are correlated prior to normalization, and these correlations persist following normalization. Moreover, phenol redcontributed to the accuracy of our antimalarial treatment classifier, further confirming that blood batch effects influenced the dataset. Lastly, lipid species were the major class of metabolites detected in our analysis (Fig. 2C) and contributed heavily to PC2 from un and protein-normalized data sets (Table S2), perhaps due to the remaining erythrocyte membranes. These results add to the overwhelming evidence of host cell and media contamination in untargeted metabolomics studies of parasites.
Following these observations, we also explored the effect of different blood batches on metabolome measurements. Because generating sufficient Plasmodium biomass for adequate biological replicates is time-intensive, many experiments require multiple batches of human blood donations. To avoid batch effects, we controlled blood batches across sample groups (Table S1). Prior to these studies, we predicted that the blood batch would have some effects on the metabolome; we did not anticipate, however, that it would be as influential as known stressors, like treatment with antimalarials with established metabolic effects (3, 5). Several results from our analysis support this observation. First, samples from either treatment group did not cluster via PCA (Fig. 3B). Second, we detected none-to-few metabolites whose levels were significantly different between conditions (zero between with and without antimalarial treatment and 1 between various blood batches). Lastly, classifiers from both treatment and blood batch predicted samples with equal accuracy (30% error rate, top predictive metabolites displayed in Fig. 3D and E). Overall, from these analyses, we concluded that sample-to-sample variation exceeded variation associated with either group. We also found 1-arachidonoyl-GPE to be significantly different in abundance across blood batches, which can be explored as a potential biomarker of host contamination. To expand on this idea, we were also able to predict a set of metabolites that are most likely to be host erythrocyte-derived (or influenced by host environment) by identifying the metabolites that are most predictive of blood batch (Fig. 3D). Additional investigations are required since these metabolites may be parasite-derived but only produced when they are in particular environments (e.g. blood batches). Going forward, it may be possible to use these metabolites to quantify host cell contribution to metabolome and assess parasite sample purity or control for host contamination during analysis.
Overall, the methodology and findings from the current study provide a basis for the use of more streamlined in vitro metabolomics approaches for the future investigation of P. falciparum biology. We suggest a set of considerations and recommendations for enhancing the accuracy parasite metabolomics (presented in Fig. 1 Table 1, and below). First, samples must be better purified away from host material. Enrichment methods, whether novel or standard, should be used to increase parasitemia, reducing the number of uninfected host cells. Second, markers of host contamination must be used to evaluate the level of host contamination and resulting data. Our studies suggest that visual detection of ghost material (via microscopy) combined with assessment of host-specific metabolite markers is an effective option to assess sample purity. Finally, data must be normalized to parasite-derived measurements to limit remaining host contamination. With these considerations,metabolomics has the potential to be a powerful tool in the study of intracellular parasites, like Plasmodium.
MATERIALS AND METHODS Parasite Cultivation
Parasite Cultivation
Laboratory-adapted P. falciparum lines were cultured in RPMI 1640 (Roswell Park Memorial Institute medium, Thermo Fisher Scientific, Waltham, MA) containing HEPES (Sigma Aldrich, St Louis, MO) supplemented with either 0.5% AlbuMAX II Lipid-rich BSA (Sigma Aldrich, St Louis, MO) and 50 mg/L hypoxanthine (Thermo Fisher Scientific, Waltham, MA) (referred to as AlbuMAX media) or 20% v/v pooled human plasma for generation of complete RPMI (referred to as cRPMI).Parasite cultures were maintained at 3% hematocrit and diluted with human red blood cells (blood batch noted in Table S1) to maintain parasitemia between 1-3%, with change of culture medium every other day (Fig. 1; Step 1). Cultures were incubated at 37oC with 5% oxygen, 5% carbon dioxide and 90% nitrogen (14). Some samples were treated with antimalarials with metabolic effects to maximize differences between groups (see below and Antimalarial treatment in Table S1).
Parasite Isolation
For isolation of sample group 1, two distinct laboratory-adapted clinical isolates of P. falciparum(BEI Resources, NIAID, NIH: Plasmodium falciparum, Strain IPC 4884/MRA-1240 and IPC 5202/MRA-1238, contributed by Didier Ménard) containing mixed stages with >50% rings were synchronized using 5% sorbitol (Sigma Aldrich, St Louis, MO) (45). The resultant early stage cultures were incubated at 37°C in AlbuMAX media to allow for the development of a schizont predominant population (see Parasite Cultivation above). After the late stage population was confirmed using microscopy, cultures were checked every one to two hours for the development of newly invaded ring stage parasites. If the parasites were treated with antimalarials, it was performed at this stage. Fourteen flasks containing early ring-stage parasites (<3 hours post invasion) were subsequently lysed from the erythrocyte membrane using 0.15% saponin, as previously described (46) (Fig. 1; Step 3). Prior to lysis, sampling of parasite material was taken for determination of erythrocyte count (hemocytometer) and parasitemia (SYBR-green based flow cytometry(47)), which contributed to parasite number determination (total erythrocytes x % parasitemia yields total parasites). Additional samples were obtained following erythrocyte lysis for protein quantification using Bradford reagent (Sigma Aldrich, St Louis, MO). A series of three wash steps were then performed using 1X PBS (Sigma Aldrich, St Louis, MO) using centrifugation at 2000 x g to remove soluble erythrocyte metabolites. Purified material was kept on ice until flash frozen using liquid nitrogen, followed by storage at −80oC until sent for analysis. This procedurewas performed five times for each parasite line to provide 10 replicates for group 1 metabolomic analysis. Additionally, matched parasites (same parasite lineage, media type, stage, blood batches, and purification methods) were also grown without drug treatment (Table S1) to generate 10 additional samples for group 1 untreated (see second comparison in Fig. 3).
For isolation of sample group 2, two Dd2-derived laboratory-adapted clones of P. falciparum(courtesyof Pradip Rathod, University of Washington, continuously cultured in the presence of antimalarial, Table S1) first underwent an initial sorbitol synchronization step as above. The resultant early stage parasites were then incubated at 37°C in cRPMI to allow for the successful transition of P. falciparum to the late trophozoite and schizont stages, occurring 24 to30 hours after initial synchronization. Next, this predominantly late stage population was enriched through magnetic purification using a MACS quad-magnet and MACS multistand (Miltenyi Biotech, Bergisch Gladbach, Germany), as previously described (18) (Fig. 1; Step 2). Briefly, parasite cultures were passed through LS columns with attached sterile syringe needles (BD Biosciences, San Jose CA) at a rate of 2-3 seconds per drop. A series of two to three column washes were performed with 5 ml of warmed cRPMI. To elute the desired material, the column was removed from the magnet prior to adding 5 ml of cRPMI. Column flow-through from 5 flasks containing late stage parasites was allowed to recover in cRPMI for 30 min at 37o C prior to saponin lysis, as described above (Fig. 1; Step 3). Determination of parasite count and protein quantification, as well as subsequent sample washing and freezing, were performed as described above for sample group 1. This procedure was performed five times for each parasite line to provide 10 samples for group 2 metabolomic analysis.
Metabolite Preparation, Analysis, and Identification
Metabolites were identified using Ultrahigh Performance Liquid Chromatography-Mass Spectroscopy (UPLC-MS) by Metabolon, Inc. (Durham, NC). All sample preparations and metabolite identifications were performed according to Metabolon, Inc, standard protocols. Briefly, double stranded DNA was quantified in all samples using the Quant-it Picogreen dsDNA AssayKit (Thermo Fisher, Waltham, MA) according to the manufacturer’s instructions and proteins were precipitated with methanol and centrifuged for extraction (Fig. 1; Step 4). Sample extracts were dried and reconstituted in solvents containing standards (see below) at fixed concentrations to ensure injection and chromatographic consistency. Waters AQUITY ultra-performance liquid chromatography (UPLC) and Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer were used for metabolite detection (Fig. 1; Step 5). Controls that were analyzed in conjunction with the experimental samples included a pooled matrix of all included samples. Internal and recovery standards were used to assess variability and to verify performance of extraction and instrumentation, as routinely performed by Metabolon, Inc.
Raw data was extracted using hardware and software developed by Metabolon, Inc. Metabolites were quantified using area-under-the-curve and identified by comparison to a library of several thousands of pre-existing entries of purified standards or recurrent unknown compounds. Each library standard was uniquely authenticated by retention time/indexes, mass to charge ratios, and chromatographic data. Named metabolites corresponded to library standards or were predicted with confidence according to Metabolon, Inc standard protocols.
Data Analysis
Following the analytical protocol outlined in (48), we first preprocessed metabolite abundances for each sample by imputing missing values with half of the lowest detectable metabolite abundance. Next, we normalized metabolite abundances by sample features, followed by normalization using metabolite features with log transformation, centering, and scaling (49). To limit inter-sample variability, metabolite abundances for each replicate were normalized to sample value for double stranded DNA, protein, or parasite number. To limit inter-metabolite variability, metabolite abundances were log transformed, centered to median (50), and scaled by standard deviation (Fig 1; Step 6). Resultant processed metabolite abundances were used for univariate and multivariate statistics, as well as classification. All analyses were conducted using R (51-59). Welch’s t-tests were used to compare group means for differential abundance determination, assuming unequal variance and normal distribution, and p-values were adjusted using a false discovery rate. The significance cutoff is 0.05. See supplementary information for code and detailed analysis.
Microscopy
Laboratory adapted P. falciparum clones (BEI Resources, NIAID, NIH: Plasmodium falciparum, Strain Patient line E/MRA-1000 or IPC 4884/MRA-1238, contributed by Didier Ménard) at >50% rings were lysed using 0.15% saponin, as previously described (46). Samples were washed twice using 1X PBS (Sigma Aldrich, St Louis, MO) and centrifugation at 2000 x g for 5 minutes. Samples were then stained on slides with either DAPI at 1:20,000 (Sigma Aldrich, St Louis, MO) and CD235a-PE antibody at 1:100 (Thermo Fisher Scientific, Waltham, MA) for fluorescence microscopy or with Giemsa stain (Sigma Aldrich, St Louis, MO) for bright field microscopy. Fluorescent images were acquired using the EVOS FL Cell Imaging System (Thermo Fisher Scientific, Waltham, MA). Bright field images were acquired using a Nikon Eclipse Ci Upright Microscope (Nikon, Melville, NY) equipped with a DMK23U274 camera (The Imaging Source, Charlotte, NC) and NIS Elements Imaging Software (Nikon, Melville, NY).
ACKNOWLEDGEMENTS
This project was supported with funding through the National Institute of Allergy and Infectious Disease R21AI119881 and the University of Virginia CHARGE Enhancement Fellowship (to JLG). MAC and VC are supported by institutional training grants (T32GM008136 and T32AI007046, respectively).
We would like to thank Dr. Jason Papin and Gregory Medlock, as well as the members of the Guler, Papin, and Petri labs at the University of Virginia for helpful discussion and feedback on experimental design and analysis. Additionally, we would like to thank Michelle Warthan (University of Virginia) for laboratory support, and Webster Santos (Virginia Tech) and Kevin Lynch (University of Virginia) for antimalarial synthesis and purification.
Footnotes
↵* Co-first authors
REFERENCES




