

Abstract
Precision medicine can be empowered by a personalized approach to patient care based on the patient’s or pathogen’s unique genomic sequence. For precision medicine, genomic findings must be robust and reproducible, and experimental data capture should adhere to FAIR guiding principles. Moreover, precision medicine requires standardized reporting that extends beyond wet lab procedures to computational methods. Rapidly developing standardization technologies can improve communication and reporting of genomic sequence data analysis steps by utilizing concepts defined in the BioCompute framework, such as error domain, usability domain, verification kit, and provenance domain. These advancements allow data provenance to be standardized and promote interoperability. Thus, a resulting bioinformatics computation instance that includes these advancements can be easily communicated, repeated and compared by scientists, regulators, test developers and clinicians. Easing the burden of doing the aforementioned tasks greatly extends the range of practical application. Advancing clinical trials, precision medicine, and regulatory submissions requires an umbrella of standards that not only fuses these elements, but also ensures efficient communication and documentation of genomic analyses. The BioCompute paradigm and the resulting BioCompute Objects (BCOs) offer that umbrella. Through standardized bundling of high-throughput sequencing studies under BCOs, regulatory agencies (e.g., FDA), test developers, researchers, and clinicians can expand collaboration to drive innovation in precision medicine, with the potential for decreasing the time and cost associated with next generation sequencing workflow exchange, reporting, and regulatory reviews.
Introduction
Precision medicine practice must capture the process of producing, sharing, and consuming genomics information. Capturing the process of genomic data generation will empower individuals, research institutes, clinical organizations, and regulatory agencies to evaluate and trust the reliability of biomarkers generated from complex analyses (e.g., presence of a specific variant). Efforts to promote data-sharing structures for genome-wide association studies (GWAS) offer additional benefits, as seen in the National Center for Biotechnology Information’s (NCBI) Database of Genotypes and Phenotypes (dbGaP)[1] and ClinVar[2] as well as LD Hub, a centralized database of GWAS results for diseases/traits[3]. Although, the importance of data sharing is well accepted, discussions related to recording, reporting, and sharing of analysis protocols are often overlooked.
The need for NGS provenance, analysis, and results is critical as we enter the clinical genomic era. The cost for High Throughput Sequencing (HTS) has fallen from $20 per base in 1990 to less than $.01 per base in 2011, creating massive data accumulation[4]. Lower costs of HTS data generation have increased the availability of data, expediting more types of analyses. In recent years, there has been a focus on novel drug development and precision medicine research to create innovative, reliable, and accurate -omics-based (i.e., genomics, transcriptomics, proteomics) tests[5]. These initiatives allow different data sources to be analyzed by a variety of methods advancing genomic analyses. Often, information about the bioinformatics pipelines used in these analyses is not reported in a format that is easily understood, comprehensive and interoperable. Without a universal standard for communicating how these data are analyzed to obtain a specific result, we quickly encounter the “tower-of-Babel problem” – a problem of diversified languages and miscommunication.
Currently, there are some standards to capture genomic sequencing information and provenance. Fast Healthcare Interoperability Resources[6,7] (FHIR) and organizations such as Global Alliance for Genomics and Health[8] (GA4GH) communicate genomic information, although these cater to specific community domains. The Common Workflow Language[9] (CWL) and Research Objects[10] (RO) capture reproducible workflows in a domain agnostic manner. The BioCompute paradigm unites these standards to provide a provenance-reporting framework for genomic sequencing data analysis in the context of FDA submissions and regulatory review in the form of a BioCompute Object (BCO)[11]. BCOs provide a new harmonizing approach designed to satisfy FAIR Data Principles allowing experimental data and protocols to be findable, accessible, interoperable and reusable[12] in the regulatory and research needs for evaluation, validation, and verification of bioinformatics pipelines[11,13,14]. The BCO also meets the NIH strategic plan for data science[15] which states that the quality of clinical data should be maintained at all stages of the research cycle where the BCO can be adjusted to fit the needs of a specific experiment from generation of the data through the entire analysis process.
This paper focuses on how the FHIR, GA4GH, CWL, and RO standards can be leveraged and harmonized by examining the BioCompute paradigm (See Fig. 1). Once established, the BioCompute framework can be utilized for other types of FDA submissions, such as large clinical trials, where data provenance in analysis datasets can be difficult to communicate.

Schematic of BioCompute Object as a frameworks for advancing regulatory science by incorporating existing standards and introducing additional concepts that include digital signature, usability domain, validation kit, and error domain.
Background
The Academy of Medical Sciences hosted a symposium on reproducibility in pre-clinical biomedical research, where a number of measures for improving reproducibility were identified, including greater openness and transparency, defined reporting guidelines, and better utilization of standards and quality control measures[16]. Issues of reproducibility have resulted in an enormous waste of research resources and have hindered progress in the life sciences, as highlighted by several high profile articles[17,18].
Researchers propose two types of reproducibility: method reproducibility and result reproducibility[19]. The first is defined as providing detailed experimental methods so others can repeat the procedure exactly; the latter, also called replicability, is defined as achieving the same results as the original experiment by closely adhering to the methods. Method reproducibility depends significantly on the completeness of the procedures researchers provide. CWL creates a similar starting point for computational methods—if scientists gather data in the same computational language, then a large part of the provenance model is standardized. As such, CWL and provenance tracking make it easier for researchers to track errors and locate deviances. Research Objects for workflows go further, describing the packaging of the method, provenance logs, and associated data and codes with richly described metadata manifests that include the experiment’s context[20].
Universally reproducible data is an outcome to aim for, but challenges remain. Without repeatability and analytics standards for NGS/HTS studies, regulatory agencies, academic researchers, pharmaceutical companies, diagnostic test developers and the FDA cannot work together to validate results and drive the emergence of new fields[21]. The lack of universally accepted standards to record a computational workflow and the subsequent analysis results in many studies that are not repeatable and therefore not usable in a clinical or practical situation. In response, various workflow management systems and bioinformatics platforms have been developed, and each has a method to track and record computational workflows, pipelines, versions, and parameters used in the studies. However, these efforts remain haphazard and require harmonization. BCOs enable reproducible data and improved information-sharing by tracking the data’s provenance and accurately documenting the trail of processes in a standard and widely applicable way.
Provenance of Data
Reproducible data requires that the data’s origin and history be captured in a standardized format. Provenance here refers to a datum’s history starting from the original source, namely, its lineage. A lineage graph can show the source of a datum in a database, data movement between databases or computational processes, or data generated from a computational process. Complementary to data lineage is a process audit. This provides a historical trail documenting a scientific study, providing snapshots of intermediary states, values for configurations and parameters, and traceability of stepwise analytical processing[22]. Such audit trails should allow an independent reviewer to effectively evaluate a computational investigation. Both types of records gather provenance information that is crucial to ensure accuracy and validity of experimental results. Modern computational workflows can now produce reams of fine-grained but not particularly useful trace records, and modern web developments make data transformation and copying easier, so gathering such material is a daunting challenge. In the molecular biology field alone, there are hundreds of public databases, but only a handful possess the “source” data; the remainder contain secondary views of the source data or views of other publications’ views[23]. Accurately collecting lineage and process records, while obtaining the appropriate granularity of the record keeping and ‘black-box’ steps remains an ordeal[24,25].
Data-provenance tracing issues have far-reaching effects on scientific work. Reproducible experiments rely on confidence in the accuracy and validity of the data, process used, and knowledge generated (final analysis product) especially after undergoing complex, multistep processes of aggregation, modeling, and analysis[26]. Computational investigations require interactions with adjacent disciplines and disparate fields to effectively analyze a large volume of relevant information. These hurdles require a solution beyond Open Data to establish Open Science in the community. Provenance needs to be preserved and shared to provide better transparency and reproducibility[27]. Complex analyses rely heavily on accurately shared data. Standards need to be established to communicate reliable genomic data between databases and other scientists, accurately reporting data provenance and process audit. In order to aid this, an active community has engaged in provenance standardization[28], e.g., culminating in the W3C PROV[29], used by FHIR and ROs, based on the idea of generating an entity target via an agent’s activity (See Fig. 2).
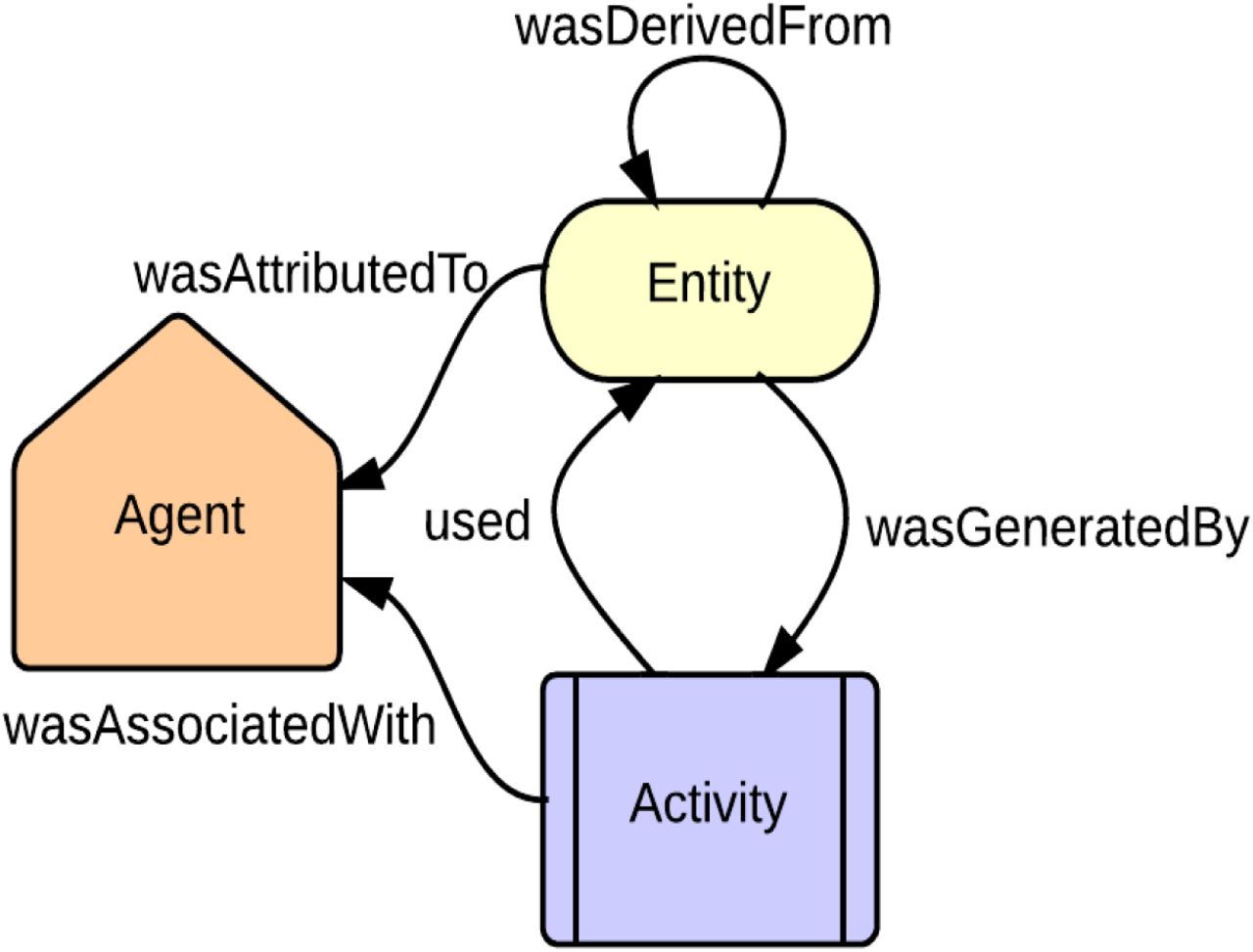
W3C Provenance Specification used in FHIR and ROS. Adapted from http://www.w3.org/TR/prov-primer/
Workflow management systems have been developed to capture analytic processes, and bioinformatic platforms have been built to capture analysis details. Together they can gather provenance information for users, and extending Provenance standards where necessary. However, these systems need greater standardization. This is where BCOs can play a key role, to ensure that provenance is recreated for regulatory approval and that standards such as PROV are appropriately adopted. By properly utilizing the fields in the Identification and Provenance domain, also known as the provenance domain, as defined by the BioCompute framework, a BCO can become a “history of what was computed.”
Key Considerations for Communication of Provenance, Analysis, and Results
Workflow Management Systems
Scientific workflows have emerged as a model to represent and manage complex distributed scientific computations[26]. Scientific workflows capture and link analysis steps and individual data transformations; each step specifies a process or computation to be executed (e.g., a software program or a web service to be invoked), and the steps are linked by the data flow and dependencies. In addition, workflows also capture the mechanisms to carry out the steps in a distributed environment, and a significant amount of work has been done to share workflows and experiments[26,30]. Workflows represent a system to capture complex analysis processes at various levels, as well as the provenance information necessary for reproducibility, result publication, and result sharing/publication [31].
Workflow management systems act to execute and monitor scientific workflows, coordinating the sequential components in a pipeline[32]. Developments in workflow management systems have led to the proposition of using workflow-centric research objects with executable components[5,20]. The use of workflow creation and management software allows researchers to utilize different resources to create complex analysis pipelines that can be executed locally, on institutional servers and on the cloud[33,34]. Extensive reviews of current workflow systems for bioinformatics are linked [33,35-37]. Ongoing systems participate in the current trend of moving from graphical systems back to script-like workflows. These systems are now executed on cloud infrastructure, high performance computing (HPC) systems, and Big Data cluster-computation frameworks, which allow for greater data reproducibility and portability (see Supplementary Info). Workflow management systems capture provenance information, but rarely in the PROV standard. Therefore, BCOs rely on existing regulatory standards like CWL to manage pipeline details, and on ROs and FHIR to unify and enhance interoperability.
Bioinformatics platforms
The dramatic increase in the use of NGS technology for patient management has rapidly increased the need to store, access, and compute sequencing reads and other NGS/biomedical data[38]. These increased requirements have led to a call for usage methods on integrated computing infrastructure, including storage and computational nodes. This kind of integration will minimize transfer costs and remove the bottlenecks found in both downstream analyses and community communication of computational analyses results[39]. For bioinformatics platforms, communication requirements include (a) recording all analysis details such as parameters and input datasets and (b) sharing analysis details so that others can understand and reproduce analyses.
To reduce unprocessed data buildup, several high-throughput[26] cloud-based infrastructures have been developed, including HIVE (High-performance Integrated Virtual Environment)[39,40] and Galaxy[41], along with commercial platforms from companies like DNAnexus (dnanexus.com), and Seven Bridges Genomics (sevenbridges.com), among others. High throughput computing (HTC) environments deliver large amounts of processing capacity over long periods of time. These are ideal environments for long-term computation projects, as with genomic research[42]. Most HTC platforms utilize distributed cloud-computing environments to support extra-large dataset storage and computation, while hosting tools and workflows for many biological analyses. Cloud-based infrastructures also reduce the “data silo” phenomenon by converting data into reproducible formats that facilitate communication (see Supplementary Info). Additionally, the National Cancer Institute has initiated the Cloud Pilots project, in order to test a distributed computing approach for the multi-level, large-scale datasets available on The Cancer Genome Atlas (TCGA) [43].
The genomic community has come to acknowledge the necessity of data sharing and communication to facilitate reproducibility and standardization [44]. Data sharing is crucial in everything from long-term clinical treatments to public health emergency response[45]. As industry policies develop, the need for voluntary and industry-wide standardization becomes undeniable. Extending bioinformatics platforms to include data provenance, standard workflow computation, and encoding results with available standards through BCO implementation will greatly support the exchange of genomic data analysis methods for regulatory review.
Regulatory Supporting Standards
Assessment of data submitted in a regulatory application requires clear communication of data provenance, computational workflows, and traceability. A reviewer must be able to verify that sequencing was done appropriately, pipelines and parameters were applied correctly, and that the final result, like an allelic difference or variant call, is valid. Because of these requirements, review of any clinical trial or any submission supported with NGS results requires considerable time and expertise. Submission of a BCO would ensure that data provenance is unambiguous and that the bioinformatics workflow is fully documented[11,15,23,46,47].
To truly understand and compare computational tests, a standard method (like BCO) requires tools to capture progress and to communicate the workflow and input/output data. As the regulatory field progresses, methods have been developed and are being continually refined to capture workflows and exchange data electronically[26]. See Figure 3 for BCO extensions to NGS analysis to support data provenance and reproducibility.

{kind=link}
{kind=link}
{kind=link}
Generic NGS platform schematic with proposed BioCompute Object integrations and extensions.
Biocompute Objects (BCOs) and Their Harmonizing Efforts
Biocompute Objects (BCOs) were conceptualized to alleviate the disparate nature of NGS computational analyses. The primary objectives of BCOs are to (a) harmonize NGS computational results and data formats and (b) encourage interoperability and success in the verification of bioinformatics protocols. Harmonizing the above standards is especially applicable to clarify genomics/workflow instance provenance for FDA submissions. Each BCO comprises information on the arguments and versions of executable programs in a pipeline, references to input/output data, a usability domain, keywords, a list of authors, and other important sources of metadata. The BCO can serve as a wrapper. Enabling BCOs to incorporate existing standards provides a universal framework for including existing advances in workflow and data specifications. What is novel about the BioCompute paradigm is the combination of existing standards with the methodologies and tools to evaluate an experiment both programmatically and empirically. The BCO takes a snapshot of an experiment’s entire computational procedure adhering to FAIR data guidelines by making it findable through the BCO portal, accessible, interoperable and maintaining it’s richness to make it reusable. Using this snapshot of the BCO, which includes the results from the experiment in the dataset (verification kit), allows any other user to run the exact experiment and produce the same results. The verification kit also allows a BCO to be assessed once someone has decided that they want to change parameters, for example, and use the same BCO. Additionally, through the use of provenance and usability domains, a knowledgeable reviewer can quickly decide if the underlying scientific principles merit approval or further review.
Discussion and Conclusions
Robust and reproducible data analysis is key to successful personalized medicine and genomic initiatives. Researchers, clinicians, administrators, and patients are all tied by the information in electronic health records (EHRs) and databases. Current systems rely on data stored with incomplete provenance records and different computing languages. This has created a cumbersome and inefficient healthcare environment.
The initiatives discussed in this review seek to make data and analyses robust and reproducible to facilitate collaboration and information sharing from data producers to data users. Increased NGS/HTS sequencing creates silos of unusable data, making standardized regulation of reproducibility more difficult. To clear the bottleneck of downstream analysis, the provenance (or origin) of data along with the analysis details (e.g., parameters, workflow versions), must be tracked to ensure accuracy and validity. The development of high-throughput cloud-based infrastructures like DNAnexus, Galaxy, HIVE, and Seven Bridges Genomics enables users to capture data provenance and store the analyses in infrastructures that allow easy user interaction.
Platform-independent provenance has largely been ignored in HTS. Emerging standards enable both representation of genomic information and linking of provenance information. By harmonizing across these standards, provenance information can be captured across both clinical and research settings extending into the conceptual experimental methods and the underlying computational workflows. There are several use cases of such work, including submission for FDA diagnostic evaluations, as is being done with the BCO effort. Such standards also enable robust and reproducible science, and facilitate open science between collaborators. At this juncture, it is imperative to lead the development of these standards to satisfy the needs of downstream consumers of genomic information.
The need to reproducibly communicate HTS/NGS computational analyses has led to collaboration among disparate industry players. Through various conferences/workshops, attention has increased exposure to standardization, tracking, and reproducibility methods[48,49]. Standards like FHIR and ROs capture the underlying data provenance to be shared in frameworks like GA4GH, enabling collaboration around reproducible data and analyses. New computing standards like Common Workflow Language (CWL) increase the scalability and reproducibility of data analysis. The BioCompute paradigm acts as a harmonizing umbrella to facilitate data submitted to regulatory agencies, increasing interoperability in the genomic field. BioCompute specifications, available at https://osf.io/h59uh/, can be used to generate BCOs by any bioinformatics platform that automatically pulls underlying data and analysis provenance into its infrastructure. Ongoing BCO pilots are currently working to streamline the flow to provide users with effortlessly reproducible bioinformatics analyses. As BCOs aim to simplify FDA approval, these pilots mirror clinical trials involving NGS data for FDA submissions. Fusing bioinformatics platforms and HTS standards to capture data and analyze provenance for BCOs make robust and reproducible analyses and results an attainable standard for the scientific community.
Disclaimer
The contributions of the authors are an informal communication and represent their own views.
Acknowledgements
We would like to recognize all the speakers and participants of the 2017 HTS-CSRS workshop who facilitated the discussion on standardizing HTS computations and analyses. The workshop was co-sponsored by FDA and GW. The comments and input during the workshop were processed and integrated into the BCO specification document and this manuscript. The participants of the 2017 HTS-CSRS workshop are listed here: https://osf.io/h59uh/wiki/2017%20HTS-CSRS%20Workshop/. This work has been funded in part by FDA (HHSF223201510129C) and The McCormick Genomic and Proteomic Center at the George Washington University.
References




