

ABSTRACT
The inactivation of the ciliary proteins polycystin 1 or 2 leads to autosomal dominant polycystic kidney disease (ADPKD), the leading genetic cause of chronic kidney disease. Both cilia signaling and interstitial inflammation play a critical role in the disease. Yet, the reciprocal interactions between immune and tubular cells are not well characterized. The transcription factor STAT3, which is suspected to fuel ADPKD progression, is involved in crosstalks between immune and non-immune cells in various tissues and is a component of the cilia proteome. Here, we explore how STAT3 intersects with cilia signaling, renal inflammation and cyst growth using conditional murine models of post-developmental Pkd1, Stat3 and cilia ablation. Our results indicate that, although primary cilia directly modulate STAT3 activation in vitro, the bulk of STAT3 activation in polycystic kidneys occurs through an indirect mechanism in which primary cilia trigger macrophage recruitment to the kidney, which in turn promotes STAT3 activation. Surprisingly, while disrupting Stat3 in Pkd1 deficient tubules slightly reduced cyst burden, it resulted in a massive infiltration of the cystic kidneys by macrophages and T cells, precluding any improvement of kidney function. Mechanistically, STAT3 represses the expression of the inflammatory chemokines CCL5 and CXCL10 in polycystic kidneys and cultured tubular cells. These results demonstrate that STAT3 is not a critical driver of cyst growth in ADPKD but plays a major role in the crosstalk between immune and tubular cells that shapes disease expression.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD), the leading genetic cause of kidney disease, accounts for 5% of patients with end-stage renal disease (ESRD). Therapies allowing patients to avoid ESRD are currently lacking. Indeed, the only drug to have shown some efficacy to date only modestly reduces kidney function decline at the expense of considerable side effects1. Thus, a better understanding of ADPKD pathophysiology is of major importance to delineate more effective therapies.
ADPKD is caused by inactivating mutations affecting either PKD1, which encodes polycystin 1, a putative orphan receptor, or PKD2, which encodes polycystin 2, a cation channel that interacts with polycystin 12. Although ADPKD patients carry heterozygous germline PKD1 (or PKD2) inactivating mutations, cyst development is caused by somatic mutations affecting the remaining functional PKD1 (or PKD2) allele in a subset of tubular cells that will proliferate to form cysts3. In adult mice, bi-allelic loss of Pkd1 in tubular cells results in a biphasic proliferative response leading first to non-cystic kidney enlargement (pre-cystic phase), which is followed by the rapid development of cysts (cystic phase)4,5. While cyst formation is the consequence of a genetic defect in tubular cells, this process is not cell-autonomous. Indeed, cystogenesis is associated with important modifications of the renal microenvironment, most importantly immune cell recruitment in the vicinity of Pkd1 deficient tubules, which, in turn, affects cyst growth and kidney function decline6–10, as well as quantitative and qualitative alterations of the extracellular matrix11,12, which are also thought to contribute to disease progression.
Primary cilia play an essential role in ADPKD4,10. The primary cilium is a microtubule-based organelle that protrudes from the apical surface of tubular cells to integrate mechanical and chemical cues delivered by the urinary flow. The Polycystin 1/2 complex localizes to the primary cilium and this localization is essential to repress cyst formation13,14. Remarkably, cilia ablation drastically suppressed both pre-cystic cell proliferation and cyst formation in Pkd1 mutant animals4,10. However, the nature of the deleterious signals delivered by cilia in this context are only starting to emerge. We and others have recently shown that cilia ablation prevents the induction of the macrophage chemoattractant CCL2 in response to Pkd1 disruption and that Ccl2 inactivation dampens renal macrophages infiltration and cyst formation in Pkd1 mutant mice7,10. While these results demonstrate that primary cilia control the communication between immune and tubular cells, the molecular network shaping the interaction between polycystin deficient tubular cells and the neighboring immune cells remain poorly understood. STAT3 is a pleiotropic transcription factor involved in a large spectrum of physiological and pathophysiologic processes15. STAT3 activation is principally mediated by its phosphorylation on tyrosine 705 by various cytosolic or membrane kinases, which promotes its nuclear accumulation and transcriptional activity. STAT3 represents an interesting candidate for immune-tubular cell communication as it stands at the crossroads of cilia signaling, immune regulation and cyst growth. Numerous studies have pinpointed STAT3 as a general driver of both pro- and anti-inflammatory crosstalk between epithelial and neighboring immune cells16,17. A recent proximity biotinylation based proteomic screen identified STAT3 as a bona fide component of the ciliary machinery18. In ADPKD and murine PKD models, phosphorylated STAT3 accumulates in the nucleus of cyst lining epithelial cells19,20. Furthermore, the cytosolic tail of polycystin 1 binds STAT3 in vitro and modulates its transcriptional activity18. Lastly, the positive effect of STAT3 inhibitory drugs in orthologous models of the disease has led to the assumption that tubular STAT3 promoted cystogenesis19,20. However, how STAT3 intersects with cilia signaling, renal inflammation and cyst growth in the disease is not understood. The limited specificity of the inhibitors used in these studies20,21 and their impact on STAT3 activation in non-tubular cells preclude definitive conclusions regarding the precise function of tubular STAT3 in polycystin deficient tubular cells.
Considering these uncertainties, we aimed to clarify (i) the function of primary cilia in STAT3 activation in tubular cells and (ii) its subsequent function in cyst growth, renal inflammation and kidney function decline.
MATERIALS AND METHODS
Mice
All animal experiments were conducted according to the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals, as well as the German and French laws for the animals welfare, and were approved by regional authorities. Mice were housed in a specific pathogen-free facility, fed ad libitum and housed at constant ambient temperature in a 12-hour day/night cycle. Breeding and genotyping were done according to standard procedures.
Pkd1flox/flox mice (B6.129S4-Pkd1tm2Ggg/J, stock number: 010671, C57BL/6 genetic background) and Ccl2-RFPflox/flox (B6.Cg-Ccl2tm1.1Pame/J, stock number: 016849, C57BL/6 genetic background) were purchased from The Jackson Laboratories, Kif3aflox/flox mice (Kif3atm1Gsn, C57BL/6 genetic background) were kindly provided by Peter Igarashi, and were crossed to Pax8rtTA (Tg(Pax8-rtTA2S*M2)1Koes)22 and TetOCre (Tg(tetO-cre)1Jaw)23 mice to generate an inducible tubule-specific Pkd1 knockout (further referred to as iPkd1ΔTub), Pkd1; Ccl2 knockout (further referred to as iPkd1ΔTub; iCcl2ΔTub) and Pkd1; Kif3a knockout (further referred to as iPkd1ΔTub; iKif3aΔTub) as previously described10. Stat3flox/flox mice (Stat3tm1Vpo1, initially kindly provided by Valeria Poli, FVB/N genetic background)24 were backcrossed for three generations with iPkd1ΔTub mice on a pure C57BL/6 genetic background. The progeny was then intercrossed to generate mice with inducible tubule-specific Pkd1 knockout (further referred as iPkd1ΔTub) or Pkd1; Stat3 knockout (further referred to as iPkd1ΔTub; iStat3ΔTub) on a common mixed FVB/N; C57BL/6 genetic background.
To induce floxed alleles recombination mice received doxycycline (Abcam, ab141091) in drinking water (2mg/mL with 5% sucrose, protected from light) from post-natal day 28 (P28) to P42. Littermates (lacking either TetOCre or Pax8rtTA) were used as controls. Experiments were conducted on males.
Plasma Analyses
Retro-orbital blood was collected from anaesthetized mice. Plasma blood urea nitrogen (BUN) and plasma creatinine was measured using a Konelab 20i analyzer (Thermo Scientific).
Morphological Analysis
Mouse kidneys were fixed in 4% paraformaldehyde, embedded in paraffin, and 4µm sections were stained with periodic acid-Schiff (PAS). Stained sections were imaged using an E800 microscope (Nikon) equipped with a Plan Neofluar 20x/0.30 NA objective and a digital Camera Dx/m/1200 (Nikon) coupled to NIS software (Laboratory Imaging Ltd). PAS stained full size kidney images were recorded using a whole slide scanner Nanozoomer 2.0 (Hamamatsu) equipped with a 20x/0.75 NA objective coupled to NDPview software (Hamamatsu).
Cortical tubular lumen fractional area was measured with ImageJ software from PAS stained full size kidney images. Interstitial infiltration score was evaluated by two independent observers in a blinded fashion assessing the overall infiltration of the whole kidney section stained with PAS.
Cell culture
Madin-Darby canine kidney (MDCK, kind gift of Prof. Kai Simons, MPI-CBG, Dresden, Germany) cells were cultured using Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (Sigma-Aldrich, F7524) and 1% Penicillin-Streptomycin (Life Technologies, 15140122). MDCK cells with a tetracycline-inducible Kif3a knockdown (further referred to as Kif3a-i), Ift88 knockdown (further referred to as Ift88-i) or control (further referred to as Luci-i) using a lentivirus-based transduction system have been previously described10. 150,000 cells/cm² were seeded with or without 1µg/mL doxycline hyclate (Abcam, ab141091) for 10 days on 12mm Transwell polycarbonate membranes (COSTAR, 3401). RNAs and proteins were extracted after 16 hours serum deprivation.
Mouse inner medullary collecting duct (mIMCD-3) cells were grown in 50% DMEM high-glucose pyruvate (Gibco, 41966052), 50% F-12 Nutrient Mixture (Gibco, 21765037) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin. mIMCD-3 cells with a stable Stat3 knockdown (further referred to as Stat3 sh1 and 2) have been already reported24. 30,000 cells/cm² mIMCD-3 were seeded. Confluent mIMCD-3 cells were serum deprived for 16 hours and then stimulated with 1µg/mL LPS (Invivogen, tlrl-3pelps). RNAs and proteins were extracted 24 hours after stimulation.
All cells were regularly tested for mycoplasma contamination and were mycoplasma free.
Quantitative real-time PCR (qRT-PCR)
Total RNAs were obtained from whole kidneys or cells using NucleoSpin® RNA Kit (Macherey Nagel) and reverse transcribed using High-Capacity cDNA Reverse Transcription kit (ThermoFisher Scientific). Quantitative PCR was performed with iTaq™ Universal SYBR® Green Supermix (Bio-Rad) on ViiA 7 Real-Time PCR system (ThermoFisher Scientific) coupled to QuantStudio™ software V1.3. Each biological replicate was measured in technical duplicates. The primers used for qRT-PCR are listed in Supplementary Table 1.
Immunohistochemistry
4µm sections of paraffin-embedded kidneys were submitted to antigen retrieval and avidin/biotin blocking (Vector, SP-2001). Sections were incubated with primary antibody followed by biotinylated antibody, HRP-labeled streptavidin (Southern Biotech, 7100-05, 1:500) and 3-3′-diamino-benzidine-tetrahydrochloride (DAB, Dako, K3468) revelation. Stained sections were imaged using an E800 microscope (Nikon) equipped with a Plan Neofluar 20x/0.30 NA objective and a digital Camera Dx/m/1200 (Nikon) coupled to NIS software (Laboratory Imaging Ltd).
Western blot
Kidneys were lyzed in 50mM Tris pH8, 200mM NaCl, 1mM EDTA, 1mM EGTA, 1mM DTT, 1% SDS buffer. Cells were lysed in modified RIPA buffer lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1%sodium laury sulfate. Nucleo-cytoplasmic fractionation was performed using NePer kit (Thermofisher). Lysis buffers were supplemented with protease and protein phosphatase inhibitors tablets (Pierce, A32959). Protein content was determined with Pierce BCA protein assay kit (Pierce, A23225). Equal amounts of protein were resolved on 4-20% gradient gels (Bio-Rad) under reducing conditions, transferred and incubated with primary and secondary antibodies and visualized using ChemiDoc™ (Bio-Rad) ismaging system coupled to Image Lab software (Bio-Rad).
Antibodies
For western blotting
pSTAT3Y705 (Cell Signaling Technology, 9145, 1:1,000), STAT3 (Cell Signaling Technology, 9132, 1:1,000), GAPDH (Millipore, MAB374, 1:5,000), α-TUBULIN (Sigma, T5168, 1:20,000), Lamin A/C (Abcam, ab108922, 1:1,000) and RELA (Cell Signaling Technology, 4764, 1:1,000).
For immunostaining
pSTAT3Y705 (Cell Signaling Technology, 9145, 1:100), STAT3α (clone D1A5, Cell Signaling Technology, 8768, 1:100), F4/80 (Clone Cl:A3-1, Bio-Rad, MCA497R, 1:100), CD3 (Abcam, ab16669; 1:100) and LY6B (Abcam, ab53457, 1:100).
Statistical Analysis
Data were expressed as means or as means ± SEM. Differences between the experimental groups were evaluated using one or two-way ANOVA as appropriate, followed when significant (P < 0.05) by the Tukey-Kramer test. When only two groups were compared, two-tailed t test or Mann–Whitney test was used as appropriate. For Kaplan Meier survival curves, Log-rang test was applied. The statistical analysis was performed using GraphPad Prism V6 software. No samples were excluded from analyses. All image analyses (immunohistochemistry, immunofluorescence) and mouse phenotypic analyses were performed in a blinded fashion.
RESULTS
Primary cilia regulate STAT3 activation in tubular cells
To investigate if STAT3 activation occurred at an early stage of the disease, we took advantage of transgenic mice allowing the inducible inactivation of Pkd1 in tubular cells upon doxycycline treatment (iPkd1Δtub). At twelve week of age (6 weeks after the completion of doxycycline treatment), tubular cell proliferation is increased and kidneys are enlarged but minimally cystic10,25. Immunolabelling of kidney sections from 12 week old iPkd1Δtub and control mice with STAT3 phospho-specific antibody directed against Tyrosine 705 revealed an increase in both tubular and interstitial phosphorylated STAT3 positive nuclei in polycystin deficient kidneys compared to controls (Figure 1A-C). Of note, most of the signal was observed in areas with mild tubular dilation and interstitial infiltration. Ablation of primary cilia in iPkd1Δtub mice through simultaneous deletion of Kif3a markedly blunted both tubular and interstitial STAT3 activation (Figure 1A-C).

(A-C) Representative images (A) and quantification (B-C) of pSTAT3Y705 immunostaining in kidneys from 12 weeks old control, iKif3aΔTub, iPkd1ΔTub, and iPkd1ΔTub; iKif3aΔTub mice (6 weeks after the completion of doxycycline treatment). Scale bar: 50µm. Each dot represents one individual mouse. One-way ANOVA followed by Tukey-Kramer test, **P<0.01, ***P<0.001.
We then investigated the mechanisms allowing cilia dependent STAT3 activation in vivo. While the identification of STAT3 as a component of the primary cilia proteome renders a direct regulation of STAT3 by primary cilia plausible, primary cilia have been shown to orchestrate an inflammatory response through the induction of CCL2 in tubular cells. In the heart, CCL2 induces macrophage recruitment, which in turn activates STAT3 in cardiomyocytes through paracrine signaling26,27. We evaluated the possibility of such a non-cell-autonomous pathway to tubular STAT3 activation in vivo. We analyzed the impact of tubule specific Ccl2 disruption on STAT3 in the polycystic kidney. We and others have previously shown that Ccl2 disruption reduced renal macrophages infiltrates and cyst burden in polycystic kidneys7,10. Indeed, western blot of kidney homogenates revealed that Ccl2 inactivation strongly reduced renal STAT3Y705 phosphorylation (Figure 2A). In line with this observation, Ccl2 inactivation also reduced the mRNA abundance of the STAT3 transcriptional targets Socs3 and Lcn2 (Figure 2B, C). Immunolabelling confirmed that Ccl2 inactivation markedly reduced both interstitial and tubular STAT3 phosphorylation (Figure 2D-F). However, residual STAT3 activation in renal tubules was detected in the kidneys of iPkd1ΔTub; iCcl2ΔTub mice, although to a level that was not statistically different from control kidneys. To investigate if primary cilia can directly modulate STAT3 activation independently of macrophage recruitment, we analyzed STAT3 phosphorylation in tetracycline-inducible Kif3a-silenced (Kif3a-i) or Ift88-silenced (Ift88-i) MDCK cells. We found that tetracycline treatment, which results in cilia ablation in both cell lines28,29, modestly but significantly reduced STAT3 phosphorylation as compared to control cells expressing an inducible shRNA targeting luciferase (Luci-i; Supplementary Figure 1A, B).

(A) Western blot of STAT3 phosphorylation in kidney lysates from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iCcl2ΔTub mice (7 weeks after the completion of doxycycline treatment). Each lane indicates one individual mouse. (B-C) Quantification of Socs3 (B) and Lcn2 (C) mRNA abundance in kidneys from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iCcl2ΔTub mice. AU: arbitrary units. (D-F) Representative images (D) and quantification (E-F) of pSTAT3Y705 immunostaining in kidneys from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iCcl2ΔTub mice. Scale bar: 50µm. (B, C, E, F) Each dot represents one individual mouse. One-way ANOVA followed by Tukey-Kramer test, ns: not significant, *P<0.05, **P<0.01, ***P<0.001.
Collectively, these results indicate that, although primary cilia are direct positive regulators of STAT3 phosphorylation on Y705 in vitro, cilia dependent STAT3 activation in polycystic kidneys is mainly mediated through paracrine mechanisms requiring CCL2 dependent macrophage recruitment.
Tubule specific Stat3 inactivation does not prevent pre-cystic kidney growth in response to Pkd1 disruption
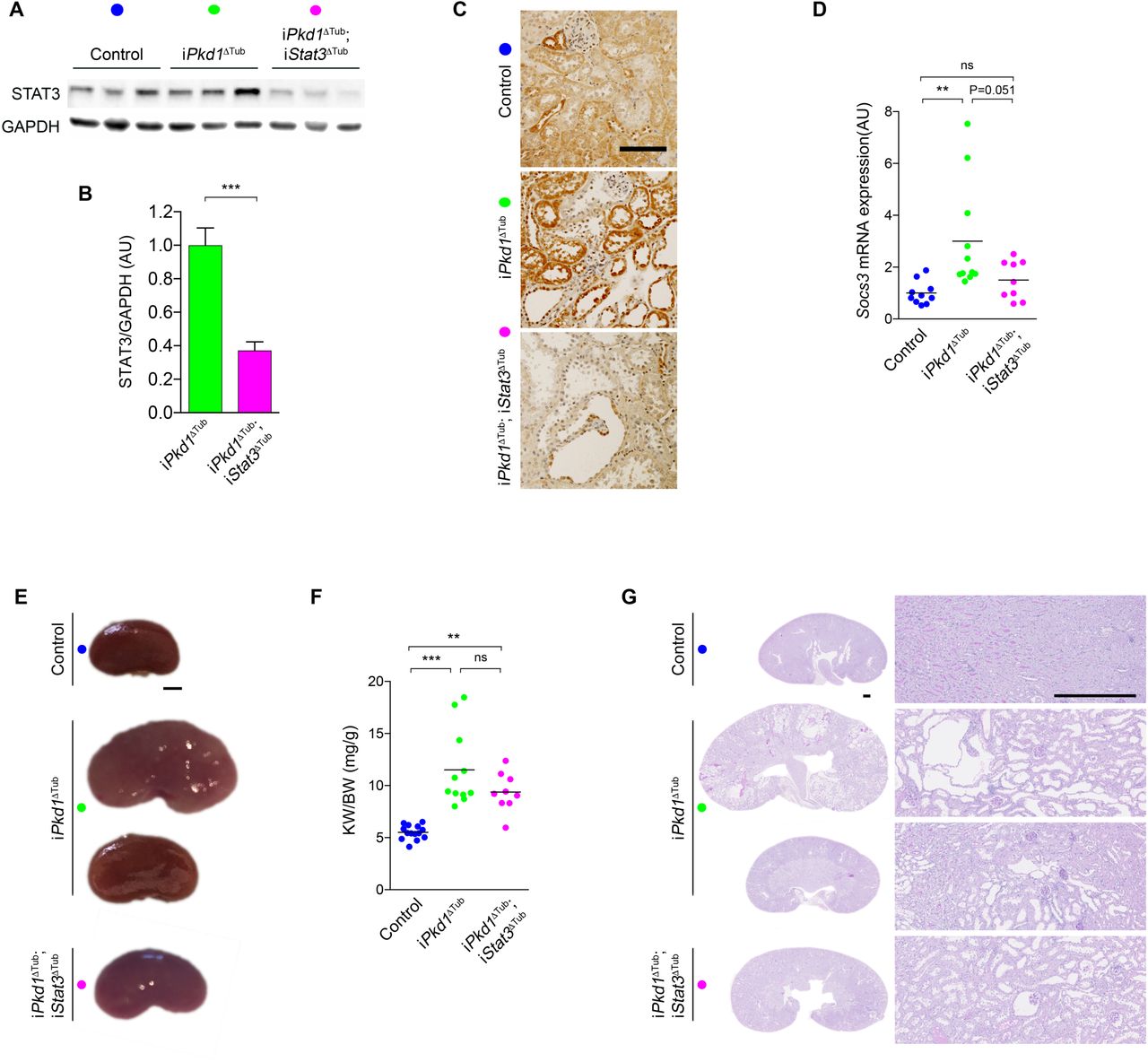
How CCL2 dependent macrophage recruitment promotes cyst growth remains unclear in ADPKD. As STAT3 activation in tubular cells has been proposed to promote cyst growth, we investigated whether STAT3 activation in renal tubules is instrumental in ADPKD progression. To this aim, we crossed iPkd1Δtub with Stat3flox/flox mice and derived mice allowing the inactivation of Pkd1 alone (iPkd1Δtub) or in association with Stat3 (iPkd1Δtub; iStat3Δtub) specifically in tubular cells upon doxycycline treatment. At 13 weeks of age (7 weeks after the completion of doxycycline treatment), western blot analysis of kidney homogenates revealed a significant reduction of STAT3 protein in kidneys from iPkd1Δtub; iStat3Δtub mice as compared to iPkd1Δtub mice (Figure 3A, B). STAT3 immunolabelling confirmed the loss of STAT3 protein in renal tubular cells from iPkd1Δtub; iStat3Δtub mice (Figure 3C). In line with this observation, Stat3 tubular inactivation in polycystin deficient mice markedly lessened Socs3 induction (Figure 3D). At this time point, both iPkd1Δtub and iPkd1Δtub; iStat3Δtub mice displayed kidney enlargement but overt polycystic kidneys was only observed in few iPkd1Δtub mice (Figure 3E-G). Although iPkd1Δtub; iStat3Δtub kidneys tend to be smaller and less variable than iPkd1Δtub kidneys, this difference did not reach statistical significance. Collectively, these results demonstrate that Stat3 is dispensable for the early kidney growth.

(A-B) Representative western blot (A) and quantification (B) of STAT3 in kidney homogenates from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice (7 weeks after the completion of doxycycline treatment). Each lane indicates one individual mouse. Bars are mean ± SEM of 6 mice per group. AU: arbitrary units. Two tailed t test, ***P<0.0001. (C) STAT3 immunostaining of kidney sections from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. Representative images of 3 mice per group. Scale bar: 100µm. (D) Quantification of Socs3 mRNA abundance in kidneys from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. (E) Representative kidneys from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. Scale bar: 2 mm. (F) Kidney weight to body weight ratio (KW/BW) at 13 weeks. (G) Representative Periodic Acid–Schiff (PAS) stained kidney sections from 13 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice at 13 weeks. Scale bar: 0.5mm. (D, F) Each dot represents one individual mouse. One-way ANOVA followed by Tukey-Kramer test, ns: not significant, **P<0.01, ***P<0.001.
Tubule specific Stat3 inactivation does not prevent cystic kidney growth nor kidney function decline in response to Pkd1 disruption
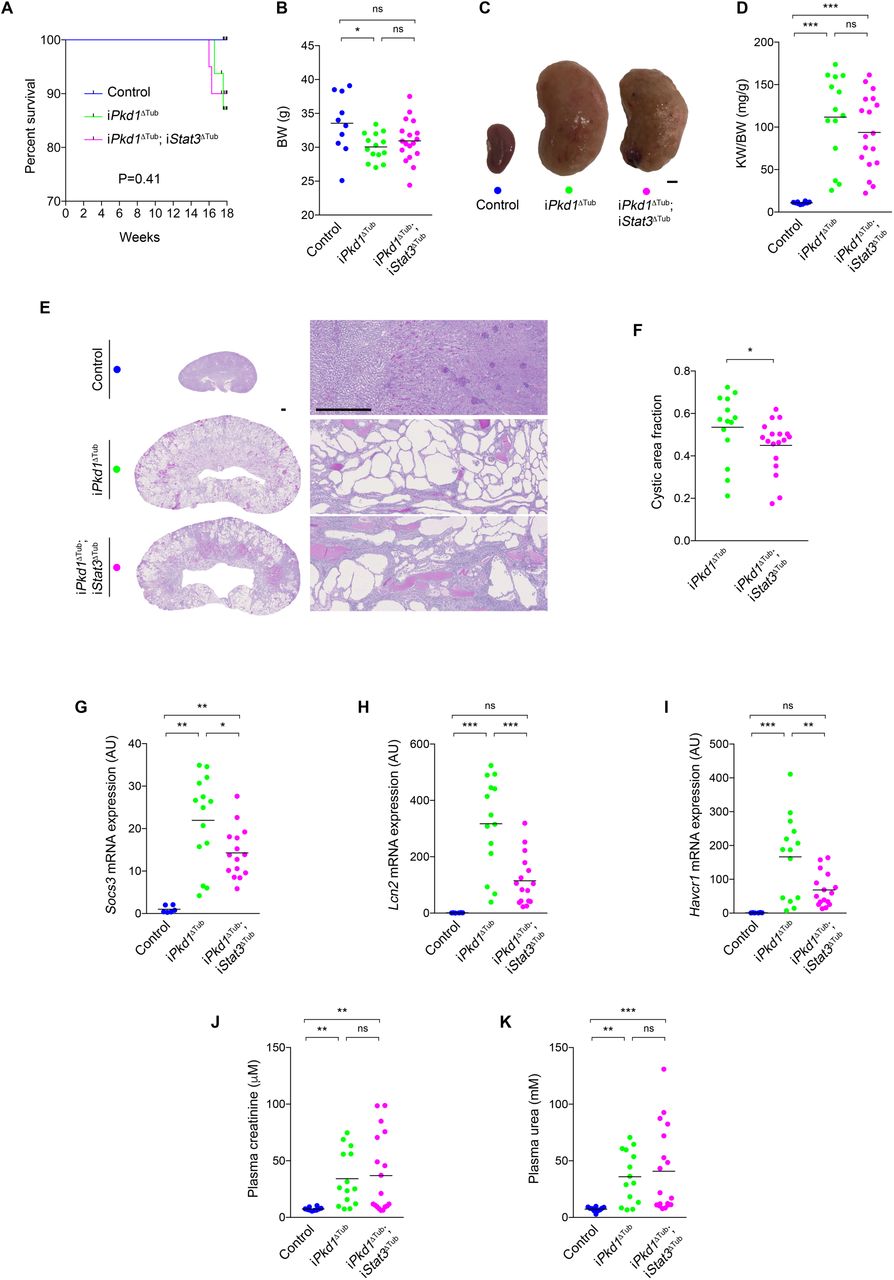
To assess the impact of loss of Stat3 function on overt cystogenesis and kidney function decline, we let iPkd1Δtub and iPkd1Δtub; iStat3Δtub mice age for 18 weeks (12 weeks after the completion of doxycycline treatment). Two over sixteen iPkd1Δtub and two over twenty iPkd1Δtub; iStat3Δtub mice but none of the control mice died before the completion of the study (between 16 and 18 weeks; Figure 4A). iPkd1Δtub and iPkd1Δtub; iStat3Δtub mice displayed a parallel modest decline in body weight in comparison to control mice at sacrifice (Figure 4B). Macroscopic inspection revealed more irregular kidneys in iPkd1Δtub; iStat3Δtub than in iPkd1Δtub mice (Figure 4C), but overall, both iPkd1Δtub and iPkd1Δtub; iStat3Δtub mice developed similar massive kidney enlargement as compared to control mice (Figure 4D). Microscopic inspection of the kidneys revealed that Stat3 deletion resulted in a slight but significant decrease of cystic area (Figure 4E, F). In line with this modest effect on cyst burden, Stat3 disruption not only reduced the expression of the STAT3 transcriptional targets Socs3 and Lcn2, but also decreased the expression of Havcr1 transcript, which encodes KIM-1, a marker of tubular injury that positively correlates with cyst burden in ADPKD patients30 (Figure 4G-I). In spite of this reduction in cyst burden both iPkd1Δtub and iPkd1Δtub; iStat3Δtub mice displayed a similar increase in plasma creatinine and urea compared to control mice (Figure 4J, K), indicating that Stat3 tubular deletion did not prevent polycystic kidney function decline. Of note, the more severe reductions in kidney function were observed in iPkd1Δtub; iStat3Δtub animals (Figure 4J, K). These results demonstrate that tubular STAT3 plays only a marginal role in cyst growth in ADPKD and that Stat3 tubular deletion does not prevent kidney function decline.

(A) Kaplan Meyer survival curves of control, iPkd1ΔTub (n=16) and iPkd1ΔTub; iStat3ΔTub (n=20) mice. (B) Body weight (BW) of 18 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub animals (12 weeks after the completion of doxycycline treatment). (C) Representative kidneys from the same animals. Scale bar: 2 mm. (D) Kidney weight to body weight ratio (KW/BW) at 18 weeks. (E-F) Representative PAS staining of kidneys sections (E) and measurement of cysts area fraction (F) from 18 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. Scale bar: 0.5 mm. Mann-Whitney test, * P<0.05. (G-I) Quantification of Socs3 (G), Lcn2 (H) and Havcr1 (I) mRNA abundance in kidneys from 18 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. (J-K) Measurement of plasma creatinine (J) and urea (K) from 18 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub animals. (B, D, F, G-K) Each dot represents one individual mouse. (B, D, G-K) One-way ANOVA followed by Tukey-Kramer test, ns: not significant, *P<0.05, **P<0.01, ***P<0.001.
Tubule specific Stat3 inactivation increases polycystic kidney inflammation
The fact that iPkd1Δtub; iStat3Δtub mice presented severe renal function impairment in spite of a reduction in cyst burden suggested that Stat3 deletion could somehow worsen kidney damage without accelerating cyst growth. Indeed, we were surprised to observe a notable increase in interstitial inflammation in iPkd1Δtub; iStat3Δtub mice (Figure 4G). Repeated semi-quantitative blinded assessment of the severity of the inflammatory infiltrates confirmed this difference (Figure 5A). Immunolabelling identified both T cells (CD3+) and macrophages (F4/80+) in the large inflammatory areas of iPkd1Δtub; iStat3Δtub mice, while neutrophils (LY6C+) were rarely observed (Figure 5B). Consistently, we observed that Stat3 inactivation was associated with a significant increase in the renal abundance of Cd3 (expressed by T cells) and Ccr2 (expressed by infiltrating macrophages) transcripts (Figure 5C, D).

(A) Representative examples of interstitial inflammation scoring and distribution of renal interstitial inflammation scores in PAS stained kidneys from 18 weeks old iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. Numbers in the bars indicate number of mice per score; P is P value from Mann-Whitney test. (B) Representative images of PAS, F4/80, CD3 and LY6C immunostaining of kidney sections from 18 weeks old iPkd1ΔTub; iStat3ΔTub. Scale bars: 1mm for upper left panel and 0.1mm for other panels. (C-H) Quantification of Cd3 (C), Ccr2 (D), Ccl2 (E), Il6 (F), Ccl5 (G) and Cxcl10 (H) mRNA abundance in kidneys from 18 weeks old control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice. Each dot represents one individual mouse. One-way ANOVA followed by Tukey-Kramer test, ns: not significant, *P<0.05, **P<0.01, ***P<0.001.
A heightened inflammatory response has been observed after STAT3 ablation in hematopoietic cells, osteoblasts or keratinocytes31,32. In macrophages, the proinflammatory consequences of Stat3 deletion are linked to an excessive activation of NF-κB signaling resulting in increased expression of NF-κB target genes33. We therefore investigated the impact of Stat3 deletion on the expression of selected NF-κB targets. While Pkd1 loss of function led to an increase in the abundance of Ccl2, Il6, Ccl5 and Cxcl10 transcripts, Stat3 inactivation amplified this phenomenon (Figure 5E-H). This was particularly evident for Ccl5 and Cxcl10, whose expression was 6 and 3 fold higher in iPkd1Δtub; iStat3Δtub than in iPkd1Δtub kidneys, respectively. In contrast, we did not detect any significant difference in Tnfa, Il1b or Cxcl3 mRNA abundance between iPkd1Δtub and iPkd1Δtub; iStat3Δtub kidneys (Supplementary Figure 2), indicating that Stat3 deletion led to the upregulation of specific inflammatory mediators. In macrophages, STAT3 anti-inflammatory effects have been linked to the transcriptional repression of the E2 ubiquitin-conjugating enzyme UB2N, which stabilizes NF-κB signaling resulting in the nuclear accumulation of the transcription factor RELA32. However, contrary to its published effect in macrophages, tubule specific Stat3 inactivation was not associated with an increase in Ub2n mRNA abundance in Pkd1 deficient kidneys, suggesting alternative mechanisms (Supplementary Figure 2).
To investigate if STAT3 directly functions as a repressor of NF-κB induced proinflammatory cytokine in tubular cells, we analyzed the impact of STAT3 depletion on the induction of Il6, Ccl2, Ccl5 and Cxcl10 expression by LPS, a major activator of NF-κB, in tubular mIMCD-3 cells. While we were not able to detect Il6 transcript in those cells, LPS treatment induced a robust induction of Ccl2, Ccl5 and Cxcl10 transcripts (Figure 6A-C). Strikingly, STAT3 depletion led to a proportional increase in the abundance of Ccl5 and Cxcl10 mRNA in response to LPS (Figure 6A, B). Ccl2 mRNA levels showed the same trend as Ccl5 and Cxl10, but statistically significant increase was restricted to the cell line with a profound STAT3 depletion (Figure 6C). Interestingly, LPS treatment led to STAT3 phosphorylation and nuclear accumulation, which was reduced by STAT3 depletion (Figure 6A, B). Contrary to macrophages, STAT3 depletion in mIMCD-3 did not increase the nuclear accumulation of RELA (Figure 6D, E). These results indicate that STAT3 dampens Ccl5 and Cxcl10 induction by LPS without affecting RELA nuclear accumulation. Collectively, these results indicate that STAT3 represses the expression of proinflammatory cytokines and restricts immune cell infiltration in ADPKD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A-C) Quantification of Ccl2 (A), Ccl5 (B) and Cxcl10 (C) mRNA abundance in mIMCD3 cells stably expressing shRNAs targeting STAT3 (sh1 and sh2) or a scramble control shRNA treated with lipopolysaccharide (LPS) or vehicle for 24 hours (24h). Bars are mean ± SEM of 8 independent experiments. Two-way paired ANOVA followed by Tukey-Kramer test, ns: not significant, *P<0.05, **P<0.01, ***P<0.001. (D) Western blot of STAT3 phosphorylation and RELA expression in whole cell lysates from scramble shRNA (scr) and STAT3 shRNA (sh1 and 2) mIMCD3 cells, 24h after LPS or vehicle treatment. Representative western blot of 3 independent experiments. (E) Western blot of STAT3 and RELA expression in nuclear extracts from mIMCD3 cells expressing the indicated shRNAs, 24h after LPS or vehicle treatment. Whole cell lysates (WCL) served as a positive control for tubulin labelling. Representative western blot of 4 independent experiments.
DISCUSSION
While multiple intrinsic defects in tubular cells homeostasis are observed in ADPKD34–36, accumulating evidence demonstrates that interstitial inflammation also plays an important role in the disease6–8,10,37,38. Yet, the nature of the factors mediating communication between tubular and immune cells and their impact on disease expression are not well-characterized. Two distinct pathways have been shown to promote macrophage accumulation in polycystic kidneys. First, polycystin deficiency triggers monocyte migration to the kidney through the induction of CCL27,10. Second, kidney injury has been shown to promote cyst progression by triggering CSF1 dependent resident macrophage proliferation37. In turn, infiltrating macrophages drive tubular cell proliferation, possibly by releasing arginine metabolites8. In this context, our results unravel an unanticipated anti-inflammatory feedback loop orchestrated by STAT3 in ADPKD.
Previous reports have documented a robust activation of STAT3 in cyst lining epithelial cells from ADPKD patients and related murine models19,20,39. While in vitro results suggested that cleaved polycystin 1 may directly activate STAT339,40, the mechanisms governing STAT3 activation in cystic tubular cells remained poorly understood. Our results indicate that the bulk of STAT3 activation in response to Pkd1 disruption in vivo occurs through a non-cell autonomous process requiring a cilia dependent induction of CCL2. Acting on its receptor CCR2, CCL2 is a major driver of macrophage recruitment, which occurs at an early step of the disease7,8,38. Thus, our results imply that infiltrating macrophages directly or indirectly promote STAT3 activation in tubular cells. Such non-cell autonomous activation of STAT3 by neighboring macrophages have been documented in distinct settings including cancers41–45, heart failure27 or pancreatitis46. Different molecular pathways have been reported, including the direct expression of paracrine activator of STAT3 such as oncostatin M27, interleukin 647, interleukin 842 and TGFβ46 or the indirect up-regulation of STAT3 activating receptors48, mirroring the multiplicity of STAT3 activation mechanisms.
In most extra-renal settings, paracrine activation of STAT3 by macrophages plays a detrimental effects in the disease, promoting tissue injury27 or tumor growth/metastasis41–45. In contrast to these situations, our data indicate that tubular Stat3 is mostly dispensable for cyst growth in ADPKD, but dampens immune cell infiltration of polycystic kidneys. The absence of a notable effect of Stat3 tubular deletion on kidney enlargement and kidney function decline in our model sharply contrasts with the positive impact of STAT3 inhibitory compounds previously reported in Pkd1 mutant mice19,20. This discrepancy may be caused by off-target effects or the consequence of inadequate target specificity of these drugs20,21,49. Alternatively, the beneficial effect of these drugs may be linked to Stat3 inhibition in non-tubular cells. Indeed, pericystic interstitial cells also display STAT3 activation in ADPKD. Importantly, different interstitial cell types have been shown to affect cyst growth, including macrophages6,8 and CD8+ T cells38. Interestingly, STAT3 activation has been reported in pericystic macrophages in ADPKD and STAT3 inhibitory compounds reduce the ability of cultured macrophages to induce tubular cell proliferation through paracrine signaling in vitro50. It is therefore plausible that the therapeutic effects of STAT3 inhibitory drugs are linked to the inhibition of STAT3 in macrophages. The proper evaluation of STAT3 functions in macrophage in ADPKD represents however a complicated task: conditional Stat3 inactivation in macrophages leads to the spontaneous development of inflammatory bowel disease precluding the use of such approach in a murine model of ADPKD51. Thus, our results do not rule out that STAT3 inhibition may have a benefit in ADPKD patients through its impact on non-tubular cells.
Unexpectedly, we observed that Stat3 deletion in renal tubules markedly increased immune cell infiltration of polycystic kidneys. Mechanistically, STAT3 depletion resulted in the up-regulation of Ccl5 and Cxcl10 transcripts in polycystic kidneys and in cultured tubular cells. While surprising, this observation is reminiscent of the well characterized anti-inflammatory functions of STAT3 in macrophages. In those cells, STAT3 activation in response to IL10 restricts NF-κB dependent proinflammatory signaling31,32. The molecular bases of this anti-inflammatory effect involves the transcriptional regulation of factor(s) that modulate(s) NF-κB signaling31. More specifically, the transcriptional repression of UB2N ubiquitin ligase by STAT3 appears to play a major role in restricting NF-κB signaling in macrophages32. Indeed, in macrophages, Stat3 inactivation increases UB2N abundance, which in turn stabilizes NF-κB signaling and promotes RELA nuclear accumulation. Of note, STAT3 anti-inflammatory functions have also been documented in non-immune cells. Indeed, the deletion of STAT3 in intestinal epithelial cells leads to an exacerbation inflammatory response after intestinal injury17. We did not fully investigate the underlying mechanisms of STAT3 anti-inflammatory function in tubular cells, but our results indicate some specificities: contrary to STAT3 anti-inflammatory functions in macrophages, the anti-inflammatory effect of STAT3 in tubular cells is not mediated by the repression of Ub2n transcript, nor is it associated with RELA nuclear accumulation. While the molecular intermediates allowing STAT3 to restrict polycystic kidney inflammation remain unclear, our results clearly expand STAT3 anti-inflammatory function to tubular cells. They further suggest that this apparently conserved function of STAT3 is related to distinct tissue specific mechanisms.
A striking aspect of our study is that tubular Stat3 disruption decoupled renal inflammation from cyst growth. Indeed, previous studies have consistently shown that reducing renal inflammation through macrophage depletion or CCL2/CCR2 inhibition led to a proportional decrease in cyst burden. Why increased macrophage infiltration does not sustain cyst growth in Stat3 deficient animals remain unclear. It may indicate a direct effect of STAT3 on tubular cell proliferation as suggested by some in vitro studies19,52. Considering the role played by CD8+ T cells in restricting cyst growth38, the massive increased in T cells infiltration induced by Stat3 inactivation could also be involved.
The identification of V2R inhibition as a valid target to reduce cyst growth in human has opened the area of specific therapy in ADPKD53,54. While this represents an important cornerstone on the road to cure the disease, V2R inhibition has only a modest efficacy and important side effects. Thus, the search for more potent and better tolerated drugs remains central to the field55. Over the last 20 years, joint research efforts allowed the identification of a large panel of pathways that are activated in growing cysts, thereby providing multiple potential target for molecular targeted therapies in ADPKD56. While concordant evidence identified STAT3 as an interesting candidate, robust evaluation of STAT3 activation mechanisms and functions in ADPKD was lacking. In this context, our study, which relay on state of the art model of tubular specific inactivation of Stat3 and Pkd1 in adult mice, clearly indicates that tubular STAT3 is not a critical driver of cyst formation. More generally, our results exemplify the critical contribution of complex genetic conditional models to the rigorous evaluations of potential therapeutic targets in ADPKD.
AUTHOR CONTRIBUTIONS
A.V and F.B designed the study; A.V, M.B, A.A, C.N and F.B performed experiments and analyzed data; A.V, E.W.K, F.T and F.B drafted and revised the manuscript; all authors approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supplementary Figure 1: Primary cilia promote STAT3 phosphorylation in tubular cells in vitro.
(A, B) Representative western blot (A) and quantification (B) of STAT3 phosphorylation in whole cell lysates from MDCK cells expressing inducible shRNA against Kif3a (Kif3a-i), Ift88 (Ift88-i) or the non-relevant control luciferase (Luci-i) after 10 days of tetracyclin treatment (+ Tet). Bars are mean ± SEM of 4 independent experiments. Paired t test, ns: not significant, *P<0.05.
Supplementary Figure 2: STAT3 disruption has a selective impact on inflammatory cytokine expression in ADPKD.
(A-D) Quantification of Tnfa (A), Il1b (B), Cx3cl1 (C), and Ub2n (D) mRNA abundance in kidneys from control, iPkd1ΔTub and iPkd1ΔTub; iStat3ΔTub mice at 18 weeks. Each dot represents one individual mouse. One-way ANOVA followed by Tukey-Kramer test, ns: not significant, **P<0.01, ***P<0.001.
ACKNOWLEDGMENTS
We thank the animal facility (LEAT, SFR Necker, INSERM US24, Paris, France), the histology facility (SFR Necker, INSERM US24, Paris, France) and the mice physiology facility (Cordelier research center, Paris, France) for technical assistance.
Amandine Viau was supported by FRM ARF20150934110, E. Wolfgang Kuehn was supported by DFG 1504/5-1, Fabiola Terzi & Frank Bienaimé were supported by Institut National de la Santé et de la Recherche Médicale, Université Paris Descartes, Assistance Publique Hôpitaux de Paris and Agence Nationale de la Recherche.
REFERENCES




