

Summary
The epidermis is a stratified epithelium in which structural and functional features are polarized across multiple cell layers. This type of polarity is essential for establishing the epidermal barrier, but how it is created and sustained is poorly understood. Previous work identified a role for the classical cadherin/filamentous-actin network in establishment of epidermal polarity. However, little is known about potential roles of the most prominent epidermal intercellular junction, the desmosome, in establishing epidermal polarity, in spite of the fact that desmosome constituents are patterned across the apical to basal cell layers. Here, we show that desmosomes and their associated intermediate filaments (IF) are key regulators of mechanical polarization in epidermis, whereby basal and suprabasal cells experience different forces that drive layer-specific functions. Uncoupling desmosomes and IF or specific targeting of apical desmosomes through depletion of the superficial desmosomal cadherin, desmoglein 1, impedes basal (stratification) and suprabasal (tight junction barrier) functions. Surprisingly, disengaging desmosomes from IF also uncouples stratification from differentiation, accelerating the expression of differentiation markers. Our data support a model in which the desmosome-IF network supports a reciprocally organized distribution of ErbB1/EGFR activity in the basal layer and mechanosensitive kinase ErbB2 activity in the suprabasal layer to ensure the proper spatiotemporal coordination of cell mechanics and the biochemical program of differentiation.
Introduction
To transition from single celled organisms to multicellular metazoans, cells evolved multiple mechanisms to form stabilized cell-cell interactions [1]. Ultimately, this process led to the development of simple epithelial sheets, which serve as a protective barrier between an organism and its environment. Additional epithelial complexity arose later in evolution, with the emergence of multilayered, stratified epithelia, including elaborate tissues like the epidermis [2]. In vertebrates, this multilayered epithelium serves as a barrier against water loss, mechanical insults, and as a physical and immune barrier to external pathogens (containing both innate and adaptive immune elements) [3, 4]. The epidermis comprises four distinct layers, basal, spinous, granular (SG), and cornified, which undergo constant regeneration. Regeneration occurs through a process whereby keratinocytes in the basal layer proliferate, at some point commit to a program of terminal differentiation, and exit the basal layer. During differentiation, cells move progressively into the suprabasal layers where they are incorporated into the epidermal barrier and ultimately slough off of the skin’s surface.
Polarity, the differential patterning of structural and functional features along an axis, is a fundamental property of all epithelial tissues and is essential to their function. Whereas simple epithelia exhibit apical to basal (apicobasal) polarization within a single cell layer, the epidermis exhibits polarization across multiple cell layers, through poorly understood mechanisms [2]. For example, barrier-essential tight junctions (TJs) are found in association with adherens junctions on the apicolateral surface of simple epithelia. In the epidermis, functional TJs localize in association with adherens junctions to the second of three SG layers [5]. Improper development or disturbed maintenance of the epidermal barrier is associated with a variety of inflammatory skin diseases (e.g., atopic dermatitis and psoriasis), impaired wound healing, and cancer [6]. In addition, simple epithelia exhibit a polarization of intercellular forces, with a region of high tension generated by actomyosin near the apical surface [7]. This region of high tension can regulate tissue morphology, homeostasis, and barrier function [7]. Moreover, loss of polarized mechanical tension is associated with cancer morphogenesis in multiple types of epithelial tissues [8]. Whether such a mechanical gradient exists in stratified epithelia and how it contributes to tissue function is not well understood.
Cell-cell adhesion complexes and their associated cytoskeletal networks are prime candidates to regulate polarized tissue mechanics. Desmosomes are calcium-dependent, intercellular adhesions that link to the intermediate filament (IF) cytoskeleton. Desmosomes comprise three main protein families: transmembrane cadherins (desmogleins and desmocollins), armadillo proteins (plakophilins and plakoglobin), and plakin proteins (desmoplakin, DP). Cadherins of adjacent cells form trans-interactions to mediate cell-cell adhesion. Intracellularly, cadherins interact with armadillo proteins, which are coupled to IF through DP. In the epidermis, desmosome cadherins display graded patterns of expression. Some cadherins such as desmoglein 3 (Dsg3) are concentrated in the proliferating basal layer, while others such as desmoglein 1 (Dsg1) are concentrated in the superficial layers. This is in contrast to the adherens junction component E-cadherin, which is expressed throughout the epidermis [9].
Since mechanical forces regulate cell behavior, their polarized distribution may drive segregation of tissue functions. Tensile (pulling) and compressive (pushing) forces are known to affect the behavior of cells within epithelia, regulating cell division, migration, tissue morphogenesis, as well as promoting cancer cell invasion [10, 11]. Moreover, the material properties of cells, tissues, and their surrounding environment, including cell and stroma stiffness (the extent to which they resist force-induced deformation) affect the morphology, proliferation, differentiation, and migration of many different cell types, including cells of the immune system [10]. It has been suggested that the biophysical properties of normal tissues can suppress inflammatory responses while aberrant tissue mechanics can stimulate inflammation [12]. Thus, understanding how the physiological biophysical properties of tissues like the epidermis are established and maintained could provide insight into common skin diseases like atopic dermatitis and psoriasis, where inflammation and perhaps tissue mechanical properties are misregulated.
There is a well-established role for filamentous-actin (F-actin)-based adhesive networks in regulating mechanics (tension and stiffness) [13, 14]. However, the contributions of the desmosome/IF network in mechanical regulation are not well understood. Our previous work elucidated a role for desmosomes in mechanical regulation by modulating the interaction between desmosomes and IF using mutant forms of DP. Through a combination of micropillar arrays and atomic force microscopy (AFM), we showed that strengthening DP-IF interactions increased cell-cell tensional forces and cell stiffness, while disrupting the interaction decreased these forces [14]. Moreover, these effects required integration with F-actin-based networks, indicating that desmosome/IF and F-actin-based networks function synergistically to regulate cell mechanics, though the mechanisms mediating this cross talk are not understood. The patterned expression of the desmosome/IF network suggests their potential role in governing the differential biophysical properties of distinct epidermal layers. This led us to the hypothesis that the spatially patterned desmosomal machinery integrates both the polarized mechanical properties of the epidermis and biochemical signaling to regulate layer-specific functions, including stratification, differentiation, and the establishment of an intact epidermal barrier.
Results
Uncoupling the desmosome/IF connection induces rearrangements of cytoskeletal/adhesive complexes
To begin addressing the role of the desmosome/IF network in orchestrating the polarized functions of the epidermis, we performed loss-of-function experiments utilizing a well-characterized mutant of the cytolinker protein DP. DP provides the connection between the desmosome core components and the IF cytoskeleton (Figure 1A). This mutant, referred to as DPNTP (Figure 1A), consists of the first 584 amino acids of DP. It retains the ability to interact with the desmosomal core but lacks the IF-binding domains, acting as a dominant negative mutant to uncouple desmosomes from the IF network [14–16]. We previously showed that expression of the uncoupling mutant in a simple epithelial A431 cell model (an epidermoid cell line) results in retraction of the IF cytoskeleton from sites of cell-cell adhesion [14].

A) Schematic depicts the interactions among the desmosomal core proteins and the intermediate filament (IF) cytoskeleton in both a control condition (left) as well as when the desmosomal core is uncoupled from IF (right). PM, plasma membrane; DP, desmoplakin; PKP, plakophilin; PG, plakoglobin; DSG, desmoglein; DSC, desmocollin.
B) Maximum projection micrographs show immunofluorescence staining of the keratin 5/14 (K5/14) IF cytoskeleton in basal cells of differentiating monolayers of NHEKs expressing either GFP (Control) or DPNTP-GFP (Uncoupled). DAPI indicates nuclei and bar is 10 μm.
C) Maximum projection micrographs show F-actin (using phalloidin) and immunofluorescence staining of vinculin near the basal/substrate interface of differentiating monolayers of NHEKs expressing either GFP (Control) or DPNTP-GFP (Uncoupled). DAPI indicates nuclei and bar is 10 μm.
D) Left, maximum projection micrographs show immunofluorescence staining of vinculin at the basal/substrate interface of differentiating NHEK ephrin colonies expressing either GFP (Control) or DPNTP-GFP (Uncoupled). DAPI indicates nuclei and bar is 10 μm. Middle, the average vinculin-positive area per cell was quantified for both cells on the edge and in the central region of the colony. The average ratio (dashed line) of edge to central cells is shown for control and DPNTP-expressing colonies. *p=0.0233, paired t-test from three independent experiments, error bars are SEM.
E) Images show the membranes (indicated by Myr-tomato) of basal cells within epidermal equivalent cultures before and after laser ablation at the indicated times. At day 1, cells expand upon ablation, indicating tension within the layer and corresponding to a positive value for measured intervertex distance and initial recoil velocity. At day 6, cells collapse upon ablation, indicating compression within the layer and corresponding to a negative value for measured intervertex distance and initial recoil velocity. Bar is 5 μm.
F) Quantification is shown for the average change in intervertex distance (Δ Distance) over time for basal cells after ablation within control and DPNTP-expressing epidermal equivalent cultures both at day 1 and day 6 of their development. *p=0.0361, **p=0.0058, two-way ANOVA with repeated measures from three independent experiments, error bars are SEM.
G) Quantification is shown for the average initial recoil velocity after ablation for basal cells within control and DPNTP-expressing epidermal equivalent cultures both at day 1 (upper) and day 6 (lower) of their development. **p<0.009, paired t-test from three independent experiments, error bar is SEM.
H) The average cell count per field is shown for control and DPNTP-expressing epidermal equivalent cultures both at day 6 of their development. Dashed line is mean of 6 independent experiments, error bars are SEM.
To determine if the desmosome/IF connection affects cell behaviors in epithelia that have the ability to stratify, we began by expressing DPNTP tagged with GFP in two established submerged models of neonatal human epidermal keratinocytes (NHEKs). Monolayers of NHEKs were induced to differentiate and stratify with either high calcium containing medium or by treatment with ephrin peptide [17] (Figure S1A and B). After 2 days of calcium-induced cell-cell adhesion in NHEK monolayers, keratin IFs closely adjoin the cell-cell interface as they are anchored there by functional desmosomes (Figure 1B). Compared with cytoplasmic GFP controls, expression of DPNTP in NHEKs resulted in loss of orthogonally anchored keratin IF bundles, indicating the successful uncoupling of the desmosome/IF connection (Figure 1B).

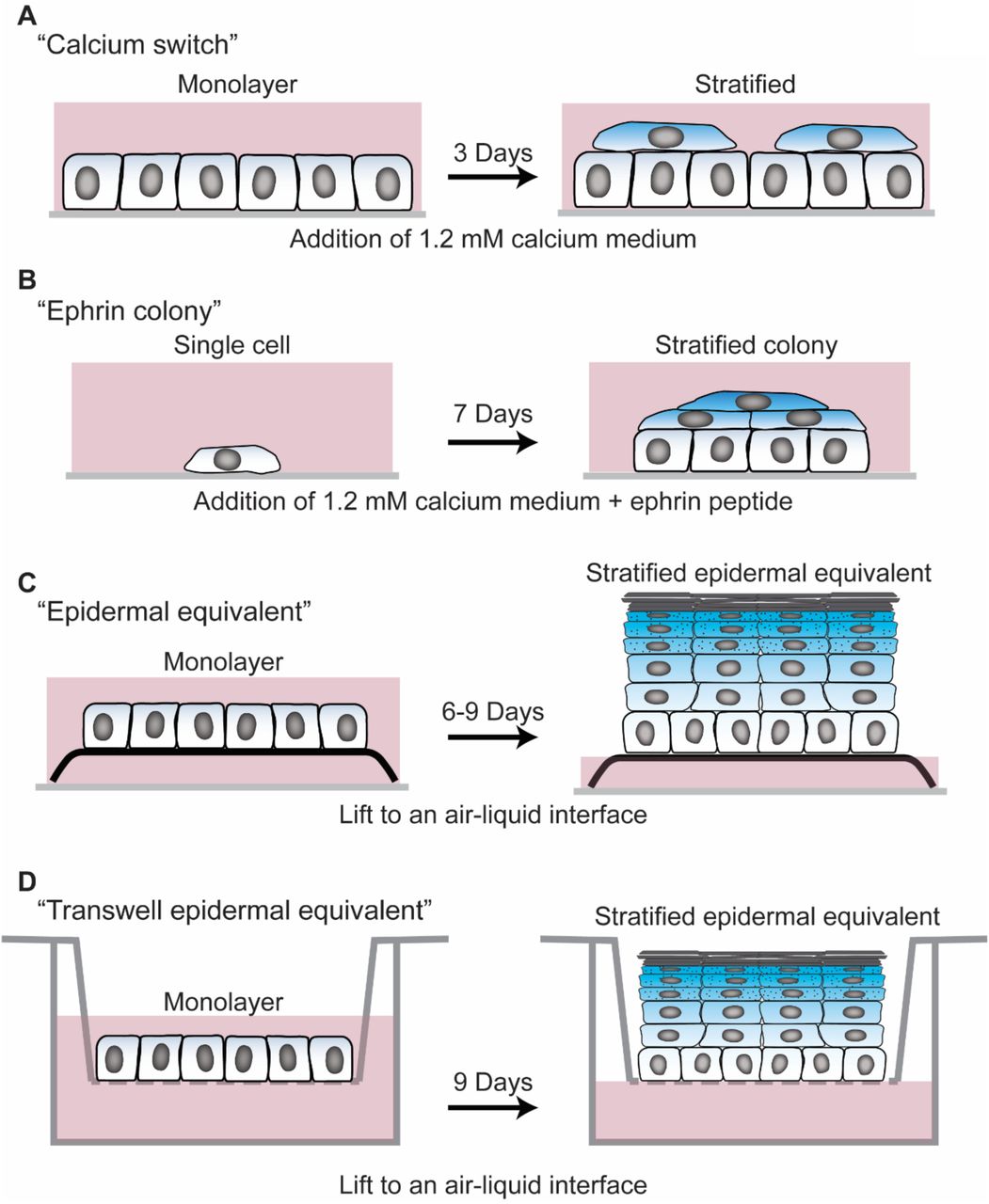
A) Calcium switch experiment- NHEKs are seeded at confluence in 0.07 mM calcium containing medium. The following day, the medium is changed to 1.2 mM calcium containing medium to induce stratification and differentiation. After 3 days, unless otherwise noted, cells were harvested for analysis.
B) Ephrin colony experiment- NHEKs are seeded at low confluence such that they are initially sparse fields of single cells that subsequently grow into individual colonies. Culture medium contains 1.2 mM calcium and 1 ug/ml ephrin-A1 peptide to induce stratification and differentiation. After 7 days, unless otherwise noted, cells were harvested for analysis.
C) Epidermal equivalent experiment- monolayers of NHEKs were seeded on fibroblast containing collagen 1 gels at confluence. Subsequently, the gels were lifted to an air-liquid interface by placing them onto a metal grid with culture medium only filling the space up to the bottom of the metal grid. Cultures were allowed to stratify and differentiate for 6-9 days prior to harvesting, unless otherwise noted.
D) Transwell epidermal equivalent experiment- monolayers of NHEKs were seeded onto transwell inserts at confluence. The cultures were exposed to an air liquid interface by removing the medium from the top chamber of the transwell. Cultures were allowed to stratify and differentiate for 9 days prior to harvesting, unless otherwise noted. Transepidermal electrical resistance measurements were performed by adding medium into the top chamber, taking measurements, then promptly removing medium to restore the air-liquid interface.
We previously showed that uncoupling the desmosome/IF connection in A431 cells was accompanied by reduced F-actin at the cell cortex near the cell-cell junctional interface [14]. A role for DP in the regulation of the actin cytoskeleton is supported by previous work as DP loss in mouse epidermal keratinocytes results in defective cortical actin cytoskeleton assembly after initiation of cell-cell contact [18, 19] and in the mouse gut, its loss induces shape and length defects in F-actin-rich microvilli [20]. Here, we show that uncoupling the desmosome/IF linkage in NHEK monolayers (Figure S1A) with DPNTP results in decreased cortical F-actin staining with a concomitant increase in observed focal adhesion-anchored F-actin stress fibers at the basal surface (Figure 1C). These data suggest a potential shift from strong cell-cell forces to enhanced cell-substrate forces upon expression of DPNTP.
This concept is easily appreciated in the context of small colonies of NHEKs, like ephrin-induced colonies (Figure S1B). In these colonies, vinculin (a component of focal adhesions and adherens junctions under tension) immunofluorescence staining indicates that the majority of large focal adhesions are near the colony edge (Figure 1D). However, uncoupling desmosomes/IF with DPNTP alters this staining pattern as many vinculin-positive substrate contacts are scattered throughout the basal surface of the cell colony, leading to a significant decrease in the enrichment of focal adhesions at the colony edge (Figure 1D). These results suggest a mechanical cell-cell to cell-substrate switch [21, 22], which is consistent with previous reports in cell culture and in drosophila where loss of cell–cell adhesion increased cell–substrate adhesion [21–23].
Uncoupling the desmosome/IF connection alters the mechanical properties of epidermal keratinocytes
In order to directly test the possibility that the desmosome/IF complex controls the mechanical properties of epidermal keratinocytes, we used laser ablation experiments in both early and late stages of in vitro morphogenesis in epidermal equivalent cultures (Figure S1C) to examine development of specific mechanical behaviors of basal cells (Figure 1E). Uncoupling the desmosome/IF linkage with DPNTP results in a significant reduction in recoil behavior at both early and late time points, indicating reduced cell-cell forces (Figure 1F and G). Interestingly, upon complete development of this epidermal model, cells within the basal layer exhibit a collapsing behavior that would be consistent with cells experiencing compressive forces due to crowding and/or tissue jamming (Figure 1E). Importantly, examination of the cell density upon completion of experiments indicated there was no significant effect of DPNTP expression on cell density (Figure 1H), suggesting the effects on mechanical behavior are not caused by altered forces due to crowding. These results are consistent with previous work in which uncoupling desmosomes from IF in A431 cells with expression of DPNTP decreased both cell-cell junctional tugging forces and cortical stiffness, which was dependent on a functional interaction with the actin cytoskeleton [14].
Uncoupling the desmosome/IF connection hinders epidermal keratinocyte stratification
In models of cell extrusion, monolayers of simple epithelial tissues maintain a constant cell density by eliminating cells that are normally fated to die. This process requires tight control of dynamic alterations in cell-cell mechanics and depends on actomyosin-generated forces [24–26]. In the case of the epidermis, cells undergo a somewhat similar process whereby they lose cell-substrate contacts as they move out of the basal layer [27], but once they reach the second layer they remain attached to the basal layer via cell-cell adhesion to form the spinous cell layer. Moreover, compressive forces in cell monolayers of simple epithelia and of epidermal keratinocytes can promote extrusion and stratification, respectively [28, 29]. Since uncoupling the desmosome/IF connection resulted in mechanical behaviors that could be consistent with a reduction in compressive forces being experienced by cells expressing DPNTP, we next examined the effects of DPNTP on stratification in NHEKs using two models of keratinocyte stratification.
To begin, we examined the effects of uncoupling the desmosome/IF linkage on the ability of ephrin-induced colonies to stratify (Figure 2A). The lateral expansion of NHEKs is suppressed upon addition of ephrin peptide, inducing colonies of cells to stratify and form multiple layers (Figure S1B) [17]. This results in the piling of cells and thus the percentage of overlapping nuclei can be used as a proxy to quantify stratification events. In control ephrin-induced colonies, an average of 25% of the nuclei were overlapping, indicating at least 25% of cells had stratified by day 7 (Figure 2B). In contrast, expression of DPNTP resulted in an average of 9% overlapping nuclei at this timepoint (Figure 2B). Since there was no significant difference in total cell density (Figure 2B), these data suggest that uncoupling the desmosome/IF linkage inhibits the stratification process.

A) Maximum projection micrographs show immunostaining of keratin 5/14 (K5/14) intermediate filaments and nuclei stained with DAPI using the indicated look-up table that represents z-depth in control and DPNTP-expressing (Uncoupled) ephrin colonies. Zooms show retraction of keratin filaments from cell-cell interfaces upon uncoupling the desmosome/intermediate filament with DPNTP. Bar is 50 μm.
B) The average total number of DAPI-stained nuclei (as shown in A) per ephrin colony (top) and the average number of overlapping DAPI-stained nuclei (bottom) are shown for control and DPNTP-expressing ephrin colonies. Examples of overlapping nuclei are indicated with arrowheads. Dashed lines indicate the mean of 5 independent experiments and error bars are SEM. *p<0.0001, paired t-test.
C) Unlabeled wild type NHEKs were mixed at known ratios with NHEKs expressing either GFP as a control or DPNTP-GFP and cultured in 1.2 mM calcium medium for 3 days. Left, representative maximum projection micrographs of the basal layer of cultures are shown using DAPI to label the total cell population. Bar is 50 μm. Middle, quantification of the average cell number per field from 3 independent experiments is shown. Right, quantification of the deviation from the predicted representation of GFP-positive cells in the basal layer is shown. Dashed lines indicate the mean of 3 independent experiments and error bars are SEM.
D) Unlabeled wild type NHEKs were mixed at known ratios with NHEKs expressing either GFP as a control or DPNTP-GFP and cultured in 1.2 mM calcium medium for 3 days. Left, representative maximum projection micrographs of the suprabasal layers of cultures are shown using plakoglobin (PG) to label the total cell population. Examples of GFP-positive suprabasal cells are indicated with arrowheads. Bar is 50 μm. Middle, quantification of the average cell number per field from 3 independent experiments is shown. Right, quantification of the deviation from the predicted representation of GFP-positive cells in the suprabasal layers is shown. Dashed lines indicate the mean of 3 independent experiments and error bars are SEM. *p=0.026, one sample t test with theoretical mean of 0.
E) Western blots showing the expression of the indicated proteins during a differentiation time course of control or DPNTP-GFP expressing ephrin colonies.
F) Quantification of the fold change (Log2-transformed) of DPNTP-expressing over control ephrin colonies at day 7 are shown for indicated protein expression. Dashed lines indicate the mean of 3-5 independent experiments and error bars are SEM. *p≤0.04, ***p=0.0003, one sample t test with theoretical mean of 0.
G) Quantification of the fold change (Log2-transformed) of DPNTP-expressing over control monolayer cultures exposed to 1.2 mM calcium medium for 3-4 days are shown for indicated protein expression. Dashed lines indicate the mean of 3-10 independent experiments and error bars are SEM. *p=0.016, **p≤0.004, one sample t test with theoretical mean of 0.
To more directly test the effects of DPNTP on the stratification process, we performed a competition assay in which fluorescently-labeled, genetically manipulated cells were mixed at known ratios with wild type cells to form a confluent monolayer (Figure S2A). Stratification was induced by switching from low calcium-to high calcium-containing medium and the fate of the labeled cells was tracked two days after initiation of stratification. We then quantified the percentage of labeled cells in the basal and suprabasal layers and compared them to the percentage of cells initially put into the assay (Figure S2B). If the genetic manipulation had no effect on the ability to compete with wild type cells to stratify, then there would be no deviation from the predicted percentage either in the basal or suprabasal layers. This was observed in control experiments where expression of cytoplasmic GFP had no significant effect on the calculated percentage of labeled cells in either the basal or suprabasal layers (Figure 2C and D). However, quantification of cells expressing the uncoupling mutant suggested a trend toward being retained in the basal layer and a significant decrease in the number of labeled cells in the suprabasal layer compared to what would be predicted (Figure 2C and D). There were no significant differences in cell density of basal or suprabasal cells in control and DPNTP-expressing conditions (Figure 2C and D). Additionally, depletion of endogenous DP using two distinct siRNA treatments resulted in a significantly decreased representation of labeled suprabasal cells while a non-targeting siRNA treatment had no significant effect (Figure S3A and B), corroborating the results obtained using the uncoupling mutant. Together, these data support a role for the desmosome/IF linkage during the stratification process such that breaking the connection impedes epidermal keratinocyte stratification.

A) Wild type NHEKs were mixed at known ratios with genetically-modified NHEKs that expressed GFP. These combined populations were switched to 1.2 mM calcium (high calcium) containing medium and allowed to stratify for 3 days. An example in which 50% of cells are expressing GFP and 50% are wild type. If stratification occurs normally, 50% of both basal and suprabasal cells will be expressing GFP. If stratification is inhibited, there will be an underrepresentation of GFP-positive cells in the suprabasal layer.
B) To quantify the percentage of cells expressing GFP, total cells were segmented (using a nuclear dye for example) and the percentage of GFP positive cells was calculated and then compared to the predicted percentage based on the initial ratio of cells plated.

A) A representative sorting assay for desmoplakin (DP) depleted cells is shown. GFP-positive cells treated with either non-targeting (siCtl) or two DP-targeting siRNA pools (siDP) were mixed with wild type cells and induced to stratify. Immunostaining for DP indicates level of knockdown and the percentage of suprabasal cells (total cells identified with plakoglobin and DAPI) that were GFP positive was calculated. Bar is 100 μm.
B) Quantification for the average number of suprabasal cells as well as the deviation from the predicted percentage of GFP positive cells in the suprabasal layer for control (siCtl) and DP-depleted (siDP1 and siDP2) conditions is shown. Dashed lines indicate the mean of 3 independent experiments and error bars are SEM. *p=0.025, **p=0.0087, one sample t test with theoretical mean of 0.
Uncoupling the desmosome/IF connection stimulates the biochemical program of epidermal keratinocyte differentiation
Generally, it is thought that the process of epidermal stratification and the program of terminal differentiation are linked [30]. Therefore, we assessed the effects of uncoupling the desmosome/IF linkage on biochemical markers of differentiation in both 2D high calcium- and ephrin-induced models (Figure S1A and B). Surprisingly, expression of DPNTP in both models resulted in a significant increase in the protein expression levels of differentiation markers including the desmosomal cadherins Dsg1 and desmocollin 1, the suprabasal cell IF protein keratin 1, and the cornified envelope component loricrin (Figure 2E-G). There were no significant effects detected on the expression of total DP or on the classical cadherin E-cadherin (Figure 2E-G), which are expressed in both basal and suprabasal cells (undifferentiated and differentiated cells, respectively). Since expression of DPNTP impedes stratification and increases biochemical markers of differentiation, these data suggest that disengaging the desmosome/IF connection uncouples the processes of differentiation and stratification.
The DPNTP-mediated effects on differentiation are dependent on ErbB2
There are four members of the ErbB family (ErbB1-4) of receptor tyrosine kinases, and all are reportedly expressed in human skin [31–33]. EGF receptor (EGFR, i.e. ErbB1) is highly active in the basal layer, where it keeps cells in an undifferentiated state [34]. We previously reported that the desmosomal cadherin Dsg1 promotes the biochemical program of differentiation through inhibition of EGFR and Erk1/2 activity [34]. Expression levels of ErbB2 and ErbB3 increase as keratinocytes differentiate [31], but their role in this process is not known. Therefore, we hypothesized that the effects of uncoupling the desmosome/IF linkage on biochemical differentiation could be mediated through EGFR. However, expression of DPNTP did not have a significant effect on either total or phosphorylated EGFR (Figure 3A). Instead, we observed a significant increase in the levels of Y877 phosphorylated ErbB2 upon expression of DPNTP (Figure 3A). This phosphorylation site is within the kinase domain and has been reported to increase ErbB2 activity [35, 36]. We therefore asked whether ErbB2 activity could be important for epidermal keratinocyte differentiation. To address this, we utilized a pharmacological approach in which we treated cells with the ErbB2 specific inhibitor TAK165 [37]. NHEKs were induced to differentiate with high calcium containing medium with either DMSO as a control or 1 μM TAK165. TAK165 treatment significantly reduced both total and phosphorylated ErbB2 (Figure 3B), indicating this concentration is effective at inhibiting ErbB2. Moreover, treatment with TAK165 resulted in a significant decrease in the protein expression levels of the differentiation associated proteins Dsg1, desmocollin 1, and loricrin, while no effects were detected for the classical cadherin E-cadherin (Figure 3B). These data indicate that ErbB2 plays a role in the differentiation process of human epidermal keratinocytes, which is consistent with the previously reported decrease in keratin 10 and filaggrin in ErbB2-deficient mouse epidermis [38].

A) Left, western blots show the expression of the indicated proteins in differentiated GFP (Control) or DPNTP-GFP expressing keratinocytes. Right, quantification of the fold change (Log2-transformed) of DPNTP-expressing over control are shown for the indicated protein expression. pErbB2 is Y877-phosphorlated ErbB2 and pEGFR is Y1068-phosphoylated EGFR. Dashed lines indicate the mean of 3-5 independent experiments and error bars are SEM. **p=0.0013, one sample t test with theoretical mean of 0.
B) Left, western blots show the expression of the indicated proteins in control (DMSO) or keratinocytes treated with an ErbB2 inhibitor (TAK165) at the indicated times after addition of 1.2 mM calcium medium. Right, quantification of the fold change (Log2-transformed) of TAK165 treated over control are shown for the indicated protein expression. Dashed lines indicate the mean of 3-5 independent experiments and error bars are SEM. *p≤0.02, **p≤0.006, one sample t test with theoretical mean of 0.
C) Left, western blots show a time course of the expression of the indicated proteins in differentiated GFP (Control) or DPNTP-GFP expressing keratinocytes either treated with DMSO as a control or the ErbB2 inhibitor TAK165. Right, quantification of the fold change (Log2-transformed) of DPNTP-expressing over GFP control keratinocytes are shown for the indicated protein expression for both DMSO and TAK165 treatment. Dashed lines indicate the mean of 3 independent experiments and error bars are SEM. *p=0.02, **p≤005, one sample t test with theoretical mean of 0.
Uncoupling the desmosome/IF linkage promoted both an increase in biochemical differentiation and phosphorylation of ErbB2. Since ErbB2 activity appears to regulate NHEK differentiation, we next assessed if the DPNTP-mediated enhancement of differentiation marker levels was dependent on ErbB2. Treatment of NHEKs with TAK165 abolished the differences seen between DPNTP and controls treated with DMSO (Figure 3C), suggesting that the DPNTP-mediated increase in expression of differentiation related proteins is dependent on ErbB2.
Uncoupling the desmosome/IF connection diminishes epidermal keratinocyte barrier function
We show that uncoupling the desmosome/IF linkage affects early morphogenic processes in the epidermis including basal cell stratification and the onset of biochemical differentiation. Next, we assessed the effects of uncoupling the desmosome/IF linkage on the ensuing development of epidermal TJ barrier function, which is attributed to the outer layers of the epidermis (Figure 4A). Previously, loss of DP in mouse epidermis was shown to modulate TJ protein expression and localization [39]. Moreover, in multiple models, TJs and their protein components have been suggested to be sensitive to mechanical input [2, 40–42]. Since uncoupling the desmosome/IF linkage affects the mechanical properties of cells, we hypothesized this could result in alterations in the TJ barrier.

A) Schematic shows the layers of the epidermis and functional tight junctions localize to the second granular layer. The expression patterns of desmoplakin (DP), keratins (K), and desmoglein 1 (Dsg1) are also shown.
B) Schematic shows how transepidermal electrical resistance (TEER) experiments were performed on transwell epidermal equivalent cultures. Electrodes were used to pass a current through the 3D culture that was grown on a transwell insert and resistance measurements were taken as an indicator of barrier function.
C) Maximum projection micrographs of day 9 transwell epidermal equivalent cultures show staining for nuclei (DAPI), F-actin (phalloidin), and immunostaining for the indicated proteins using a look-up table indicating z-depth. Bar is 25 μm.
D) Fluorescence micrographs show the expression of GFP (Control) and DPNTP-GFP in the granular layer of day 9 transwell epidermal equivalent cultures that were also immunostained for the tight junction component ZO1. Magenta dotted line delineates DPNTP-GFP high expressing cells. Nuclei are stained with DAPI. Bar is 25 μm.
E) Quantification of resistance measurements from TEER experiments performed on a time course of control (GFP) and DPNTP-GFP expressing transwell epidermal equivalent cultures. *p=0.0125, Sidak’s multiple comparison test from 7 independent experiments. Data are presented as mean ± SEM.
To address this, we generated human epidermal equivalent cultures using a porous transwell system (Figure S1D) in order to perform transepidermal electrical resistance (TEER) measurements on the developing cultures as a means of assessing barrier function (Figure 4B). We found that altering cell mechanics in this 3D system using an activator of actomyosin contractility (CN01, a Rho activator) had an effect on the organization of F-actin in all layers of the cultures and significantly inhibited barrier function (Figure S4A-B). In addition, we performed immunostaining of the TJ protein ZO1 (Figure 4C) as an indicator of the functional TJs found in the SG layer of the epidermis [2, 5]. ZO1 localizes to cell-cell interfaces in the SG layer in control GFP-expressing cultures (Figure 4C), an area with strong F-actin staining. However, when desmosomes are uncoupled from IF with DPNTP, there is a cell autonomous loss of ZO1 staining in the SG layer (Figure 4D), suggesting a potentially impaired barrier. We do not observe altered ZO1 staining in the basal or spinous layers upon expression of DPNTP (Figure S5), suggesting layer-specific mechanisms. TEER measurements of the developing barrier were significantly reduced in DPNTP-expressing cultures compared with GFP controls (Figure 4E), indicating expression of DPNTP reduces epidermal barrier function.
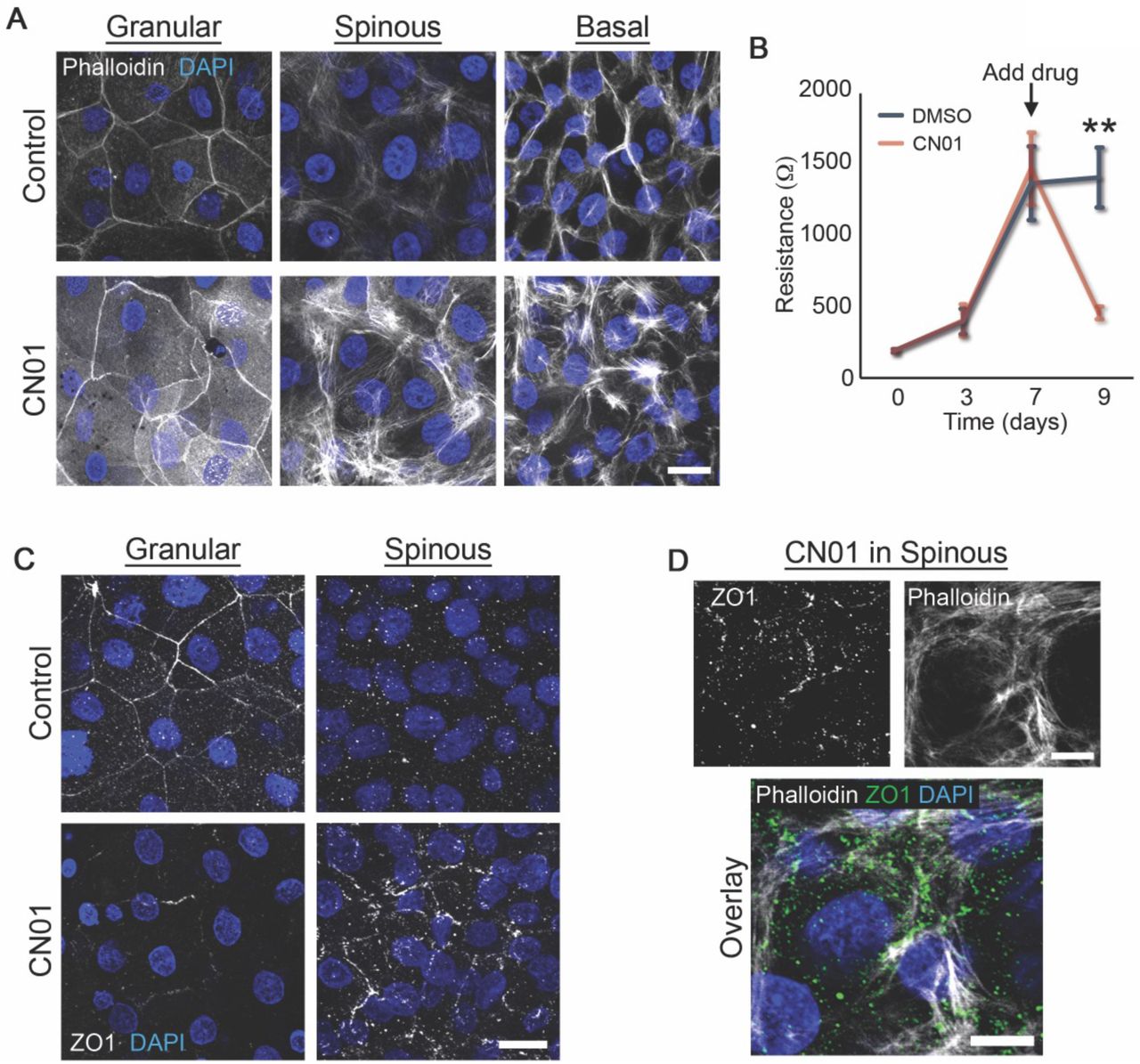
A) Maximum projection micrographs of day 9 transwell epidermal equivalent cultures that were treated with either DMSO (Control) or a Rho activator (CN01, 1 unit/mL) at day 7-9 show staining for nuclei (DAPI) and F-actin (Phalloidin) within the indicated cell layers. Bar is 20 μm.
B) Quantification of resistance measurements from TEER experiments performed on a time course of transwell epidermal equivalent cultures that were treated with either DMSO or a Rho activator (CN01, 1 unit/mL) at day 7. Data are presented as mean ± SEM. **p=0.0029, paired t test from 6 independent experiments.
C) Maximum projection micrographs of day 9 transwell epidermal equivalent cultures that were treated with either DMSO (Control) or a Rho activator (CN01, 1 unit/mL) at day 7-9 show immunostaining for the tight junction protein ZO1 and staining for nuclei (DAPI) within the indicated cell layers. Bar is 20 μm.
D) Maximum projection micrographs of day 9 transwell epidermal equivalent cultures that were treated with a Rho activator (CN01, 1 unit/mL) at day 7-9 show immunostaining for the tight junction protein ZO1, staining for nuclei (DAPI), and F-actin (Phalloidin) within the spinous cell layer. Bar is 10 μm.

Fluorescence micrographs show the expression of GFP (Control) and DPNTP-GFP in the spinous and basal layer of day 9 transwell epidermal equivalent cultures (same as Figure 4D) that were immunostained for the tight junction component ZO1. Nuclei are stained with DAPI. Bar is 25 μm.
Dsg1 regulates a tension gradient and TJ proteins in epidermal equivalents
Since DP is expressed throughout all the layers of the epidermis, the loss of function induced by expression of DPNTP would affect desmosomes in all layers. Since we hypothesize the patterning of the desmosome/IF components drives polarized, layer-specific epidermal functions, we next took a more targeted approach, ablating the function of the specific desmosomal cadherin Dsg1. Dsg1 is expressed first as basal cells commit to differentiate and its expression progressively increases so that it is most concentrated in the superficial layers where functional TJs form (Figure 4A and C). We previously demonstrated that Dsg1 controls F-actin remodeling and cell mechanics to promote early epidermal morphogenetic events including basal cell delamination [43].
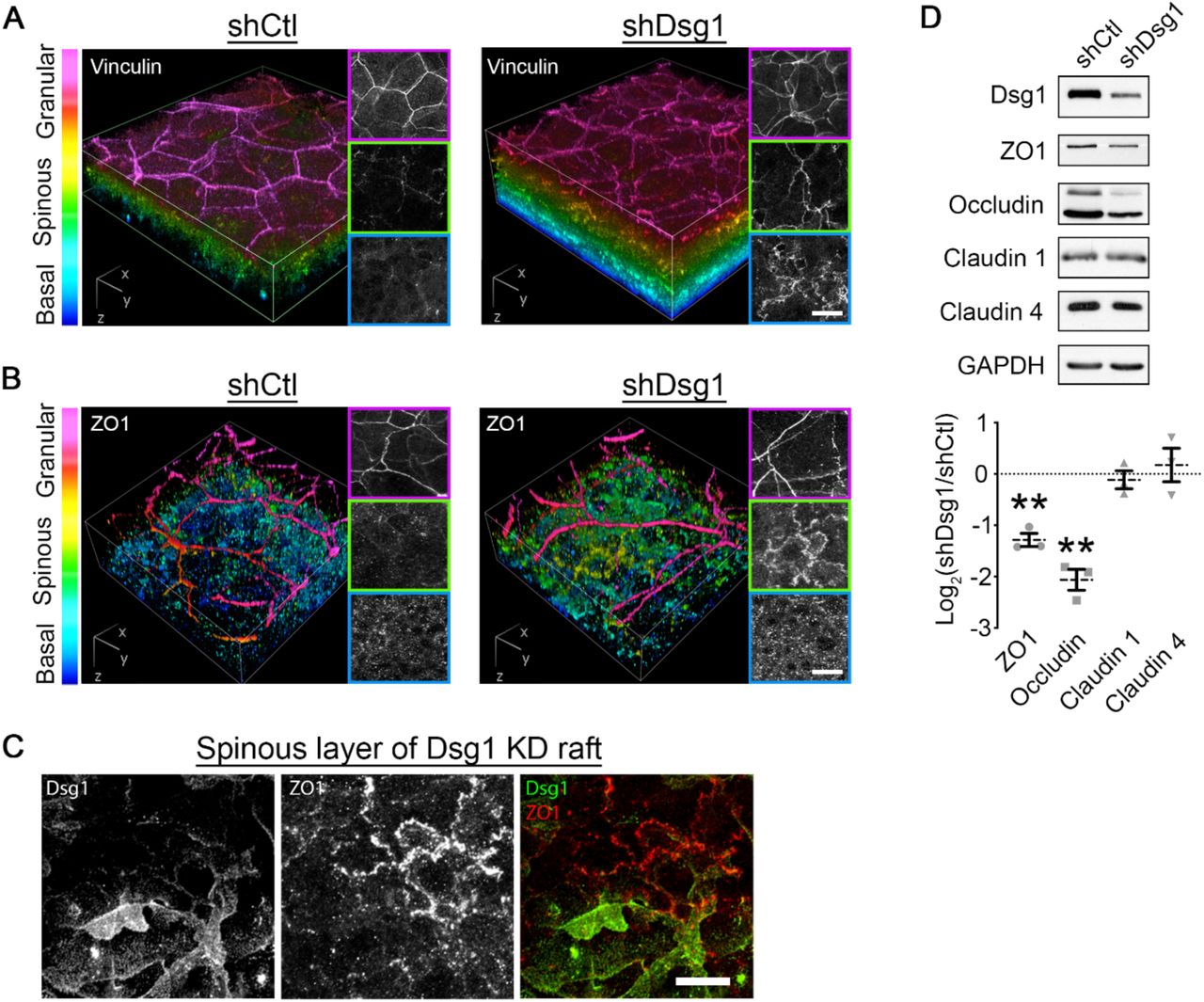
To address whether Dsg1 regulates mechanical forces and cell behaviors in the upper layers, where TJs form, we examined the distribution of the tension sensitive cell-cell junction component vinculin in a 3D epidermal equivalent model (Figure S1C). In control cultures infected with nontargeting shRNA, vinculin immunostaining was restricted to the SG layer where tension is thought to be high [2]. While control cultures exhibited a steep gradient of vinculin border staining along the apical to basal axis, this staining pattern was attenuated in Dsg1-deficient cultures, with increased cell-cell border localized vinculin in both the spinous and basal layers (Figure 5A). These data suggest that Dsg1 loss shifts the gradient of forces within the epidermal culture, similar to results obtained in E-cadherin-deficient mouse epidermis [2].

A) 3D renderings of whole mount immunostaining of tension-sensitive vinculin in control (shCtl) and Dsg1-depleted (shDsg1) day 6 epidermal equivalent cultures are presented with a look-up table that depicts z-depth in the indicated colors. Right panels show representative vinculin staining per layer. Bar is 20 μm.
B) 3D renderings of whole mount immunostaining of tight junction component ZO1 in control (shCtl) and Dsg1-depleted (shDsg1) day 6 epidermal equivalent cultures are presented with a look-up table that depicts z-depth in the indicated colors. Right panels show representative ZO1 staining per layer. Bar is 20 μm.
C) Representative micrographs of spinous layer in Dsg1-depeleted day 6 epidermal equivalent cultures showing both ZO1 and Dsg1 immunostaining. Bar is 20 μm.
D) Upper, western blot analysis from control (shCtl) and Dsg1-depleted (shDsg1) day 6 epidermal equivalent cultures show the protein expression of the indicated tight junction proteins with GAPDH shown as a loading control. Lower, quantification of the fold change (Log2-transformed) of Dsg1-depleted over control cultures are shown for the indicated proteins. Dashed lines indicate the mean of 3 independent experiments and error bars are SEM. **p<0.01, one sample t test with theoretical mean of 0.
Since TJs are thought to be regulated by a tension gradient in the epidermis [2], we next assessed the staining of the TJ component ZO1 in epidermal equivalent cultures. ZO1 immunolabeling in control cultures shows specific localization to the SG layer (Figure 5B), consistent with the reported location where functional epidermal TJs form [5]. Similar to the effects observed with vinculin redistribution, Dsg1 depletion led to a shift in this staining pattern (Figure 5B). A shift in the ZO1 staining pattern was also observed upon treatment with a Rho activator, whereby ZO1 similarly localized prematurely to cell-cell interfaces in the spinous layer (Figure S4C and D). The effect of Dsg1 on ZO1 appears to be cell autonomous, as mosaic expression of Dsg1 in the spinous layer altered cell-cell contact localized ZO1 specifically in Dsg1-depleted cells (Figure 5C). Moreover, biochemical analysis of the TJ proteins ZO1 and occludin indicates Dsg1 depletion results in a decrease in their expression (Figure 5D), while no effects were detected for claudin 1 or claudin 4. Together, these data suggest that Dsg1 controls the distribution of forces within the epidermis to restrict TJ protein localization to the SG layer.
ErbB2 as a potential mediator of Dsg1-mediated effects on epidermal polarity and barrier function
Since the epidermis is polarized across multiple layers, specific signaling platforms and intercellular junctions are present in only certain layers. Dsg1 promotes the transition of the undifferentiated basal phenotype to a differentiated suprabasal one through modulation of EGFR/Erk activity [34] and promotion of delamination [43]. As ErbB2 is also an important regulator of differentiation (Fig. 3), we determined the relative expression and localization of EGFR and ErbB2 in human epidermis. ErbB2 localization is highly enriched in the SG2 layer of human epidermis (Figure 6A), the specific layer where functional TJs form [5]. The expression of ErbB2 and its phosphorylation status at Y877 both increase as human epidermal equivalent cultures develop (Figure 6B). Moreover, ErbB2 has been reported to modulate the polarized distribution of ZO1 in a simple epithelial model of MDCK cells [44]. This suggested to us that ErbB2 may play a role Dsg1-mediated regulation of TJs within the epidermis.

A) Left, representative micrographs of transverse sections of human epidermis immunostained for the indicated proteins are shown. Zoom is of area in dashed box. SG, stratum granulosum. Bar is 20 μm. Right, line scan analysis shows the average relative level of fluorescence intensity from 3 independent samples for the indicated proteins as a function of distance from the most superficial living epidermal layer (stratum granulosum, SG).
B) Representative western blots of a differentiation time course of human epidermal equivalent cultures showing the total EGFR and Y1068-phosphorylated EGFR (pEGFR) as well as total ErbB2 and Y877-phosphorylated ErbB2 (pErbB2). GAPDH is used as a loading control.
C) Left, representative western blots of day 6 human epidermal equivalent cultures expressing either a non-targeting shRNA (shCtl) or an shRNA targeting Dsg1 (shDsg1) showing Dsg1 protein level, total ErbB2, and Y877-phosphorylated ErbB2 (pErbB2). GAPDH is used as a loading control. Right, quantification of the fold change (Log2-transformed) of Dsg1-depleted over control cultures are shown for the indicated proteins. Dashed lines indicate the mean of at least 4 independent experiments and error bars are SEM. **p<0.01, one sample t test with theoretical mean of 0.
D) Micrographs of transverse sections of day 6 human epidermal equivalent cultures expressing either a non-targeting shRNA (shCtl) or an shRNA targeting Dsg1 (shDsg1) immunostained for Dsg1 and ErbB2 are shown. Dotted line marks the bottom of the basal layer. Bar is 20 μm. Lower, quantification of ErbB2 immunofluorescence intensity expressed as a ratio of the membrane localized signal over the cytoplasmic localized signal. Dashed lines indicate the mean of 5 independent experiments and error bars are SEM. **p=0.008, paired t test.
E) Day 9 transwell epidermal equivalent cultures were treated with either DMSO or the ErbB2 inhibitor TAK165 (1 μM) for the last 48 hours prior to harvesting. Maximum projection micrographs show staining for nuclei (DAPI), F-actin (phalloidin), and immunostaining for the tight junction protein ZO1 using a look-up table that indicates z-depth. Bar is 25 μm.
F) Day 9 transwell epidermal equivalent cultures were treated with either DMSO or the ErbB2 inhibitor TAK165 (1 μM) for the last 48 hours prior to harvesting. Left, western blots showing the expression of the indicated tight junction proteins for samples treated with either DMSO (Control) or TAK165. GAPDH and α-tubulin are used as loading controls. Right, quantification of the fold change (Log2-transformed) of TAK165-treated over control cultures are shown for the indicated proteins. Dashed lines indicate the mean of 3 independent experiments and error bars are SEM. *p=0.025, **p<0.01, one sample t test with theoretical mean of 0.
G) Quantification of resistance measurements from TEER experiments performed on a time course of transwell epidermal equivalent cultures that were treated with either DMSO or the ErbB2 inhibitor TAK165 (1 μM) on day 7 (as indicated) for the last 48 hours. *p=0.0325, paired t test from 5 independent experiments. Data are presented as mean ± SEM.
To examine the effects of Dsg1 on ErbB2 function, we performed Dsg1 depletion experiments. Loss of Dsg1 resulted in a biochemical decrease in both total and Y877 phosphorylated ErbB2 as well as a loss of ErbB2 at sites of cell-cell contact in the upper layers of human epidermal equivalent cultures (Figure 6C and D). These data suggest that Dsg1 indeed regulates ErbB2. We then assessed the potential role of ErbB2 in regulating TJ protein expression and function. Since we showed that ErbB2 is important for the process of epidermal keratinocyte differentiation, we allowed human transwell epidermal equivalent cultures to form for 7 days and then treated with either DMSO as a control or the TAK165 inhibitor for 2 additional days. TAK165 treatment altered overall ZO1 and F-actin staining (Figure 6E) with aberrant punctate ZO1 staining in the spinous layer (Figure S6), similar to the results seen upon Dsg1 depletion (Figure 5B and C). Additionally, compared to controls, TAK165 significantly reduced the expression of TJ protein components including ZO1, claudin 4, and occludin (Figure 6F). Finally, inhibiting ErbB2 with TAK165 significantly diminished the forming barrier assessed using TEER measurements (Figure 6G). These data support the idea Dsg1 regulates the epidermal barrier by patterning ErbB function and suggest Dsg1 may have distinct basal vs. suprabasal functions.

3D renderings of whole mount immunostaining of tight junction component ZO1 in control (DMSO) and TAK165 treated day 9 epidermal equivalent cultures are presented with a look-up table that depicts z-depth in the indicated colors. Right panels show representative ZO1 immunostaining and F-actin (phalloidin staining) in the spinous layer. Cultures were allow to grow normally until day 7 and then treated with DMSO or TAK165 for 2 subsequent days. Bar is 20 μm.
Discussion
Apicobasal polarity is an intrinsic property of epithelial tissues that permits discrimination between interior and exterior compartments by establishing a functional barrier near the apical, external-facing surface. In contrast to simple epithelia, in which single cells in an epithelial sheet are polarized in an apicobasal fashion, in the epidermis apicobasal polarity is organized across multiple layers [9]. Here, we demonstrate that the desmosome/IF network is a critical player in establishing the polarized structure and function of the epidermis. Our evidence suggests that desmosomes contribute to both the early and late states of morphogenesis: interfering with desmosome-IF interactions alters basal cell mechanics to inhibit stratification and alters superficial cell mechanics to impair TJ organization. Mechanistically, desmosomes support a reciprocally organized gradient of ErbB1/EGFR activity in the basal layer and ErbB2 activity in the suprabasal layer to ensure the proper spatiotemporal coordination of cell mechanics and the biochemical program of differentiation.
Alterations in both basal and superficial cell mechanics in response to desmosome impairment are consistent with a switch from strong cell-cell forces to enhanced cell-substrate forces (basal layer) or enhanced intercellular tension in the cells below the TJ layer (Figure 7). For instance, in small colonies of differentiating keratinocytes the desmosome/IF DP uncoupling mutant resulted in an increase in vinculin-positive, cell-substrate contacts and associated F-actin stress fibers, previously documented to signal a switch from strong cell-cell forces to enhanced cell-substrate forces [21–23]. At the same time, a decrease in cell-cell forces is supported by laser ablation experiments of early stage epidermal cultures and by our previous finding that expression of this mutant in A431 cells reduced cell-cell tugging forces [14]. Likewise, in superficial layers, interfering with superficial desmosome-IF interactions through knockdown of Dsg perturbed the normally restricted pattern of vinculin in the high-tension layer where TJs form, such that higher tension cell-cell junctions occurred in the lower layers. This is consistent with previous work showing impaired desmosome function results in increased molecular indicators of tension experienced by adherens junctions in mouse epidermis [39].

Working model of the effects of the desmosome/intermediate filament (IF) network on early and late epidermal morphogenetic events. During early morphogenesis, the desmosome/IF network regulates stratification by altering the mechanical properties of basal cells. Uncoupling the desmosome/IF linkage results in reduced compressive forces, which normally promote stratification. Additionally, basal cells in which the desmosome/IF linkage has been uncoupled exhibit increased focal contacts and associated F-actin stress fibers, which likely contribute to a decreased ability to stratify. In the upper layers of the epidermis, the desmosome/IF network controls the restricted localization of the tight junction component ZO1 as well as the tension-sensitive adherens junction component vinculin (vinc). Loss of desmoglein 1 (Dsg1) containing desmosomes results in premature localization of vinculin and ZO1 to layers lower than the granular, suggesting a downward shift in distribution of cell-cell forces. Additionally, the mechanically-sensitive kinase ErbB2 is lost from the granular layer upon disruption of Dsg1, potentially due to the downward shift in cell forces. Our data show that ErbB2 is a regulator of tight junction components and of the epidermal barrier. Therefore, the ability of the desmosome/IF network to regulate layer-specific mechanical forces drives layer specific functions: 1) compression regulating stratification and differentiation in the basal layer and 2) a cell-cell junction tension gradient that modulates the epidermal tight junction barrier of the granular layer through ErbB2. DP, desmoplakin; K, keratin.
How does interfering with basal cell mechanics affect an epidermal keratinocyte’s commitment to differentiate? In the epidermis, compressive forces have been linked to epidermal fate specification [45]. In vitro, differentiating keratinocytes exhibit a transient decrease in cortical tension that is associated with the onset of expression of differentiation markers and subsequent exit of cells from the basal layer in the process of delamination [46]. Here we show that as normal human epidermal cultures progress from early to later stages of epidermal development, cells transition from a high-tension state to a compressive state. Both of these states are prevented when IF are uncoupled from desmosomes. Thus, uncoupling IF from the desmosome reduces compressive forces within the basal layer that have been associated with differentiation. Consistent with this, cells failed to stratify into the superficial layers when IF were uncoupled from desmosomes. Based on our previous observation showing that the actomyosin contractile system is required for the altered mechanics observed when the desmosome/IF connection is impaired [14], these data support the idea that desmosomes help coordinate changes in cortical tension and adhesion necessary to drive stratification (Figure 7).
The process of keratinocyte stratification is generally associated with the onset of a terminal program of differentiation. Therefore, it was initially surprising that while uncoupling IF from desmosomes impedes stratification, the biochemical program of differentiation is accelerated. This observation is not without precedent, as inhibition of actomyosin contractility was shown to uncouple morphogenesis and fate specification of inner cell mass cells in the developing mouse embryo [47]. We propose that uncoupling the desmosome/IF linkages induces mechanical changes (i.e. reduced cortical tension at cell-cell contacts) that are sufficient to initiate the process of differentiation, but due to altered cell-cell adhesion and increased cell-substrate adhesion they fail to stratify (Figure 7). This model is reminiscent of loss of E-cadherin inducing a retention of differentiated keratinocytes within the basal layer of the epidermis [46].
It is well known that in simple epithelia, TJs are regulated by mechanical forces. Junctional tension, like that produced by actomyosin forces, recruits the TJ components ZO1 and occludin to promote barrier function in simple polarized epithelia [40, 42]. Here we show that interfering with the superficially concentrated desmosomal cadherin Dsg1 in 3D epidermal cultures, shifts the localization of force-sensitive vinculin and ZO1 to more basal layers. Coupled with previous work showing the importance of E-cadherin in the TJ barrier [2], these data are consistent with a model in which loss of Dsg1 induces a transfer of forces experienced to non-desmosomal junctions and premature mechanosensitive recruitment of junctional proteins to cell-cell interfaces in spinous layers (Figure 7).
This idea is in accordance with our previous finding that expression of Dsg1 redistributed molecular tension in keratinocyte monolayers through rearrangements of cortical F-actin mediated by Arp2/3-dependent actin polymerization [43]. In the context of early morphogenesis, these tension-redistributing forces help promote basal cell delamination by reducing tension on E-cadherin [43]. Here, we show that in the context of TJ organization, Dsg1 helps restrict tension to the superficial TJ layer. Collectively, these observations suggest that Dsg1’s expression gradient from low in the basal layer to high in the superficial TJ layer, helps pattern layer-specific mechanical forces.
Molecular tension required for TJ organization depends on actomyosin contractility, perturbation of which negatively impacts TJs and increases their permeability [48–50]. However, too much force can also disrupt TJ barrier function. For example, subjecting lung epithelial monolayers as well as intact lung tissue to large external stretch is sufficient to induce a reduction in barrier function [51]. Here, we provide evidence that pharmacological activation of actomyosin contractility disrupts the epidermal barrier. On the other hand, disruption of the desmosome/IF linkage, which decreases cell-cell forces, is sufficient to reduce barrier function of transwell epidermal equivalent cultures. Therefore, the fine tuning of mechanical input is critical to TJ barrier function and this tuning requires desmosome/IF attachment. This suggests that there may be epidermal layer-specific properties that drive the polarized formation of functional TJs.
Our previous work demonstrated that the onset of differentiation stimulated by Dsg1 expression in basal cells depends on its ability to dampen EGFR/MAPK signaling that is active in basal layer [34, 52]. While total EGFR levels appear consistent over the cell layers, here we show that basal EGFR activity is balanced by a reciprocal increase in the activity of the related receptor tyrosine kinase ErbB2/Her2. ErbB2 begins to be expressed as EGFR activity wanes, and it is highly enriched to the SG2 layer where functional TJs form. Inhibiting ErbB2 disrupts the epidermal barrier. Together with the observations that ErbB2 is sensitive to both changes in stiffness [53] and tension induced by stretch [54], these data support a model in which Dsg1 patterns mechanical signals in an apicobasal manner to control the epidermal TJ barrier.
Like other receptor tyrosine kinases, ErbB2 is often associated with proliferation and cancer progression [55, 56], and can also have a negative impact on epithelial permeability [57]. On the other hand, ErbB2 has also been linked in a positive way to integrity of the airway epithelial barrier, suggesting its functions are context dependent [58]. As EGFR has been shown to affect epidermal barrier function, it is possible that ErbB2 coordinates with this or other members of the ErbB family to regulate TJ function [2]. In addition to supporting apical TJ formation, here we also connect ErbB2 expression and activity with differentiation, showing that it contributes to the observed acceleration of the biochemical program of differentiation in DP-IF uncoupled cultures. Interestingly in zebrafish heart development, ErbB2 plays a dual role, promoting both cardiomyocyte proliferation and differentiation [59].
We previously reported that Dsg1 associates with Erbin to dampen EGFR/MAPK. Erbin has also been shown to regulate ErbB2 protein stability in the context of breast cancer cells [60]. This raises the possibility that Dsg1-dependent recruitment of Erbin to the plasma membrane dampens EGFR activity while contributing to stabilization of ErbB2 to promote differentiation and TJ assembly in the superficial epidermal layers.
Collectively, the data presented here expose the desmosome/IF network as an essential element of the machinery in epidermis that patterns epidermal layer-specific mechanical properties. In the basal layer, this network affects compressive forces that coordinate the processes of stratification and differentiation, and in the upper layer, the network integrates mechanosensitive cytoskeletal and signaling components to maintain the TJ barrier. We identify ErbB2 as a critical regulator of both keratinocyte differentiation and barrier function, placing it in a key position to control polarized epidermal functions downstream of the desmosome/IF network.
Materials and methods
Antibodies and reagents
The following primary antibodies were used: chicken anti-plakoglobin 1407 and 1408 (Aves Laboratories); rabbit anti-loricrin, rabbit anti-keratin 1, rabbit anti-keratin 5, and rabbit anti-keratin 14 (gifts from J. Segre, National Human Genome Research Institute); rabbit NW6 and NW161 anti-desmoplakin (Green laboratory); mouse DM1α anti-α-tubulin (Sigma-Aldrich, T6199); rabbit anti-GAPDH (Sigma-Aldrich, G9545); mouse JL8 anti-GFP (Clontech, 632381); mouse P124 anti-desmoglein 1 (Progen, 651111); mouse U100 anti-desmocollin 1a/b (Progen, 65192); mouse anti-vinculin (Sigma-Aldrich, V9131); mouse C4 anti-actin (EMD-Millipore, MAB1501); mouse HECD1 anti-E-cadherin (gift from M. Takeichi and O. Abe, Riken Center for Developmental Biology, Kobe, Japan); rabbit anti-EGF Receptor (4261), rabbit anti-phospho-EGF Receptor Tyr1068 (2234), rabbit anti-HER2/ErbB2 (2165), and anti-phospho-HER2/ErbB2 Tyr877 (2241) were from Cell Signaling Technology; mouse anti-ZO1 (BD BioSciences, 610967); rabbit anti-claudin 1 (51-9000), rabbit anti-claudin 4 (36-4800), and mouse anti-occludin (33-1500) were from Thermo Fisher Scientific. Secondary antibodies included goat anti-mouse, -rabbit, and -chicken HRP (Kirkegaard Perry Labs); goat anti-mouse, -rabbit and -chicken conjugated with Alexa Fluor 488, 568, or 647 nm (Thermo Fisher Scientific, 1:300 IF); goat anti-mouse, -rabbit, and -chicken conjugated with Alexa Fluor 488, 568, or 647 nm (Thermo Fisher Scientific); goat anti-mouse IgG1 isotype conjugated with Alexa Fluor 488, 568, or 647 nm (Thermo Fisher Scientific); and goat anti-mouse IgG2a isotype conjugated with Alexa Fluor 488, 568, or 647 (Thermo Fisher Scientific). Alexa Fluor 488, 568, or 647 nm phalloidin (Thermo Fisher Scientific) was used to stain filamentous actin and 4′,6-Diamido-2-Phenylindole (DAPI, Sigma-Aldrich) to stain nuclei.
Western blot analysis
Whole cell lysates were generated using Urea Sample Buffer (8 M urea, 1% SDS, 60 mM Tris, pH 6.8, 5% β-mercaptoethanol, 10% glycerol). Proteins were separated by SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked with either 5% milk or 1% BSA in PBS or TBS with or without 0.05% Tween. Primary and secondary antibodies were incubated in this blocking solution. Immunoreactive proteins were visualized using chemiluminescence. Densitometry analysis was performed using ImageJ.
Cell culture
Primary normal human epidermal keratinocytes (NHEKs) were isolated from neonatal foreskin provided by the Northwestern University Skin Biology and Disease Resource-based Center (SBDRC) as previously described [61]. Cells were maintained in growth medium (M154 media supplemented with 0.07 mM CaCl2, human keratinocyte growth supplement (HKGS), and gentamicin/amphotericin B solution (G/A); Thermo Fisher Scientific). NHEKs were induced to differentiate by addition of 1.2 mM CaCl2 to growth medium with or without addition of 1 μg/mL ephrin A1-FC peptide (R&D Systems) for the indicated times. NHEKs were used to generate 3D epidermal equivalent cultures as previously described previously [61]. Pharmacological treatments included 1 unit/ml Rho Activator I (CN01; Cytoskeleton, Denver, CO), 1 mM TAK165 (Mubritinib, Selleck Chemicals), and dimethyl sulfoxide (DMSO, Sigma-Aldrich).
NHEKs were transfected with either siGENOME Non-Targeting siRNA Pool #2 (siCtl) or siGENOME SMARTpool siRNA D-019800-17 DSP (Dharmacon, Lafayette, CO) (siDP1) or stealth siRNA oligonucleotides 5′-CAGGGCUCUGUCUUCUGCCUCUGAA-3′ from Life Technologies (siDP2) using the Amaxa Nucleofector System (Lonza) according to manufacturer’s instructions. NHEKs were suspended in Ingenio Electroporation Solution (Mirus) with siRNA (final concentration 50 nM) and electroporated using program X-001.
Viral transduction
DPNTP-GFP was cloned from pEGFP-N1-DPNTP into pLZRS using the standard Gateway/TOPO cloning protocol. pLZRS-miR Dsg1 (shDsg1) was generated as previously described (Getsios et al., 2009). LZRS–NTshRNA (shCtl) was generated with the following sequences inserted: NTshRNA-fwd 5′–GTATCTCTTCATAGCCTTAAA–3′ and NTshRNA-rev 5′–TTTAAGGCTATGAAGAGATAC–3′. Keratinocytes were transduced with retroviral supernatants produced from Phoenix cells (provided by G. Nolan, Stanford University, Stanford, CA) as previously described (Simpson, Kojima, & Getsios, 2010). Briefly, Phoenix cells transiently transfected with retroviral pLZRS cDNA constructs were harvested at 70% confluency for 24 hours at 32°C. Supernatants were collected and concentrated using Centricon Plus-20 columns (EMD Millipore). Infection of keratinocytes was done at 15% cell confluence with incubation at 32°C for 1.5 hours in M154 media containing 4 μg/ml polybrene (Sigma-Aldrich, H9268) and retrovirus supernatants. Lentivirus (pLVX myristoylated tdTomato, Clontech) transduction of keratinocytes utilized virus obtained from the Northwestern University SBDRC. Briefly, keratinocytes were plated at 30% confluence and the next day treated with 1 μg/ml polybrene plus lentivirus and incubated at 37°C overnight followed by washing
Immunofluorescence/Microscopy
For immunofluorescence analysis, NHEKs cultured on glass coverslips were fixed either in anhydrous ice-cold methanol for 3 min on ice or 4% paraformaldehyde solution for 15 min at room temperature. Cells were permeabilized and blocked with 0.25% Triton X-100 and 5% goat serum for 30 minutes and then processed for immunofluorescence. For skin biopsies, paraffin-embedded sections were baked overnight at 60°C and deparaffinized using xylene/ethanol. After permeabilization with 0.5% Triton X-100 in PBS, antigen retrieval was performed using 0.01 M citrate buffer, pH 6.0. All samples were mounted onto glass slides or using glass coverslips with ProLong Gold antifade reagent (Thermo Fisher).
Apotome images were acquired using ZEN 2.3 software (Carl Zeiss) with an epifluorescence microscope system (Axio Imager Z2, Carl Zeiss) fitted with an X-Cite 120 LED Boost System, an Apotome.2 slide module, Axiocam 503 Mono digital camera, and a Plan-Apochromat 40x/1.4 or Plan-Apochromat 63x/1.4 objective(Carl Zeiss). Confocal z-stacks (z-step size of 0.24-0.5 μm) were acquired using a Nikon A1R confocal laser microscope equipped with GaAsP detectors or a Nikon W1 Spinning Disk Confocal with a 95B prime Photometrics camera and a 60× Plan-Apochromat objective lambda with a NA of 1.4 and run by NIS-Elements software (Nikon). NIS-Elements (version 5.02) was used to generate 3D reconstructions of z-stacks using the Volume Viewer tool with z-depth coding blending (rainbow contrast lookup table) or ImageJ was used to generate color coded z-projections using Temporal-Color Code (Fire lookup table). All fluorescence intensity-based quantification was performed using ImageJ.
Laser ablation
NHEKs were transduced with pLVX myristoylated tdTomato (myr-tomato) lentivirus to allow tracking cell outlines and 24 hours after they were transduced with either pLZRS GFP or DPNTP-GFP retrovirus. These cells were then used to generate 3D epidermal equivalent cultures as described above. At the indicated timepoints, sufficient medium was added to a culture dish to re-submerge the epidermal equivalent to perform imaging with a water dipping objective and secured using a harp anchor. The culture was allowed to equilibrate for 20 minutes before imaging and only imaged for 1 hour each. Two-photon laser ablation was used to assess intercellular forces. Briefly, ablation was performed on a Nikon A1R-MP+ multiphoton microscope running Elements version 4.50 and equipped with an Apo LWD 25× 1.10W objective. Cells were maintained at 37 °C and 5% CO2. Images of myr-tomato and GFP were obtained at a rate of 1 frame per second for 2 s before and 45 s after ablation using 4% laser power at 970 nm. Ablation was performed using 40% laser power at a scan speed of 512. The distance between the cell–cell vertices over time was measured using the Manual Tracking plugin in ImageJ. Distance curves were then generated using Excel software. Initial recoil measurements were calculated using Prism 8 software as previously described [62].
Sorting/stratification assay
Wild type NHEKs were mixed at known ratios with genetically-modified NHEKs that were transduced with GFP, DPNTP-GFP, or GFP with subsequent nucleofection with siRNA. These combined populations were switched to M154 medium containing 1.2 mM calcium, HKGS, and G/A. Cultures were allowed to stratify for 3 days before fixation using 4% paraformaldehyde. Samples were processed for immunostaining as above for plakoglobin to visualize all cell outlines as well as DAPI to visualize nuclei. Confocal z-stacks were obtained and processed in ImageJ to contain either only basal cells or suprabasal cells by selecting a subset of z-frames. The total number of cells per field was the quantified using DAPI and plakoglobin staining. The subset of the total population that expressed GFP was then calculated and compared to the percentage of cells predicted to express GFP based on the number of wild type and GFP expressing cells put into the system. This was compared for basal as well as suprabasal cells.
Transepidermal electrical resistance
Costar 24 mm Transwell 0.4 μm pore inserts (Costar, 3450) were coated with CELLstart CTS Substrate (Thermo Fisher Scientific, A10142-01) according to manufacturer. NHEKs were seeded at confluence in Defined PCT Epidermal Keratinocyte Medium, (CnT-07, Zenbio) and submerged in CnT-07 for 2 days with medium in both upper and lower chambers. On the third day, CnT-07 was replaced with CnT-Prime 3D Barrier Culture Medium (CnT-PR-3D, Zenbio) with cells submerged. The following day, “Day 0” TEER measurements were taken and medium was removed from the upper chamber to expose NHEKs to an air-liquid interface. Subsequent TEER measurements were taken every other day/ at indicated times by adding pre-warmed (37°C) CnT-3D-PR to the top chamber. Measurements were made with an EVOM epithelial volt/ohm meter (World Precision Instruments, Sarasota, FL, USA). Measurements were taken in triplicate per sample and averaged. Medium was removed promptly from the top chamber as soon as TEER measurements were taken.
Statistical Analysis
For all assays, at least three independent experiments were carried out and exact numbers are found in the figure legends. Independent experiments were considered as those performed with NHEKs derived from individual patient materials. Data in all graphs are presented as means and error bars represent standard error of the mean (SEM). Graphs were generated using Excel or Prism software. Statistical analyses were performed using Prism software and are specifically indicated in figure legends. P values less than 0.05 were considered statistically significant.
Acknowledgments
We thank Alpha Yap, Andrew Kowalczyk, Lisa Godsel, and Quinn Roth-Carter for helpful feedback on the manuscript. Research reported in this publication was supported by Northwestern University Skin Biology & Diseases Resource-Based Center of the National Institutes of Health under award number P30AR075049. Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. Multiphoton microscopy was performed on a Nikon A1R multiphoton microscope, acquired through the support of NIH 1S10OD010398-01. This work was supported by NIH grants R01 AR041836, R37 AR043380, with partial support from R01 CA228196, the J.L. Mayberry Endowment to K.J.G. and the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. J.A.B. was supported by NIH K01 AR075087 and T32 AR060710.
Footnotes
Lead contact: Kathleen J. Green
Declaration of Interests The authors declare no competing interests.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}