

Abstract
As of February 3, 2020, the 2019 Novel Coronavirus (2019-nCoV) spread to 27 countries with 362 deaths and more than 17000 confirmed cases. 2019-nCoV is being compared to the infamous SARS coronavirus outbreak. Between November 2002 and July 2003, SARS resulted in 8098 confirmed cases worldwide with a 9.6% death rate and 774 deaths. Mainland China alone suffered 349 deaths and 5327 confirmed cases. Though 2019-nCoV has a death rate of 2.2% as of 3 February, the 17489 confirmed cases in a few weeks (December 8, 2019 to February 3, 2020) are alarming. Cases are likely under-reported given the comparatively longer incubation period. Such outbreaks demand rapid elucidation and analysis of the virus genomic sequence for timely treatment plans. We classify the 2019-nCoV using MLDSP and MLDSP-GUI, alignment-free methods that use Machine Learning (ML) and Digital Signal Processing (DSP) for genome analyses. Genomic sequences were mapped into their respective genomic signals (discrete numeric series) using a two-dimensional numerical representation (Chaos Game Representation). The magnitude spectra were computed by applying Discrete Fourier Transform on the genomic signals. The Pearson Correlation Coefficient was used to calculate a pairwise distance matrix. The feature vectors were constructed from the distance matrix and used as an input to the supervised machine learning algorithms. 10-fold cross-validation was applied to compute the average classification accuracy scores. The trained classifier models were used to predict the labels of 29 2019-nCoV sequences. The classification strategy used over 5000 genomes and tested associations at taxonomic levels of realm to species. From our machine learning-based alignment-free analyses using MLDSP-GUI, we corroborate the current hypothesis of a bat origin and classify 2019-nCoV as Sarbecovirus, within Betacoronavirus.
1. Introduction
Coronaviruses are single-stranded positive-sense RNA viruses that are known to contain some of the largest viral genomes, up to around 32 kbp in length [1–5]. After increases in the number of coronavirus genome sequences available following efforts to investigate the diversity in the wild, the family Coronaviridae now contains four genera (International Committee on Taxonomy of Viruses, [6]). While those species that belong to the genera Alphacoronavirus and Betacoronavirus can infect mammalian hosts, those in Gammacoronavirus and the recently defined Deltacoronavirus mainly infect avian species [4,7–9]. Phylogenetic studies have revealed a complex evolutionary history, with coronaviruses thought to have ancient origins and recent crossover events that can lead to cross-species infection [8,10–12]. Some of the largest sources of diversity for coronaviruses belong to the strains that infect bats and birds, providing a reservoir in wild animals for recombination and mutation that may enable cross-species transmission into other mammals and humans [4,7,8,10,13].
Like other RNA viruses, coronavirus genomes are known to have genomic plasticity, and this can be attributed to several major factors. RNA-dependent RNA polymerases (RdRp) have high mutation rates, reaching from 1 in 1000 to 1 in 10000 nucleotides during replication [7,14,15]. Coronaviruses are also known to use a template switching mechanism which can contribute to high rates of homologous RNA recombination between their viral genomes [9,16–20]. Furthermore, the large size of coronavirus genomes is thought to be able to accommodate mutations to genes [7]. These factors help contribute to the plasticity and diversity of coronavirus genomes today.
The highly pathogenic human coronaviruses, Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) belong to lineage B (sub-genus Sarbecovirus) and lineage C (sub-genus Merbecovirus) of Betacoronavirus, respectively [9,21–23]. Both result from zoonotic transmission to humans and lead to symptoms of viral pneumonia, including fever, breathing difficulties, and more [24,25]. Recently, an unidentified pneumonia disease with similar symptoms caused an outbreak in Wuhan and is thought to have started from a local fresh seafood market [26–30]. This was later attributed to a novel coronavirus deemed 2019-nCoV and represents the third major zoonotic human coronavirus of this century [31]. As of February 3, confirmed cases have risen to 17489 globally, with infections reported in 27 countries [32]. As a result, the World Health Organization set the risk assessment to “Very High” for China, where the bulk of the cases are contained and “High” for regional and global levels [33]. Initiatives to identify the source of transmission and possible intermediate animal vectors have commenced since the genome sequence became publicly available.
From analyses employing whole genome to viral protein-based comparisons, the 2019-nCoV strain is thought to belong to lineage B (Sarbecovirus) of Betacoronavirus. From phylogenetic analysis of the RdRp protein, spike proteins, and full genomes of 2019-nCoV and other coronaviruses, it was found that 2019-nCoV is most closely related to two bat SARS-like coronaviruses, bat-SL-CoVZXC21 and bat-SL-CoVZC45, found in Chinese horseshoe bats Rhinolophus sinicus [12,34–38]. Along with the phylogenetic data, the genome organization of 2019-nCoV was found to be typical of lineage B (Sarbecovirus) Betacoronaviruses [34]. From phylogenetic analysis of full genome alignment and similarity plots, it was found that 2019-nCoV has the highest similarity to the bat coronavirus RaTG13 [39]. Close associations to bat coronavirus RaTG13 and two bat SARS-like CoVs (ZC45 and ZXC21) are also supported in alignment-based phylogenetic analyses [39]. Within the 2019-nCoV strains, over 99% sequence similarity and a lack of diversity within these strains suggest a common lineage and source, with support for recent emergence of the human strain [12,31]. There is still ongoing debate whether the 2019-nCoV strain arose following recombination with previously identified bat and unknown coronaviruses [40] or arose independently as a new lineage to infect humans [39]. In combination with the identification that the angiotensin converting enzyme 2 (ACE2) protein is a receptor for 2019-nCoV, as it is for SARS and other Sarbecovirus strains, the hypothesis that 2019-nCoV originated from bats is deemed very likely [12,34,36,39,42–45].
All analyses performed thus far have been alignment-based and rely on the annotations of the viral genes. Though alignment-based methods are significantly successful in finding sequence similarities, they can be challenging in many cases [46,47]. It is realistically impossible to analyze thousands of complete genomes using alignment-based methods due to the heavy computation time. Moreover, the alignment demands the sequences to be continuously homologous which is not always the case. Alignment-free methods [48–52] have been proposed in the past as an alternative to address the limitations of the alignment-based methods. Comparative genomics beyond alignment-based approaches have benefited from the computational power of machine learning. Machine learning-based alignment-free methods have also been used successfully for a variety of problems including virus classification [50–52]. An alignment-free approach [50] was proposed for subtype classification of HIV-1 genomes and achieved ∼ 97% classification accuracy. MLDSP, with the use of a broad range of 1D numerical representations of DNA sequences, has also achieved very high levels of classification accuracy with viruses. Even rapidly evolving, plastic genomes of viruses such as Influenza and Dengue are classified down to the level of strain and subtype, respectively with 100% classification accuracy. MLDSP-GUI [52] provides an option to use 2D Chaos Game Representation (CGR) [53] as numerical representation of DNA sequences. CGR’s have a longstanding use in species classification with identification of biases in sequence composition [49,52,53]. MLDSP-GUI has shown 100% classification accuracy for Flavivirus genus to species classification using 2D CGR as numerical representation [52]. MLDSP and MLDSP-GUI have shown the ability to identify the genomic signatures (a species-specific pattern known to be pervasive throughout the genome) with species level accuracy that can be used for sequence (dis)similarity analyses. In this study, we use MLDSP [51] and MLDSP-GUI [52] with CGR as a numerical representation of DNA sequences to assess the classification of 2019-nCoV from the perspective of machine learning-based alignment-free whole genome comparison of genomic signatures. Timely identification of novel sequences at the time of such epidemic events is of immense importance. This alignment-free approach is ultra-fast and scalable enough to accommodate thousands of complete genomes to provide rapid, thorough analysis. This paper sets a benchmark and demonstrates how alignment-free methods have progressed over the years and are now able to find genomic dissimilarities at a very fine levels. Using MLDSP and MLDSP-GUI, we confirm that the 2019-nCoV belongs to the Betacoronavirus and its genomic similarity to the sub-genus Sarbecovirus corroborates a possible bat origin.
2. Materials and Methods
The Wuhan seafood market pneumonia virus (2019-nCoV virus) isolate Wuhan-Hu-1 complete reference genome of 29903 bp was downloaded from the NCBI database on January 23, 2020. Also, we downloaded all of the available 29 sequences of 2019-nCoV and the bat Betacoronavirus RaTG13 from the GISAID platform and two additional sequences (bat-SL-CoVZC45, and bat-SL-CoVZXC21) from the NCBI on January 27, 2019. We downloaded all of the available viral sequences from the Virus-Host DB (14688 sequences available on January 14, 2020). Virus-Host DB covers the sequences from the NCBI RefSeq (release 96, September 9, 2019), and GenBank (release 233.0, August 15, 2019). All sequences shorter than 2000 bp and longer than 50000 bp were ignored to address possible issues arising from sequence length bias.
ML-DSP [51] and MLDSP-GUI [52] were used as the machine learning-based alignment-free methods for complete genome analyses. As MLDSP-GUI is an extension of the ML-DSP methodology, we will refer to the method hereafter as MLDSP-GUI. Each genomic sequence is mapped into its respective genomic signal (a discrete numeric sequence) using a numerical representation. For this study, we use a two-dimensional k-mer (oligomers of length k) based numerical representation known as Chaos Game Representation (CGR) [53]. The k-mer value 7 is used for all the experiments. The value k = 7 achieved the highest accuracy scores for the HIV-1 subtype classification [50] and this value could be relevant for other virus related analyses. The magnitude spectra are then calculated by applying Discrete Fourier Transform (DFT) to the genomic signals [51]. A pairwise distance matrix is then computed using the Pearson Correlation Coefficient (PCC) [54] as a distance measure between magnitude spectra. The distance matrix is used to generate the 3D Molecular Distance Maps (MoDMap3D) [55] by applying the classical Multi-Dimensional Scaling (MDS) [56]. MoDMap3D represents an estimation of the relationship among sequences based on the genomic distances between the sequences. The feature vectors are constructed from the columns of the distance matrix and are used as an input to train six supervised-learning based classification models (Linear Discriminant, Linear SVM, Quadratic SVM, Fine KNN, Subspace Discriminant, and Subspace KNN) [51]. A 10-fold cross-validation is used to train, and test the classification models and the average of 10 runs is reported as the classification accuracy. The trained machine learning models are then used to test the 2019-nCoV sequences. The unweighted pair group method with arithmetic mean (UPGMA) phylogenetic tree is also computed using the pairwise distance matrix.
For validation of MLDSP-GUI results using CGR as a numerical representation, another statistical method that is reliant on genomic signatures, Spearman’s rank correlation coefficient [57–60], is used. The frequency of each k-mer is calculated in each genome. Due to differences in genome length between species, proportional frequencies are computed by dividing each k-mer frequency by the length of the respective sequence. To determine whether there is a correlation between k-mer frequencies in 2019-nCoV and specific taxonomic groups, a Spearman’s rank correlation coefficient test is conducted for k = 1 to k = 7.
3. Results
Table 1 provides the details of three datasets Test-1, Test-2, Test-3a and Test-3b used for analyses with MLDSP-GUI. Each dataset’s composition (clusters with number of sequences), the respective sequence length statistics, and results of MLDSP-GUI after applying 10-fold cross-validation as classification accuracy scores are shown.
Classification accuracy scores of viral sequences at different levels of taxonomy.
As shown in Table 1, for the first test (Test-1), we organized the dataset of sequences into 12 clusters (11 families, and Riboviria realm). Only the families with at least 100 sequences were considered. The Riboviria cluster contains all families that belong to the realm Riboviria. For the clusters with more than 500 sequences, we selected 500 sequences at random. Our method can handle all of the available 14668 sequences, but using imbalanced clusters, in regard to the number of sequences, can introduce an unwanted bias. After filtering out the sequences, our pre-processed dataset is left with 3273 sequences organized into 12 clusters (Adenoviridae, Anelloviridae, Caudovirales, Geminiviridae, Genomoviridae, Microviridae, Ortervirales, Papillomaviridae, Parvoviridae, Polydnaviridae, Polyomaviridae, and Riboviria). We used MLDSP-GUI with CGR as the numerical representation at k = 7. The maximum classification accuracy of 94.9% is obtained using the Quadratic SVM model. The respective MoDMap3D is shown in Figure 1(a). All six classification models trained on 3273 sequences were used to classify (predict the label of) the 29 2019-nCoV sequences. All of our machine learning-based models correctly predicted and confirmed the label as Riboviria for all 29 sequences (Table 2).
Predicted taxonomic labels of 29 2019-nCoV sequences.

MoDMap3D of (a) 3273 viral sequences from Test-1 representing 11 viral families and realm Riboviria, (b) 2779 viral sequences from Test-2 classifying 12 viral families of realm Riboviria, (c) 208 Coronaviridae sequences from Test-3a classified into genera.
Test-1 classified the 2019-nCoV virus as belonging to the realm Riboviria. The second test (Test-2) is designed to classify 2019-nCoV among the families of the Riboviria realm. We completed the dataset pre-processing using the same rules as in Test-1 and obtained a dataset of 2779 sequences placed into the 12 families (Betaflexiviridae, Bromoviridae, Caliciviridae, Coronaviridae, Flaviviridae, Peribunyaviridae, Phenuiviridae, Picornaviridae, Potyviridae, Reoviridae, Rhabdoviridae, and Secoviridae), see Table 1. MLDSP-GUI with CGR at k = 7 as the numerical representation was used for the classification of the dataset in Test-2. The maximum classification accuracy of 93.1% is obtained using the Quadratic SVM model. The respective MoDMap3D is shown in Figure 1(b). All six classification models trained on 2779 sequences were used to classify (predict the label of) the 29 2019-nCoV sequences. All of our machine learning-based models predicted the label as Coronaviridae for all 29 sequences (Table 2).
The third test (Test-3a) is designed to classify the 2019-nCov sequences at the genus level. We considered 208 Coronaviridae sequences available under four genera (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) (Table 1). MLDSP-GUI with CGR at k = 7 as the numerical representation was used for the classification of the dataset in Test-3a. The maximum classification accuracy of 98.1% is obtained using the Linear Discriminant model and the respective MoDMap3D is shown in Figure 1(c). All six classification models trained on 208 sequences were used to classify (predict the label of) the 29 2019-nCoV sequences. All of our machine learning-based models predicted the label as Betacoronavirus for all 29 sequences (Table 2). To verify that the correct prediction is not an artifact of possible bias because of larger Betacoronavirus cluster, we did a secondary Test-3b with cluster size limited to the size of smallest cluster (after removing the Gammacoronavirus because it just had 9 sequences). The maximum classification accuracy of 100% is obtained using the Linear Discriminant model for Test-3b. All six classification models trained on 60 sequences were used to classify the 29 2019-nCoV sequences. All of our machine learning-based models predicted the label as Betacoronavirus for all 29 sequences (Table 2). This secondary test showed that the possible bias is not significant enough to have any impact on the classification performance.
Given confirmation that the 2019-nCoV belongs to the Betacoronavirus genus, there now is a question of its origin and relation to the other viruses of the same genus. To examine this question, we preprocessed our dataset from our third test to keep the sub-clusters of the Betacoronavirus with at least 10 sequences (Test-4). This gives 124 sequences placed into four clusters (Embecovirus, Merbecovirus, Nobecovirus, Sarbecovirus) (Table 3). The maximum classification accuracy of 98.4% with CGR at k = 7 as the numerical representation is obtained using the Quadratic SVM model. The respective MoDMap3D is shown in Figure 2(a). All six classifiers trained on 124 sequences predicted the label as Sarbecovirus, when used to predict the labels of 29 2019-nCoV sequences. For Test-5, we added 2019-nCoV with 29 sequences as the fifth cluster, see Table 3. The maximum classification accuracy of 98.7% with CGR at k = 7 as the numerical representation is obtained using the Subspace Discriminant model. The respective MoDMap3D is shown in Figure 2(b). In the MoDMap3D plot from Test-5, 2019-nCoV sequences are placed in a single distinct cluster, see Figure 2(b). As visually suggested by the MoDMap3D (Figure 2(b)), the average inter-cluster distances confirm that the 2019-nCoV sequences are closest to the Sarbecovirus (average distance 0.0556), followed by Merbecovirus (0.0746), Embecovirus (0.0914), and Nobecovirus (0.0916). The three closest sequences based on the average distances from all 2019-nCoV sequences are RaTG13 (0.0203), bat-SL-CoVZC45 (0.0418), and bat-SL-CoVZXC21 (0.0428).
Genus to sub-genus classification accuracy scores of Betacoronavirus.

MoDMap3D of (a) 124 Betacoronavirus sequences from Test-4 classified into sub-genera, (b) 153 viral sequences from Test-5 classified into 4 sub-genera and 2019-nCoV, (c) 76 viral sequences from Test 6 classified into Sarbecovirus and 2019-nCoV.
For Test-6, we classified Sarbecovirus (47 sequences) and 2019-nCoV (29 sequences) clusters and achieved separation of the two clusters visually apparent in the MoDMap3D, see Figure 2(c). Quantitatively, using 10-fold cross-validation, all six of our classifiers report 100% classification accuracy. We generated a phylogenetic tree based on all pairwise distances for the dataset in Test-6 that shows the separation of the two clusters and relationships within the clusters (Figure 3). As observed in Test-5, the phylogenetic tree shows that the 2019-nCoV sequences are closer to the bat Betacoronavirus RaTG13 sequence collected from a bat host.

The UPGMA phylogenetic tree using the Pearson Correlation Coefficient generated pairwise distance matrix shows 2019-nCoV (Red) sequences proximal to the bat Betacoronavirus RaTG13 (Blue) and bat SARS-like coronaviruses ZC45/ZXC21 (Green) in a distinct lineage from the rest of Sarbecovirus sequences
Figure 4 shows the Chaos Game Representation (CGR) plots of different sequences from the four different genera (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus) of the family Coronaviridae. The CGR plots visually suggests and the pairwise distances confirm that the genomic signature of the 2019-nCoV Wuhan-Hu-1 (Figure 4(a)) is closer to the genomic signature of the BetaCov-RaTG13 (Figure 4(b); distance: 0.0204), followed by the genomic signatures of bat-SL-CoVZC45 (Figure 4(c); distance: 0.0417), bat-SL-CoVZXC21(Figure 4(d); distance: 0.0428), Alphacoronavirus /DQ811787 PRCV ISU-1 (Figure 4(e); distance: 0.0672), Gammacoronavirus / Infectious bronchitis virus NGA /A116E7/2006/FN430415(Figure 4(f); distance: 0.0791), and Deltacoronavirus / PDCoV / USA / Illinois121 /2014/KJ481931 (Figure 4(g); distance: 0.0851).

Chaos Game Representation (CGR) plots at k = 7 of (a) 2019-nCoV / Wuhan seafood market pneumonia virus isolate Wuhan-Hu-1/MN908947.3, (b) Betacoronavirus / CoV / Bat / Yunnan / RaTG13 /EPI_ISL_402131, (c) Betacoronavirus / Bat SARS-like coronavirus isolate bat-SL-CoVZC45 /MG772933.1, (d) Betacoronavirus / Bat SARS-like coronavirus isolate bat-SL-CoVZXC21 /MG772934.1, (e) Alphacoronavirus /DQ811787 PRCV ISU-1, (f) Gammacoronavirus / Infectious bronchitis virus NGA /A116E7/2006/FN430415, and (g) Deltacoronavirus / PDCoV / USA / Illinois121 /2014/KJ481931. Chaos plot vertices are assigned top left Cytosine, top right Guanine, bottom left Adenine and bottom right Thymine.
The Spearman’s rank correlation coefficient tests were used to further confirm the ML-DSP findings. The first test in Figure 5 shows the 2019-nCoV being compared to the four genera; Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. The 2019-nCoV showed the highest k-mer frequency correlation to Betacoronavirus at k = 7 (Table 4), which is consistent with the ML-DSP results in Test-3 (Table 2). The 2019-nCoV was then compared to all sub-genera within the Betacoronavirus genus: Embecovirus, Merbecovirus, Nobecovirs and Sarbecovirus seen in Figure 6. The Spearman’s rank test was again consistent with the ML-DSP results seen in Table 3, as the k-mer frequencies at k = 7 showed the highest correlation to the sub-genus Sarbecovirus (Table 4). These tests confirm the findings in ML-DSP and are consistent with the 2019-nCoV virus as part of the sub-genus Sarbecovirus.

Hexbin scatterplots of the proportional k-mer (k = 7) frequencies of the 2019-nCoV sequences versus the four genera: (a) Alphacoronavirus, ρ = 0.7; (b) Betacoronavirus, ρ = 0.74; (c) Gammacoronavirus, ρ = 0.63 and (d) Deltacoronavirus, ρ = 0.6. The color of each hexagonal bin in the plot represents the number of points (in natural logarithm scale) overlapping at that position. All ρ values resulted in p-values < 10−5 for the correlation test. By visually inspecting each hexbin scatterplot, the degree of correlation is displayed by the variation in spread between the points. Hexagonal points that are closer together and less dispersed as seen in (b) are more strongly correlated and have less deviation.
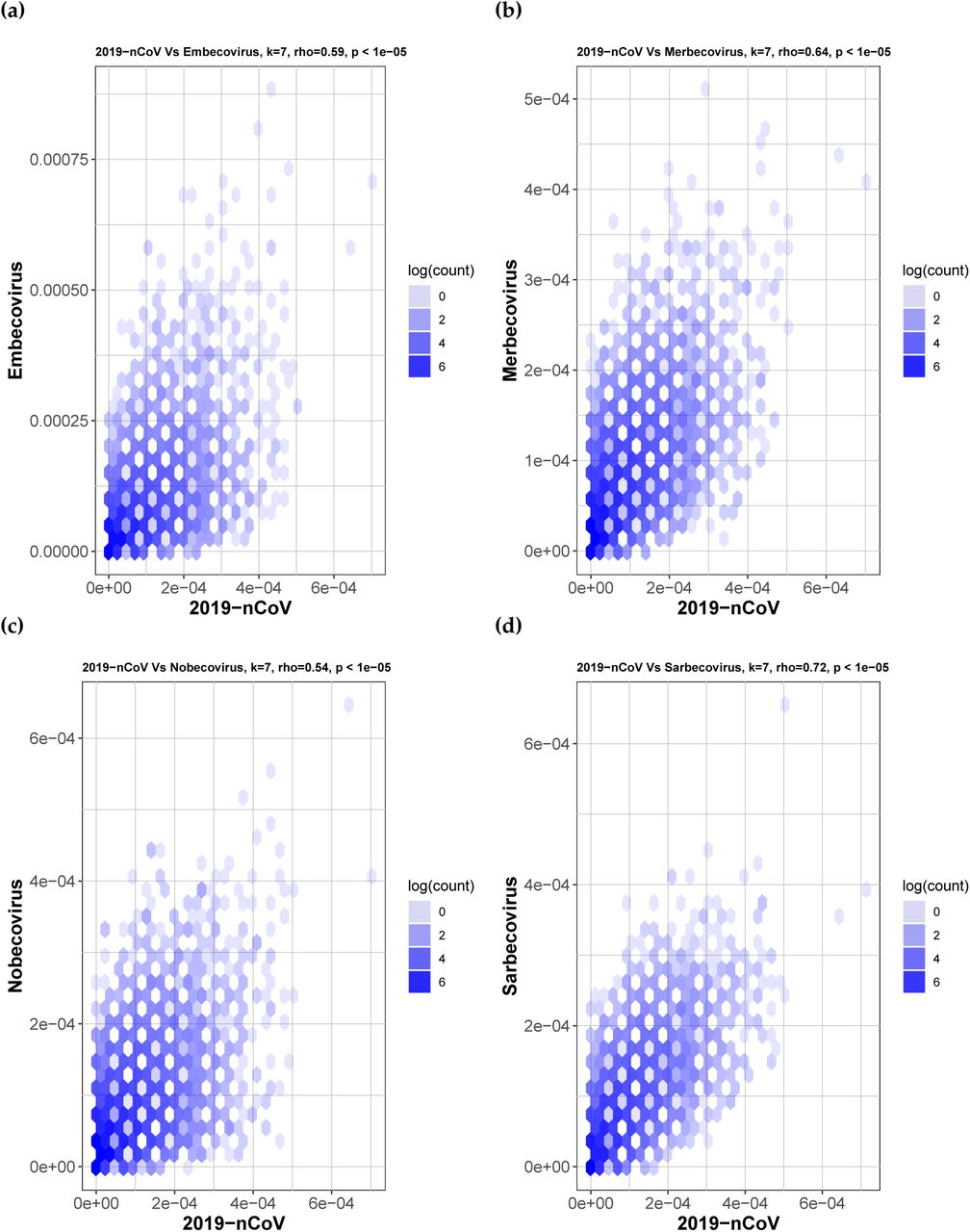
Hexbin scatterplots of the proportional k-mer (k = 7) frequencies of the 2019-nCoV sequences versus the four sub-genera: (a) Embecovirus, ρ = 0.59; (b) Merbecovirus, ρ = 0.64; (c) Nobecovirus, ρ = 0.54 and (d) Sarbecovirus, ρ = 0.72. The color of each hexagonal bin in the plot represents the number of points (in natural logarithm scale) overlapping at that position. All ρ values resulted in p-values < 10−5 for the correlation test. By visually inspecting each hexbin scatterplot, the degree of correlation is displayed by the variation in spread between the points. Hexagonal points that are closer together and less dispersed as seen in (d) are more strongly correlated and have less deviation.
4. Discussion
Prior work elucidating the evolutionary history of the Wuhan 2019-nCoV virus had suggested an origin from bats prior to zoonotic transmission [12,34,36,39,42,61]. Most early cases of individuals infected with 2019-nCoV had contact with the Huanan South China Seafood Market [26–31]. Human-to-human transmission is confirmed, further highlighting the need for continued intervention [34,61–63]. Still, the early 2019-nCoV genomes that have been sequenced and uploaded are over 99% similar, suggesting these infections result from a recent cross-species event [12,31,41].
These prior analyses relied upon alignment-based methods to identify relationships between 2019-nCoV and other coronaviruses with nucleotide and amino acid sequence similarities. When analyzing the conserved replicase domains of ORF1ab for coronavirus species classification, nearly 94% of amino acid residues were identical to SARS-CoV, yet overall genome similarity was only around 70%, confirming that 2019-nCoV was genetically different [63]. Within the RdRp region, it was found that another bat coronavirus, RaTG13, was the closest relative to 2019-nCoV and formed a distinct lineage from other bat SARS-like coronaviruses [39,41]. Other groups found that two bat SARS-like coronaviruses, bat-SL-CoVZC45 and bat-SL-CoVZXC21, were also closely related to 2019-nCoV [12,34–38]. There is a consensus that these three bat viruses are most similar to 2019-nCoV, however, whether or not 2019-nCoV arose from a recombination event is still unknown [39–41].
Regardless of the stance on recombination, current consensus holds that the hypothesis of 2019-nCoV originating from bats is highly likely. Bats have been identified as a reservoir of mammalian viruses and cross-species transmission to other mammals, including humans [4,7,8,10,13,64–66]. Prior to intermediary cross-species infection, the coronaviruses SARS-CoV and MERS-CoV were also thought to have originated in bats [24,25,35,68,69]. Many novel SARS-like coronaviruses have been discovered in bats across China, and even in European, African and other Asian countries [35,70–76]. With widespread geographic coverage, SARS-like coronaviruses have likely been present in bats for a long period of time and novel strains of these coronaviruses can arise through recombination [4]. Whether or not 2019-nCoV was transmitted directly from bats, or from intermediary hosts, is still unknown, and will require identification of 2019-nCoV in species other than humans, notably from the wet market and surrounding area it is thought to have originated from [30]. While bats have been reported to have been sold at the Huanan market, at this time, it is still unknown if there were intermediary hosts involved prior to transmission to humans [27,31,34,40,77]. Snakes had been proposed as an intermediary host for 2019-nCoV based on relative synonymous codon usage bias studies between viruses and their hosts [40], however, this claim has been disputed [78]. China CDC released information about environmental sampling in the market and indicated that 33 of 585 samples had evidence of 2019-nCoV, with 31 of these positive samples taken from the location where wildlife booths were concentrated, suggesting possible wildlife origin [79,80]. Detection of SARS-CoV in Himalyan palm civets and horseshoe bats identified 29 nucleotide sequences that helped trace the origins of SARS-CoV isolates in humans to these intermediary species [13,24,39,76]. Sampling additional animals at the market and wildlife in the surrounding area may help elucidate whether intermediary species were involved or not, as was possible with the SARS-CoV.
Viral outbreaks like nCoV-2019 demand timely analysis of genomic sequences to guide the research in the right direction. This problem being time-sensitive requires quick sequence similarity comparison against thousands of known sequences to narrow down the candidates of possible origin. Alignment-based methods are known to be time-consuming and can be challenging in cases where homologous sequence continuity cannot be ensured. It is challenging (and sometimes impossible) for alignment-based methods to compare a large number of sequences that are too different in their composition. Alignment-free methods have been used successfully in the past to address the limitations of the alignment-based methods [49–52]. The alignment-free approach is quick and can handle a large number of sequences. Moreover, even the sequences coming from different regions with different compositions can be easily compared quantitatively, with equally meaningful results as when comparing homologous/similar sequences. We use MLDSP-GUI (a variant of MLDSP with additional features), a machine learning-based alignment-free method successfully used in the past for sequence comparisons and analyses [51]. The main advantage alignment-free methodology offers is the ability to analyze large datasets rapidly. In this study, we not only confirm the taxonomy of 2019-nCoV but also show how to perform a thorough analysis rapidly when a novel unclassified sequence is presented. We start with the highest taxonomic level, train the classification models on the available complete genomes, test the novel unknown sequences to predict the label among the labels of the training dataset, move to the next taxonomic level, and repeat the whole process to the lowest taxonomic label.
Test-1 starts at the highest available level and classifies the viral sequences to the 11 families and Riboviria realm (Table 1). There is only one realm available in the viral taxonomy, so all of the families that belong to the realm Riboviria are placed into a single cluster and a random collection of 500 sequences are selected. No realm is defined for the remaining 11 families. The objective is to train the classification models with the known viral genomes and then predict the labels of the 2019-nCoV virus sequences. The maximum classification accuracy score of 95% was obtained using the Quadratic SVM model. This test demonstrates that MLDSP-GUI can distinguish between different viral families. The trained models are then used to predict the labels of 29 2019-nCoV sequences. As expected, all classification models correctly predict that the 2019-nCoV sequences belong to the Riboviria realm, see Table 2. Test-2 is composed of 12 families from the Riboviria, see Table 1, and the goal is to test if MLDSP-GUI is sensitive enough to classify the sequences at the next lower taxonomic level. It should be noted that as we move down the taxonomic levels, sequences become much more similar to one another and the classification problem becomes challenging. MLDSP-GUI is still able to distinguish between the sequences within the Riboviria realm with a maximum classification accuracy of 91.1% obtained using the Linear Discriminant classification model. When 2019-nCoV sequences are tested using the models trained on Test-2, all of the models correctly predict the 2019-nCoV sequences as Coronaviridae (Table 2). Test-3a moves down another taxonomic level and classifies the Coronaviridae family to four genera (Alphacoronavirus, Betacoronavirus, Deltacoronavirus, Gammacoronavirus), see Table 1. MLDSP-GUI distinguishes sequences at the genus level with a maximum classification accuracy score of 98%, obtained using the Linear Discriminant model. This is a very high accuracy rate considering that no alignment is involved and the sequences are very similar. All trained classification models correctly predict the 2019-nCoV as Betacoronavirus, see Table 2. Test-3a has Betacoronavirus as the largest cluster and it can be argued that the higher accuracy could be a result of this bias. To avoid bias, we did an additional test removing the smallest cluster Gammacoronavirus and limiting the size of remaining three clusters to the size of the cluster with the minimum number of sequences i.e. 20 with Test-3b. MLDSP-GUI obtains 100% classification accuracy for this additional test and still predicts all of the 2019-nCoV sequences as Betacoronavirus. These tests confirm that the 2019-nCoV are from the genus Betacoronavirus.
Sequences become very similar at lower taxonomic levels (sub-genera and species). Test-4, Test-5, and Test-6 investigate within the genus Betacoronavirus for sub-genus classification. Test-4 is designed to classify Betacoronavirus into the four sub-genera (Embecovirus, Merbecovirus, Nobecovirus, Sarbecovirus), see Table 3. MLDSP-GUI distinguishes sequences at the sub-genus level with a maximum classification accuracy score of 98.4%, obtained using the Quadratic SVM model. All of the classification models trained on the dataset in Test-4 predicted the label of all 29 2019-nCoV sequences as Sarbecovirus. This suggests substantial similarity between 2019-nCoV and the Sarbecovirus sequences. Test-5 and Test-6 (see Table 3) are designed to verify that 2019-nCoV sequences can be differentiated from the known species in the Betacoronavirus genus. MLDSP-GUI achieved a maximum classification score of 98.7% for Test-5 and 100% for Test-6 using Subspace Discriminant classification model. This shows that although 2019-nCoV and Sarbecovirus are closer on the basis of genomic similarity (Test-4), they are still distinguishable from known species. Therefore, these results suggest that 2019-nCoV may represent a genetically distinct species of Sarbecovirus. All 2019-nCoV virues are visually seen in MoDMap3D generated from Test-5 (see Figure 2(b)) as a closely packed cluster and it supports a fact that there is 99% similarity among these sequences [12,31]. The MoDMap3D generated from the Test-5 (Figure 2(b)) visually suggests and the average distances from 2019-nCoV sequences to all other sequences confirm that the 2019-nCoV sequences are most proximal to the RaTG13 (distance: 0.0203), followed by the bat-SL-CoVZC45 (0.0418), and bat-SL-CoVZX21 (0.0428). To confirm this proximity, a UPGMA phylogenetic tree is computed from the PCC-based pairwise distance matrix of sequences in Test-6, see Figure 3. The phylogenetic tree placed the RaTG13 sequence closest to the 2019-nCoV sequences, followed by the bat-SL-CoVZC45 and bat-SL-CoVZX21 sequences. This closer proximity represents the smaller genetic distances between these sequences and aligns with the visual sequence relationships shown in the MoDMap3D of Figure 2(b). The closeness of 2019-nCoV with the sequences from the Betacoronavirus genus (especially sub-genus Sarbecovirus) is further verified using the quantitative analyses based on the Spearman’s rank correlation coefficient tests.
Spearman’s rank correlation coefficient [57–60] tests use the frequencies of oligonucleotide segments and compares them to the total number of segments at different k-mer lengths to measure the degree of correlation between two sets of genomic sequences. Spearman’s ρ value provides the degree of correlation between the two groups and their k-mer frequencies. The 2019-nCoV virus was compared to all genera under the Coronaviridae family and the k-mer frequencies showed the strongest correlation to the genus Betacoronavirus, and more specifically Sarbecovirus. The Spearman tests corroborate that the 2019-nCoV virus is part of the Sarbecovirus sub-genus, as shown by CGR and ML-DSP. When analyzing sub-genera, it could be hard to classify at lower k values due to the short oligonucleotide frequencies not capturing enough information to highlight the distinctions. Therefore despite the Spearman’s rank correlation coefficient providing results for k = 1 to k = 7, the higher k-mer lengths provided more accurate results, and k = 7 was used.
Attributes of the 2019-nCoV genomic signature are consistent with previously reported mechanisms of innate immunity operating in bats as a host reservoir for coronaviruses. Vertebrate genomes are known to have an under-representation of CG dinucleotides in their genomes, otherwise known as CG suppression [81,82]. This feature is thought to have been due to the accumulation of spontaneous deamination mutations of methyl-cytosines over time [81]. As viruses are obligate parasites, evolution of viral genomes is intimately tied to the biology of their hosts [83]. As host cells develop strategies such as RNA interference and restriction-modification systems to prevent and limit viral infections, viruses will continue to counteract these strategies [82–84]. Dinucleotide composition and biases are pervasive across the genome and make up a part of the organism’s genomic signature [83]. These host genomes have evolutionary pressures that shape the host genomic signature, such as the pressure to eliminate CG dinucleotides within protein coding genes in humans [82]. Viral genomes have been shown to mimic the same patterns of the hosts, including single-stranded positive-sense RNA viruses, which suggests that many RNA viruses can evolve to mimic the same features of their host’s genes and genomic signature [81–85]. As genomic composition, specifically in mRNA, can be used as a way of discriminating self vs non-self RNA, the viral genomes are likely shaped by the same pressures that influence the host genome [82]. One such pressure on DNA and RNA is the APOBEC family of enzymes, members of which are known to cause G to A mutations [85–87]. While these enzymes primarily work on DNA, it has been demonstrated that these enzymes can also target RNA viral genomes [86]. The APOBEC enzymes therefore have RNA editing capability and may help contribute to the innate defence system against various RNA viruses [85]. This could therefore have a direct impact on the genomic signature of RNA viruses. Additional mammalian mechanisms for inhibiting viral RNA have been highlighted for retroviruses with the actions of zinc-finger antiviral protein (ZAP) [81]. ZAP targets CG dinucleotide sequences, and in vertebrate host cells with the CG suppression in host genomes, this can serve as a mechanism for the distinction of self vs non-self RNA and inhibitory consequences [81]. Coronaviruses have A/U rich and C/G poor genomes, which over time may have been, in part, a product of cytidine deamination and selection against CG dinucleotides [88–90]. This is consistent with the fact that bats serve as a reservoir for many coronaviruses and that bats have been observed to have some of the largest and most diverse arrays of APOBEC genes in mammals [66,67]. The Spearman’s rank correlation data and the patterns observed in the CGR images from Figure 4, of the coronavirus genomes, including 2019-nCoV identify patterns such as CG underepresentation, also present in vertebrate and importantly, bat host genomes.
With human-to-human transmission confirmed and concerns for possible asymptomatic transmission, there is a strong need for continued intervention to prevent the spread of the virus [33,34,61–63]. Due to the high amino acid similarities between 2019-nCoV and SARS-CoV main protease essential for viral replication and processing, anticoronaviral drugs targeting this protein and other potential drugs have been identified using virtual docking to the protease for treatment of 2019-nCoV [29,44,45,91–94]. The human ACE2 receptor has also been identified as the potential receptor for 2019-nCoV and represents a potential target for treatment [42,43].
MLDSP-GUI is an ultra-fast, alignment-free method as is evidenced by the time-performance of MLDSP-GUI for Test-1 to Test-6 given in Figure 7. MLDSP-GUI took just 10.55 seconds to compute a pairwise distance matrix (including reading sequences, computing magnitude spectra using DFT, and calculating the distance matrix using PCC combined) for the Test-1 (largest dataset used in this study with 3273 complete genomes). All of the tests combined (Test-1 to Test-6) are doable in under 10 minutes including the computationally heavy 10-fold cross-validation, and testing of 29 2019-nCoV sequences.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Time performance of MLDSP-GUI for Test1 to Test-6 (in seconds).
For any novel sequence, when thorough and time-sensitive complete genome analysis is required to narrow down the possibilities to the lowest taxonomic level possible, alignment-free methods provide a reliable and dramatically faster option. From our machine learning-based alignment-free analyses using MLDSP-GUI, we corroborate the current hypothesis of a bat origin for 2019-nCoV and classify 2019-nCoV as sub-genus Sarbecovirus, within Betacoronavirus.
5. Conclusions
We confirm current taxonomic classification of the 2019-nCoV virus through consideration of a two-dimensional genomic signature representation enabling alignment-free comparative genomics with a machine learning-based classification approach. Quantitative evidence supports classification of 2019-nCoV within the Betacoronavirus genus proximal to the Sarbecovirus sub-genus. This study provides a proof of concept that alignment-free methods are capable of accurate prediction of taxonomy of yet unclassified new sequences. Timely identification of taxonomic classification at critical periods exemplified by viral outbreaks is important and alignment-free approaches to comparative genomics complement the challenges and time-consuming attributes of alignment-based analyses. We corroborate the current hypothesis of a bat origin and classify 2019-nCoV as Sarbecovirus, within Betacoronavirus.
Supplementary Materials
The following are available as Supplementary material, Table S1: rho values for k = 1 to k = 7 from the Spearman’s rank correlation coefficient test., Table S2: Accession IDs of the Sequences downloaded from the GISAID, Accession IDs of sequences used in Test-1 to Test-6.
Author Contributions
Conceptualization, G.S.R.; methodology, G.S.R. and L.K.; statistical analyses, G.S.R. and C.P.E.d.S. and H.E.; software, G.S.R. and L.K.; validation, G.S.R.; formal analysis, G.S.R., M.P.M.S and K.A.H.; investigation, G.S.R., M.P.M.S and K.A.H.; resources, G.S.R.; data curation, G.S.R.; writing—original draft preparation, G.S.R and M.P.M.S writing—review and editing, G.S.R., M.P.M.S., H.E., C.P.E.d.S., K.A.H. and L.K.; visualization, G.S.R.; supervision, K.A.H and L.K.; project administration, K.A.H. and L.K.; funding acquisition, K.A.H. and L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NSERC (Natural Science and Engineering Research Council of Canada) Discovery Grants R2824A01 to L.K., and R3511A12 to K.A.H.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors are appreciative of the review of a manuscript draft by Hailie Pavanel
Abbreviations
The following abbreviations are used in this manuscript:
- DFT
- Discrete Fourier Transform
- MDS
- Classical MultiDimensional Scaling MoDMap3D Molecular Distance Map
- PCC
- Pearson Correlation Coefficient
References
- 1.↵
- 2.
- 3.
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.
- 18.
- 19.
- 20.↵
- 21.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.
- 60.↵
- 61.↵
- 62.
- 63.↵
- 64.↵
- 65.
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.
- 90.↵
- 91.↵
- 92.
- 93.
- 94.↵




