

Abstract
SARS-CoV-2 is a betacoronavirus that is responsible for the COVID-19 pandemic. The genome of SARS-CoV-2 was reported recently, but its transcriptomic architecture is unknown. Utilizing two complementary sequencing techniques, we here present a high-resolution map of the SARS-CoV-2 transcriptome and epitranscriptome. DNA nanoball sequencing shows that the transcriptome is highly complex owing to numerous recombination events, both canonical and noncanonical. In addition to the genomic RNA and subgenomic RNAs common in all coronaviruses, SARS-CoV-2 produces a large number of transcripts encoding unknown ORFs with fusion, deletion, and/or frameshift. Using nanopore direct RNA sequencing, we further find at least 41 RNA modification sites on viral transcripts, with the most frequent motif being AAGAA. Modified RNAs have shorter poly(A) tails than unmodified RNAs, suggesting a link between the internal modification and the 3′ tail. Functional investigation of the unknown ORFs and RNA modifications discovered in this study will open new directions to our understanding of the life cycle and pathogenicity of SARS-CoV-2.
Highlights
We provide a high-resolution map of SARS-CoV-2 transcriptome and epitranscriptome using nanopore direct RNA sequencing and DNA nanoball sequencing.
The transcriptome is highly complex owing to numerous recombination events, both canonical and noncanonical.
In addition to the genomic and subgenomic RNAs common in all coronaviruses, SARS-CoV-2 produces transcripts encoding unknown ORFs.
We discover at least 41 potential RNA modification sites with an AAGAA motif.
Main Text
Coronavirus disease 19 (COVID-19) is caused by a novel coronavirus designated as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1,2. Like other coronaviruses (order Nidovirales, family Coronaviridae, subfamily Coronavirinae), SARS-CoV-2 is an enveloped virus with a positive-sense, single-stranded RNA genome of ~30 kb. SARS-CoV-2 belongs to the genus betacoronavirus, together with SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) (with 78% and 50% homology, respectively)3. Coronaviruses (CoVs) were thought to primarily cause enzootic infections in birds and mammals. But, the recurring outbreaks of SARS, MERS, and now COVID-19 have clearly demonstrated the remarkable ability of CoVs to cross species barriers and transmit between humans4.
CoVs carry the largest genomes (26-32 kb) among all RNA virus families (Fig. 1). Each viral transcripts have a 5′-cap structure and a 3′ poly(A) tail5,6. Upon cell entry, the genomic RNA is translated to produce nonstructural proteins (nsps) from two open reading frames (ORFs), ORF1a and ORF1b. The ORF1a produces polypeptide 1a (pp1a, 440-500 kDa) that is cleaved into 11 nsps. The −1 ribosome frameshift occurs immediately upstream of the ORF1a stop codon, which allows continued translation of ORF1b, yielding a large polypeptide (pp1ab, 740-810 kDa) which is cleaved into 16 nsps. The proteolytic cleavage is mediated by viral proteases nsp3 and nsp5 that harbor a papain-like protease domain and a 3C-like protease domain, respectively.
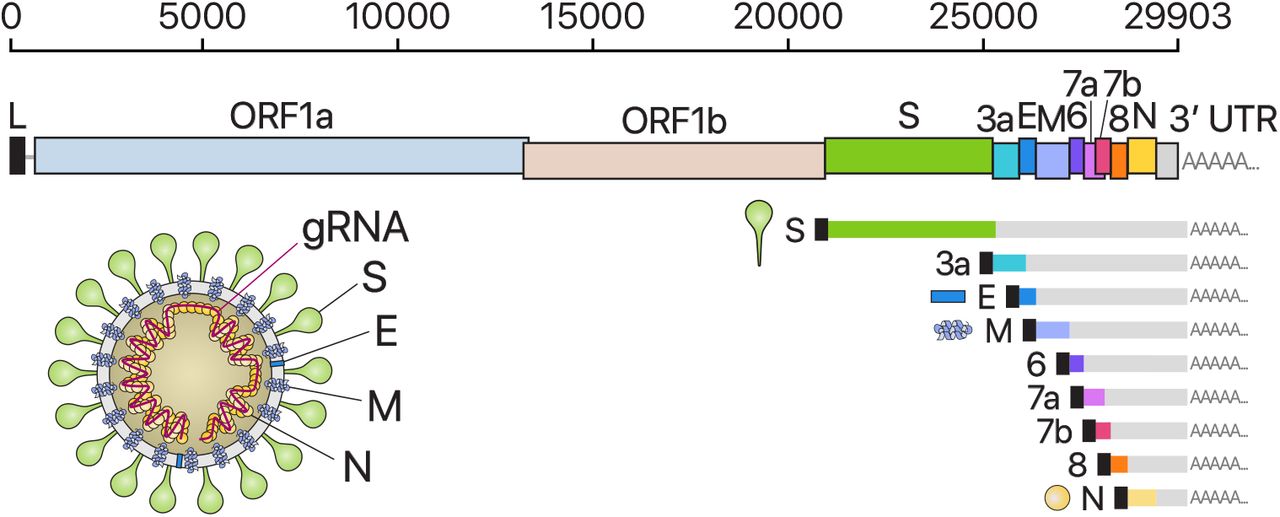
From the full-length genomic RNA (29,903 nt) which also serves as an mRNA, ORF1a and ORF1b are translated. In addition to the genomic RNA, nine major subgenomic RNAs are produced. The sizes of the boxes representing small accessory proteins are bigger than the actual size of the ORF for better visualization. The black box indicates leader sequence. Note that ORF10 is not included here because our data show no evidence for ORF10 expression.
The viral genome is also used as the template for replication and transcription, which is mediated by nsp12 harboring RNA-dependent RNA polymerase (RdRP) activity7,8. Negative-strand RNA intermediates are generated to serve as the templates for the synthesis of positive-sense genomic RNA (gRNA) and subgenomic RNAs (sgRNAs). The gRNA is packaged by the structural proteins to assemble progeny virions. Shorter sgRNAs encode conserved structural proteins (spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N)) and several accessory proteins. SARS-CoV-2 is known to have 6 accessory proteins (3a, 6, 7a, 7b, 8, and 10) according to the current annotation (NCBI Reference Sequence: NC_045512.2). But the ORFs have not yet been experimentally verified for expression. Therefore, it is currently unclear which accessory genes are actually expressed from this compact genome.
Transcription of coronaviral RNA occurs following the well-characterized recombination by template switching during negative-strand RNA synthesis. Each coronaviral RNA contains the common 5′ “leader” sequence of 70-100 nt fused to the “body” sequence from the 3′ end of the genome5,7 (Fig. 1A). The fusion of the leader and the body occurs during negative-strand synthesis at short motifs called transcription-regulating sequences (TRSs) that are located immediately adjacent to ORFs. TRSs include a conserved 6-7 nt core sequence (CS) surrounded by variable sequences (5′TRS and 3′TRS). The CS of the leader of gRNA (CS-L) can base-pair with the CS in the body of the nascent negative-sense RNA (complementary CS-B, cCS-B), which allows template switching and the leader-body fusion during negative-strand synthesis. The replication and gene regulation mechanisms have been studied in other coronaviruses. However, it is unclear whether the general mechanisms also apply to SARS-CoV-2 and if there are any unknown components in the SARS-CoV-2 transcriptome. For the development of diagnostic and therapeutic tools and the understanding of this new virus, it is critical to define the organization of the SARS-CoV-2 genome.
Deep sequencing technologies offer powerful means to investigate viral transcriptome. The “sequencing-by-synthesis (SBS)” methods such as the Illumina and MGI platforms confer high accuracy and coverage. But they are limited by short read length (200-400 nt) so the fragmented sequences should be re-assembled computationally, during which the haplotype information is lost. More recently introduced is the nanopore-based direct RNA sequencing (DRS) approach. While nanopore DRS is limited in sequencing accuracy, it enables long-read sequencing, which would be particularly useful for the analysis of long nested CoV transcripts. Moreover, because DRS does not require reverse transcription to generate cDNA, the RNA modification information can be detected directly during sequencing. Numerous RNA modifications have been found to control eukaryotic RNAs and viral RNAs9. Terminal RNA modifications such as RNA tailing also plays a critical role in cellular and viral RNA regulation10.
In this study, we combined two complementary sequencing approaches, DRS and SBS. We unambiguously map the sgRNAs, ORFs, and TRSs of SARS-CoV-2. Additionally, we find numerous unconventional recombination events that are distinct from canonical TRS-mediated joining. We further discover RNA modification sites and measure the poly(A) tail length of gRNAs and sgRNAs.
To delineate the SARS-CoV-2 transcriptome, we first performed DRS runs on a MinION nanopore sequencer using total RNA extracted from Vero cells infected with SARS-CoV-2 (BetaCoV/Korea/KCDC03/2020). The virus was isolated from a patient who was diagnosed of COVID-19 on January 26, 2020 after traveling from Wuhan, China3. We obtained 879,679 reads from infected cells (corresponding to a throughput of 1.9 Gb) (Fig. 2A). The majority (65.4%) of the reads mapped to SARS-CoV-2, indicating that viral transcripts dominate the transcriptome while the host gene expression is strongly suppressed. Although Nanopore DRS has the 3′ bias due to directional sequencing from the 3′-ends of RNAs, approximately half of the viral reads still contained the 5′ leader.

A, Read counts from nanopore direct RNA sequencing of total RNA from Vero cells infected with SARS-CoV-2. “Leader+” indicates the viral reads that contain the 5′ end leader sequence. “No leader” denotes the viral reads lacking the leader sequence. “Nuclear” reads match to mRNAs from the nuclear chromosome while “mitochondrial” reads are derived from the mitochondrial genome. “Control” indicates quality control RNA for nanopore sequencing. B, Genome coverage of the nanopore direct RNA sequencing data shown in the panel A. The stepwise reduction in coverage corresponds to the borders expected for the canonical sgRNAs. The smaller inner plot magnifies the 5′ part of the genome. C, Read counts from DNA nanoball sequencing of total RNA from Vero cells infected with SARS-CoV-2. D, Genome coverage of the DNA nanoball sequencing data shown in the panel C.
The SARS-CoV-2 genome was fully covered, missing only 12 nt from the 5′ end (Fig. 2B). The longest tags (111 reads) correspond to the full-length gRNA (Fig. 2B). The coverage of the 3′ side of the viral genome is substantially higher than that of the 5′ side, which reflects the nested sgRNAs. This is also partly due to the 3′ bias of directional DRS technique. The presence of the leader sequence (72 nt) in viral RNAs results in a prominent coverage peak at the 5′ end, as expected. We could also clearly detect vertical drops in the coverage, whose positions correspond to the leader-body junction in sgRNAs. All known sgRNAs are supported by DRS reads, with an exception of ORF10 (see below).
In addition, we observed unexpected reads reflecting noncanonical recombination events. Such fusion transcripts result in the increased coverage towards the 5′ end (Fig. 2B, inner box). Early studies on coronavirus mouse hepatitis virus reported that recombination occurs frequently11–13. Some viral RNAs contain the 5′ and 3′ proximal sequences resulting from “illegitimate” recombination events.
To further validate sgRNAs and their junction sites, we performed DNA nanoball sequencing (DNBseq) and obtained 305,065,029 reads (Fig. 2C). The results are overall consistent with the DRS data. The leader-body junctions are frequently sequenced, giving rise to a sharp peak at the 5′ end in the coverage plot (Fig. 2D). The 3′ end exhibits a high coverage as expected for the nested transcripts.
The depth of DNB sequencing allowed us to confirm and examine the junctions on an unprecedented scale. We mapped the 5′ and 3′ sites at the recombination junctions and estimated the recombination frequency by counting the reads spanning the junctions (Fig. 3A). The leader represents the most prominent 5′ site, as expected (Fig. 3A, red asterisk on the x-axis). The known TRSs are detected as the top 3′ sites (Fig. 3A, red dots on the y-axis).
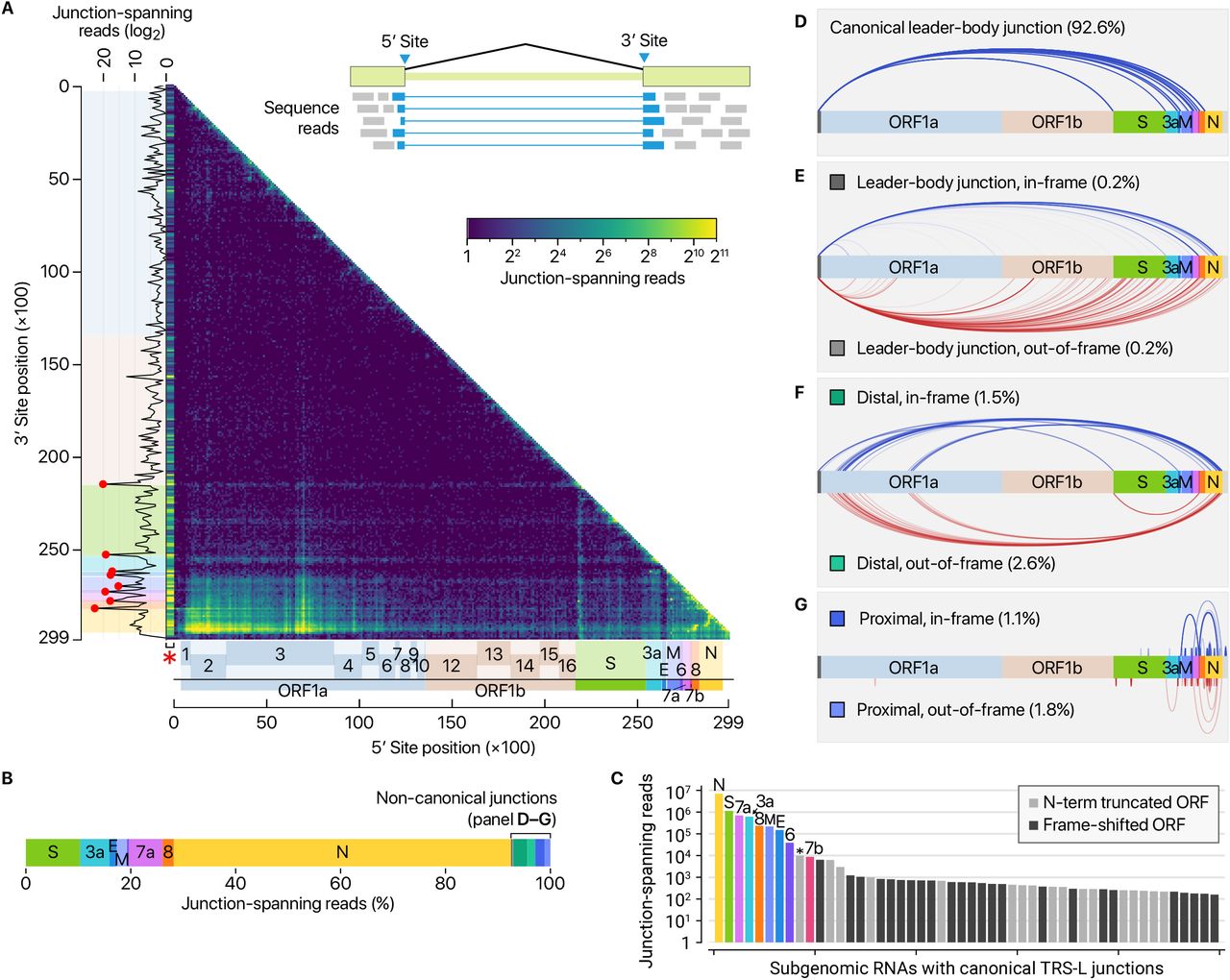
A, Positions of recombination sites determined by using junction-spanning reads from DNBseq data. The x-axis and y-axis show the positions of the 5′ sites and their joined 3′ sites, respectively. The frequency of the recombination was estimated by the read counts of the junction-spanning reads. The red asterisk on the x-axis is to indicate that the leader sequence. Please note that the left-most bins containing the leader TRS was expanded horizontally on this heatmap to improve visualization. The red dots on the sub-plot alongside the y-axis denote local peaks which coincide with the 5′ end of the body of each sgRNA. B-C, Transcript abundance was estimated by counting the DNBseq reads that span the junction of the corresponding RNA. In panel C, the asterisk indicates an ORF beginning at 27,825 which encodes the 7b protein with an N-terminal truncation of 23 amino acid. The grey bars denote minor transcripts that encode proteins with an N-terminal truncation compared with the corresponding overlapping transcript. The black bars indicate minor transcripts that encode proteins in a different reading frame from the overlapping major mRNA. D, Canonical recombination. E, Recombination between the leader TRS and a noncanonical site in the body. F, Long-distance “distal” recombination. G, “Proximal” recombination yielding local deletion between proximal sites (20-5,000 nt distance).
These results confirm that SARS-CoV-2 uses the canonical TRS-mediated mechanism for discontinuous transcription to produce major sgRNAs (Fig. 3B). Quantitative comparison of the junction-spanning reads shows that the N mRNA is the most abundantly expressed transcript, followed by S, 7a, 3a, 8, M, E, 6, and 7b (Fig. 3C). It is important to note that ORF10 is represented by only one read in DNB data (0.000009 % of viral junction-spanning reads) and that it was not supported at all by DRS data. ORF10 does not show significant homology to known proteins. Thus, ORF10 is unlikely to be expressed, and the annotation of ORF10 should be reconsidered. Taken together, SARS-CoV-2 expresses nine canonical sgRNAs (S, 3a, E, M, 6, 7a, 7b, 8, and N) together with the gRNA (Fig. 1 and Fig. 3C).
In addition to the canonical mRNAs with expected structure and length (Fig. 3B–D), our results show many minor recombination sites (Fig. 3E–G). There are three main types of such recombinant events. The RNAs in the first cluster have the leader combined with the body in the middle of ORFs or UTRs (Fig. 3E, “leader-body junction”). The second cluster shows a long-distance splitting between sequences that do not have similarity to the leader (Fig. 3F, “distal”). The last group undergoes proximal recombination which leads to smaller local deletions, mainly in structural and accessory genes, including S (Fig. 3G, “proximal”).
Of note, the junctions in these noncanonical transcripts do not contain a known TRS, indicating that at least some of these transcripts are generated through a different mechanism(s). It was previously shown in other coronaviruses that transcripts with partial sequences are produced11–13. These RNAs are considered as parasites that compete for viral proteins, hence referred to as “defective interfering RNAs” (DI-RNAs)14. Similar sgRNAs have also been described in a recent sequencing analysis on alphacoronavirus HCoV-229E15, suggesting this mechanism may be conserved among coronaviruses. While this may be due to erroneous replicase activity, it remains an open question if the recombination has an active role in the viral life cycle and evolution. Although individual RNA species are not abundant, the combined read numbers are often comparable to the levels of accessory transcripts. Most of the transcripts have coding potential to yield proteins. A notable example is the 7b protein with an N-terminal truncation that may be produced at a level similar to the annotated full-length 7b (Fig. 3C, asterisk). Many transcripts (that belong to the “distal” cluster) encode the upstream part of ORF1a, including nsp1, nsp2, and truncated nsp3, which may change the stoichiometry between nsps (Fig. 3F). Frame-shifted ORFs may also generate short peptides that are different from known viral proteins (Fig. 3B). It will be interesting in the future to examine if these unknown ORFs are actually translated and yield functional products.
As nanopore DRS is based on single-molecule detection of RNA, it offers a unique opportunity to examine multiple epitranscriptomic features of individual RNA molecules. We recently developed a software to measure the length of poly(A) tail from DRS data. Using this software, we confirm that, like other CoVs, SARS-CoV-2 RNAs carry poly(A) tails (Fig. 4A–B). The tail is likely to be critical for both translation and replication. We further find that the tail of viral RNAs are 28-71 nt in length (10th and 90th percentiles, median 47 nt). The full-length viral RNA has a relatively longer tail (~55 nt) than sgRNAs (~45 nt). Notably, sgRNAs have two tail populations: a minor peak at ~30 nt and a major peak at ~45 nt. Wu and colleagues previously observed that the poly(A) tail length of bovine CoV mRNAs change during infection: from ~45 nt immediately after virus entry to ~65 nt at 6-9 h.p.i. and ~30 nt at 120-144 h.p.i.16.

A-B, Poly(A) tail length distribution of viral transcripts. C, Poly(A) tail length distribution of quality control RNA which has a 30-nt poly(A) tail, host mRNAs from the nuclear chromosome, host RNAs from the mitochondrial chromosome.
Thus, the short tails of ~30 nt observed in this study may represent aged RNAs that are prone to decay. Viral RNAs exhibit a homogenous length distribution unlike host nuclear genome-encoded mRNAs (Fig. 4C). The viral RNAs show a similar length distribution to mitochondrial chromosome-encoded RNAs whose tail is generated by MTPAP17. It was recently shown that HCoV-229E nsp8 has an adenylyltransferase activity, which may extend poly(A) tail of viral RNA18. Given that poly(A) tail is constantly targeted by host deadenylases, it will be interesting to investigate the regulation of viral RNA tailing.
Next, we examined the epitranscriptomic landscape of SARS-CoV-2 by using the DRS data. Viral RNA modification was first described more than 40 years ago19. N6-methyladenosine (m6A) is the most widely used modification 20–24, but other modifications have also been reported on viral RNAs, including cytosine methylation (5mC), 2′-O-methylation (Nm), deamination, and terminal uridylation. In a recent analysis of HCoV-229E using DRS, modification calling suggested frequent 5mC signal across viral RNAs15. But since no direct control group was included in the analysis, the proposed modification needs validation. To unambiguously investigate the modifications, we generated negative control RNAs by in vitro transcription of the viral sequences and performed a DRS run on these unmodified control (SFig. 1A). The partially overlapping control RNAs are ~2.1 kb or ~4.4 kb each and cover the entire length of the genome (SFig. 1B). Detection using pre-trained models reported numerous signal level changes corresponding to 5mC modification sites even with the unmodified controls (SFig. 1C). We obtained highly comparable results from the viral RNAs from infected cells (SFig. 1D), clearly demonstrating that the 5mC sites detected without a control are likely to be false positives.

A, Read counts from nanopore direct RNA sequencing of in vitro transcribed (IVT) RNAs that have viral sequences. “Control” indicates quality control RNA for nanopore sequencing. B, The 15 partially overlapping patches cover the entire genome (blue rectangles at the bottom). Each RNA is ~2.1 kb in length. One fragment marked with a green rectangle is longer than others (~4.4 kb) to circumvent difficulties in the PCR amplification. The sequenced reads were downsampled so that every region is equally covered. The resulting balanced coverage is shown in the chart at the top. C, Detected 5mC modification from in vitro transcribed unmodified RNAs by the “alternative base detection” mode in Tombo. Black dots indicate the sites that satisfy the estimated false discovery rate cut-off calculated using unmodified yeast ENO2 mRNA15. D, Comparison between the sites called from unmodified IVT products and those from viral RNAs expressed in Vero cells.
We, however, noticed intriguing differences in the ionic current (called “squiggles”) between negative control and viral transcripts. At least 41 sites displayed substantial differences (over 20% frequency), indicating potential RNA modifications (Fig. 5). Notably, some of the sites showed different frequencies depending on the sgRNA species (Fig. 5A–B). Figures 5A–C show an example that is modified more heavily on the S RNA than the N RNA while Figure S2 A–C presents a site that is modified frequently on the ORF8 RNA compared with the S RNA. Moreover, the dwell time of the modified base is longer than that of the unmodified base (Fig. 5D), suggesting that the modification interferes with the passing of RNA molecules through the pore.
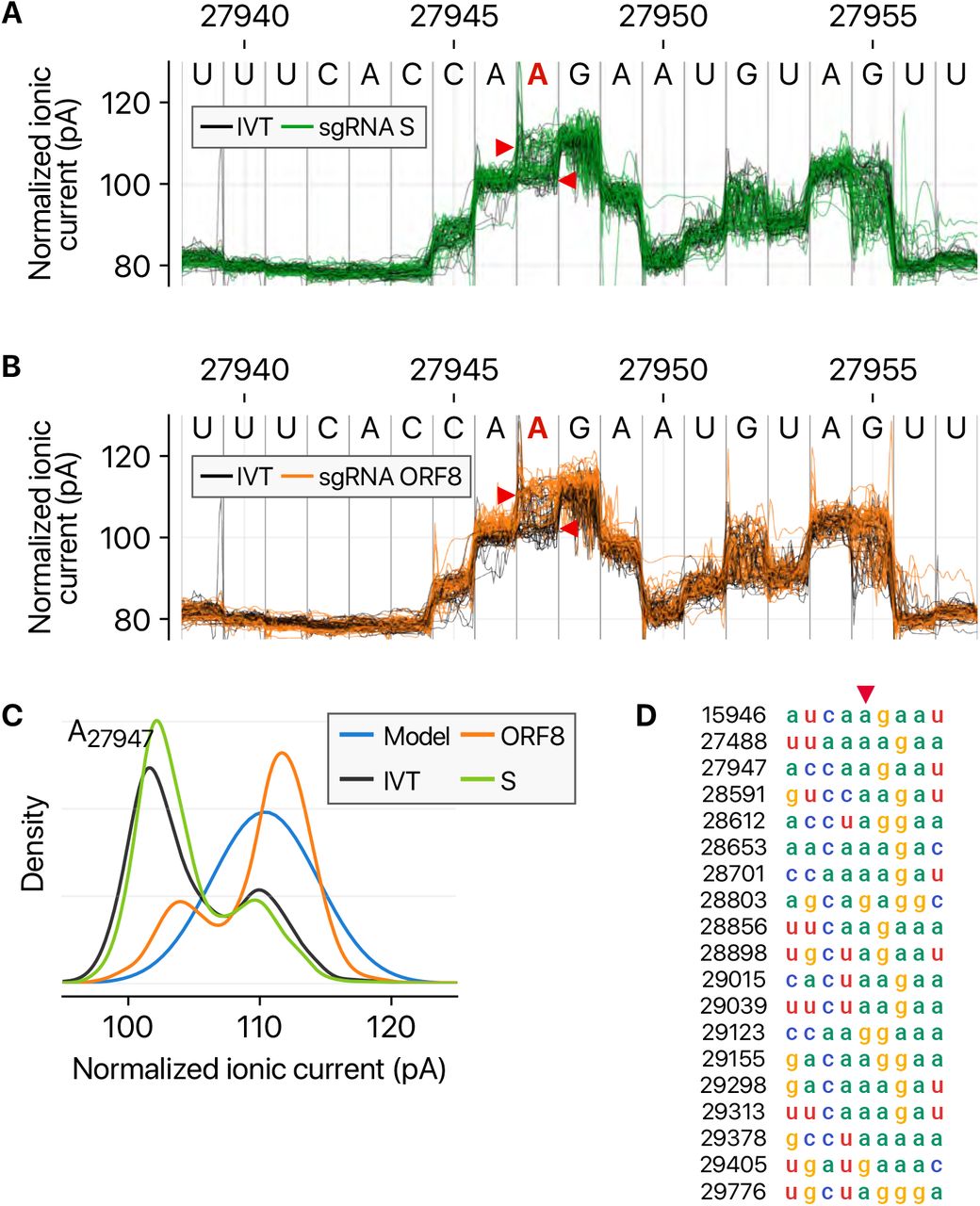
A, Ionic current levels near the genomic position 27,947 in the viral S RNA (green lines) and IVT control RNA (black lines). B, Ionic current levels for the identical region in the viral ORF8 RNA (orange lines) and IVT control RNA (black lines). C, Signal distributions at the position 27,947 in the different RNAs. The blue line shows the standard model used for modification detections without controls (“alternative base detection” and “de novo” modes) in Tombo. D, Sequence alignment of the surrounding sequence near the detected modification sites with AAGAA-like motif. Base positions on the left hand side correspond to the genomic coordinates denoted with red arrowhead.
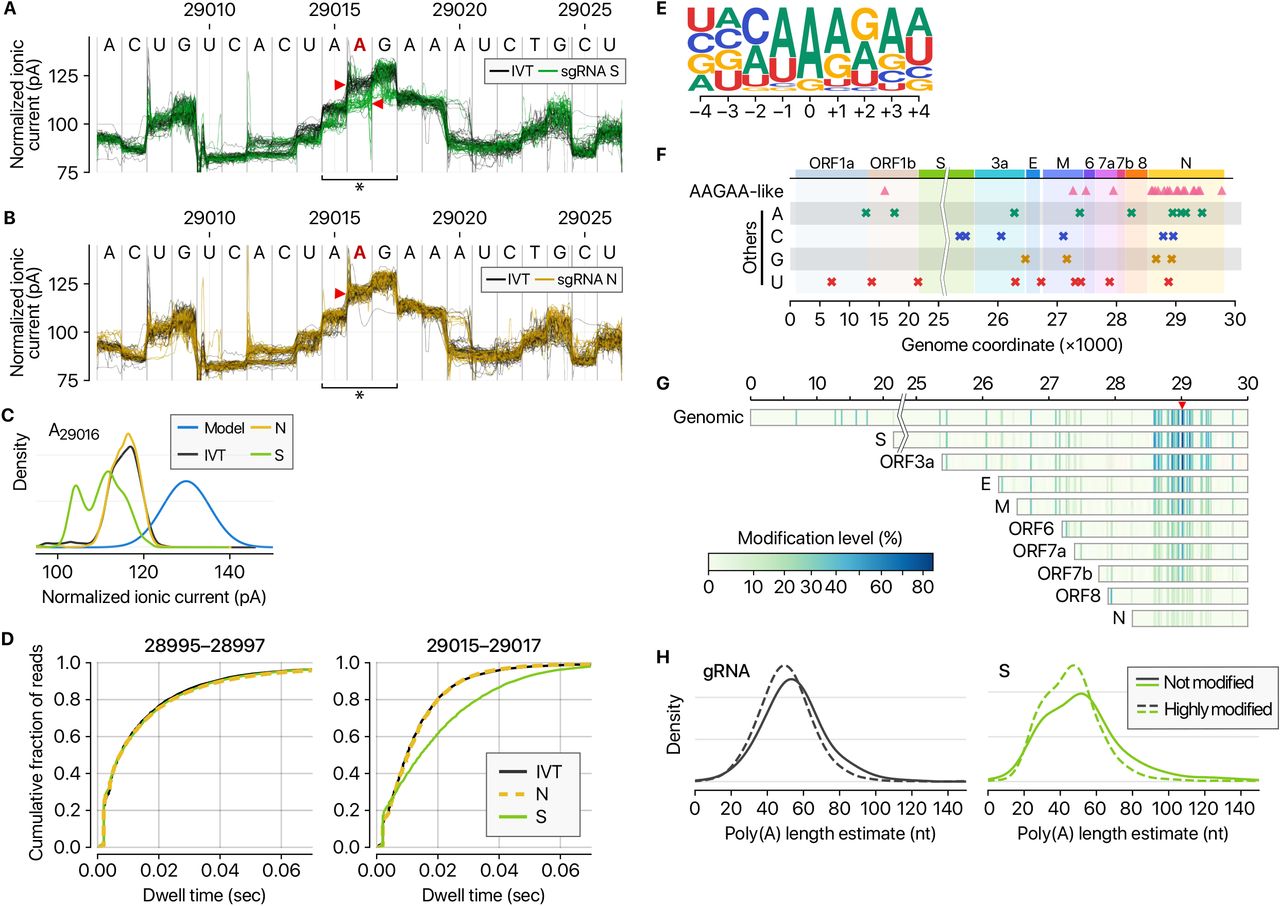
A, Distinct ionic current signals (“squiggles”) from viral S transcript (green lines) and in vitro transcribed control (IVT, black lines) indicate frequent RNA modification at the genomic position 29,016 on the S sgRNA. B, The ionic current signals from viral N transcript at the genomic position 29,016 (yellow lines) are similar to those from IVT control RNA (black lines), indicating that modification is not frequent on the N sgRNA. C, Ionic current distribution at A29016. Blue line shows the signal distribution in the standard model of Tombo 1.5. D, Dwell time difference supports RNA modification. The dwell time of the region 29,015-29,017 of the S RNA (right) is significantly longer than those of IVT control and N RNAs. On the contrary, the neighboring region 28,995-28,997 does not show the difference among IVT, N, and S RNA (left). E, Position-specific base frequency of a motif enriched in the frequently modified sites. F, Genomic location of modification sites with the AAGAA-type motif (top row) and the others grouped by the detected nucleotide base. G, Location and modification levels in different RNA species. Longer transcripts are generally modified more frequently than short sgRNAs. H, Modified viral RNAs carry shorter poly(A) tails. (Left) Poly(A) length distribution of gRNA. (Right) Poly(A) length distribution of the S mRNA.
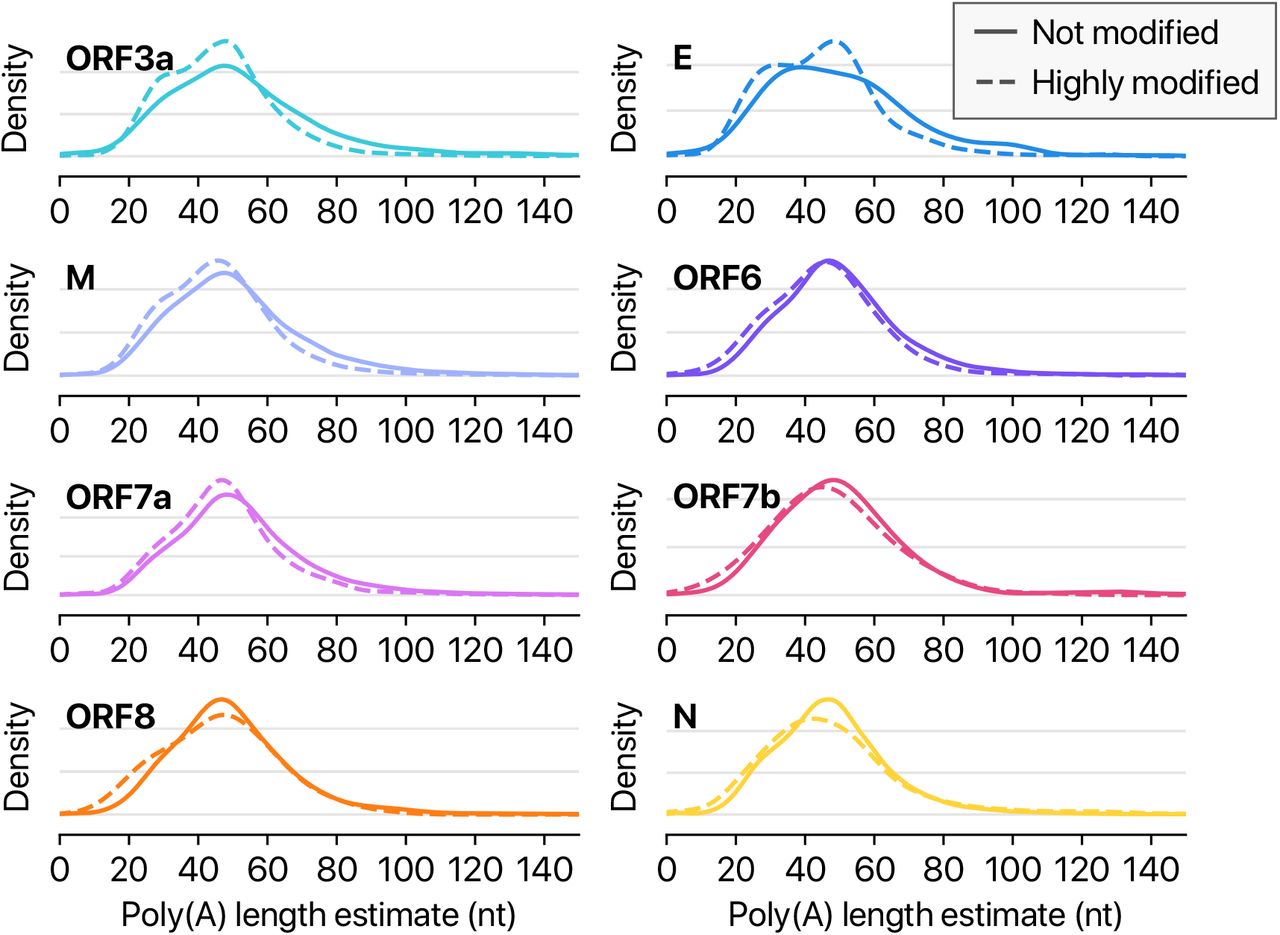
Among the 41 potential modification sites, the most frequently observed motif is ‘AAGAA’ (Fig. 5E and SFig. 2D). The modification sites with AAGAA-type motif are found throughout the viral genome, but particularly enriched in genomic positions 28,500-29,500 (Fig. 5F). Long viral transcripts (gRNA, S, 3a, E, and M) are more frequently modified than shorter RNAs (6, 7a, 7b, 8, and N) (Fig. 5G), suggesting a modification mechanism that is specific for certain RNA species.
Since the single-molecule based DRS allows a simultaneous detection of multiple features on individual molecules, we cross-examined the poly(A) tail length and internal modification sites. Interestingly, modified RNA molecules have shorter poly(A) tails than unmodified ones (Fig. 5H and SFig. 3). These results suggest a link between internal modification and 3′ end tail. Since poly(A) tail plays an important role in RNA turnover, it is tempting to speculate that the observed internal modification is involved in viral RNA stability control. It is also plausible that RNA modification is a mechanism to evade host immune response. The type of modification(s) is yet to be identified although we can exclude METTL3-mediated m6A (for lack of consensus motif RRACH), ADAR-mediated deamination (for lack of A-to-G sequence change in the DNBseq data), and m1A (for lack of the evidence for RT stop). Our finding implicates a hidden layer of CoV regulation. It will be interesting in the future to identify the chemical nature, enzymology, and biological functions(s) of the modification(s).

Poly(A) tail length distribution of each viral transcript other than shown in Fig. 5.
In this study, we delineate the transcriptomic and epitranscriptomic architecture of SARS-CoV-2. Unambiguous mapping of the expressed sgRNAs and ORFs is prerequisite for the functional investigation of viral proteins, replication mechanism, and host-viral interactions involved in pathogenicity.
In-depth analysis of the joint reads revealed a highly complex landscape of viral RNA synthesis. Like other RNA viruses, CoVs undergo frequent recombination which may allow rapid evolution to change their host/tissue specificity and drug sensitivity. It will be worth testing if the ORFs found in this study may serve as accessory proteins that modulate viral replication and host immune response. The RNA modifications may also contribute to viral survival and innate immune response in infected tissues. Our data provide a rich resource and open new directions to investigate the mechanisms underlying the pathogenicity of SARS-CoV-2.
Methods
SARS-Cov-2 sample preparation
SARS-CoV-2 viral RNA was prepared by extracting total RNA from Vero cells infected with BetaCoV/Korea/KCDC03/2020, at a multiplicity of infection (MOI) of 0.05, and cultured in DMEM supplemented with 2% fetal bovine serum and penicillin-strepamycin. The virus is the fourth passage and not plaque-isolated. Cells were harvested at 24 hours post-infection and washed once with PBS before adding TRIzol. Viral culture was conducted in a biosafety level-3 facility.
In vitro transcription
Total RNA from SARS-CoV-2-infected Vero cell was extracted by using TRIzol (Invitrogen) followed by DNaseI (Takara) treatment. Reverse transcription (SuperScript IV Reverse Transcriptase [Invitrogen]) was done with virus-specific RT primers. Templates for in vitro transcription were prepared by PCR (Q5® High-Fidelity DNA Polymerase [NEB]) with virus-specific PCR primers followed by in vitro transcription (MEGAscript™ T7 Transcription Kit [Invitrogen]). The oligonucleotides used in this study were listed in Supplementary Table 1.
Nanopore direct RNA sequencing
For nanopore sequencing on non-infected and SARS-CoV-2-infected Vero cells, each 4 μg of DNase I (Takara)-treated total RNA in 8 μl was used for library preparation following the manufacturer’s instruction (the Oxford Nanopore DRS protocol, SQK-RNA002) with minor adaptations. 20 U of SUPERase-In RNase inhibitor (Ambion, 20 U/μl) was added to both adapter ligation steps. SuperScript IV Reverse Transcriptase (Invitrogen) was adopted instead of SuperScript III, and the reaction time of reverse transcription was lengthened by 2 hours. The library was loaded on FLO-MIN106D flow cell followed by 42 hours sequencing run on MinION device (ONT).
For nanopore sequencing on SARS-CoV-2 RNA fragments by in vitro transcription, the same method was applied except for a total 2 μg of fragment RNAs and 30 minutes reaction time of reverse transcription.
DNBseq RNA sequencing
Total RNA from SARS-CoV-2-infected Vero cell was extracted by using TRIzol (Invitrogen) followed by DNaseI (Takara) treatment. Dynabeads® mRNA Purification Kit (Invitrogen) was applied to 1 μg of total RNA for rRNA depletion. RNA-seq library for 250 bp insert size was constructed following the manufacturer’s instruction (MGIEasy RNA Directional Library Prep Set). The library was loaded on MGISEQ-200RS Sequencing flow cell with MGISEQ-200RS High-throughput Sequencing Kit (PE 100), and the library was run on DNBSEQ-G50RS (paired-end run, 100 × 100 cycles).
Ethics Statement
This study was carried out in accordance with the biosafety guideline by the KCDC. The Institutional Biosafety Committee of Seoul National University approved the protocol used in these studies (SNUIBC-200219-10).
Funding
This work was supported by IBS-R008-D1 of Institute for Basic Science from the Ministry of Science, ICT and Future Planning of Korea (D.K., H.C. and V.N.K.), BK21 Research Fellowship from the Ministry of Education of Korea (D.K.), the New Faculty Startup Fund from Seoul National University (H.C.).
Author Contributions
H.C, J.Y.L, and V.N.K. designed the study. D.K., S.S.Y., and J.W.K. performed molecular and cell biological experiments. H.C. carried out computational analyses. H.C., J.Y.L, and V.N.K. wrote the manuscript.
Competing Interests statements
The authors declare no competing interests.
Accession Numbers
The sequencing data were deposited into the Open Science Framework (OSF) with an accession number doi:10.17605/OSF.IO/8F6N9.
Acknowledgements
We thank members of our laboratories for discussion and help, especially Dr. Junghye Roe, Eun-jin Chang, and Inhye Park.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}