

Abstract
DNA methylation is found throughout all domains of life, yet the extent and function of DNA methylation differ between eukaryotes. Many strains of the plant pathogenic fungus Zymoseptoria tritici appeared to lack cytosine DNA methylation (5mC) because gene amplification followed by Repeat-Induced Point mutation (RIP) resulted in the inactivation of the Ztdim2 DNA methyltransferase gene. 5mC is, however, present in closely related sister species. We demonstrate that inactivation of Ztdim2 occurred recently as some Z. tritici isolates carry a functional Ztdim2 gene. Moreover, we show that Ztdim2 inactivation occurred by a different path than previously hypothesized. We mapped the genome-wide distribution of 5mC in strains with and without functional Ztdim2. Presence of functional Ztdim2 correlated with high levels of 5mC in transposable elements (TEs), suggesting a role in genome defense. We identified low levels of 5mC in strains carrying inactive Ztdim2 alleles, suggesting that 5mC is maintained over time, presumably by an active Ztdnmt5 gene. Integration of a functional Ztdim2 allele in strains with mutated Ztdim2 restored normal 5mC levels, demonstrating de novo cytosine methylation activity of Ztdim2. To assess the importance of 5mC for genome evolution, we performed an evolution experiment, comparing genomes of strains with high levels of 5mC to genomes of strains lacking Ztdim2. We found that the presence of Ztdim2 alters nucleotide composition by promoting C to T transitions (C→T) specifically at CpA (CA) sites during mitosis, likely contributing to TE inactivation. Our results show that dense 5mC at TEs is a polymorphic trait in Z. tritici populations that can impact genome evolution.
Significance Cytosine DNA methylation (5mC) is known to silence transposable elements in fungi and thereby appears to contribute to genome stability. The genomes of plant pathogenic fungi are highly diverse, differing substantially in transposon content and distribution. Here, we show extensive differences of 5mC levels within a single species of an important wheat pathogen that were caused by inactivation of the DNA methyltransferase ZtDim2 in the majority of studied isolates. Presence of widespread 5mC increased point mutation rates in regions with active or mutated transposable elements during mitosis. The mutation pattern is dependent on the presence of ZtDim2 and resembles a mitotic version of Repeat-Induced Point mutation (RIP). Thus, loss of 5mC may represent an evolutionary trade-off offering adaptive potential at the cost of transposon control.
Introduction
DNA methylation is an important process for epigenetic regulation, and functions range from dynamic control of gene expression to transposon silencing and the maintenance of genome integrity (1, 2). Although DNA methylation has been detected on both cytosines and adenines in eukaryotes, cytosine DNA methylation (5mC) has been the focus of most studies so far. In mammals, cytosine methylation is mainly found in “CpGs” (cytosine followed by guanine; here abbreviated CG), while non-CpG methylation (CHG or CHH, where H is any nucleotide other than G) is commonly detected in plants and fungi (3, 4). Various DNA methyltransferases (DNMTs) are involved in the establishment and maintenance of DNA methylation but the distribution and number of enzymes involved is highly variable in different kingdoms (5). In mammals and plants, enzymes of the DNMT1/MET1 class are maintenance methyltransferases that detect hemi-methylated DNA sequences, for example after replication (6–8). De novo methyltransferases, like DNMT3a and DNMT3b in mammals (9) and DRM in plants (10) act on sequences that are free of methylation, presumably by recognition of specific motifs or patterns (9, 11). Although DNMTs are often classified as maintenance or de novo enzymes their function is not necessarily limited to one or the other (12, 13).
In fungi, four classes of DNMTs have been identified. Basidiomycetes have DNMT1 homologs that appear to be classical maintenance DNMTs (4, 14). In the Ascomycete Neurospora crassa, DNA methylation is mediated by a single enzyme, DIM-2 (15). While the conserved DIM2 class of enzymes shows limited sequence similarity to DNMT1/MET1 maintenance DNMTs, it is a class specific to fungi (16). Other fungal proteins resembling DNMT1 include Ascobolus immersus Masc1 and N. crassa RID (17, 18), involved in “Methylation Induced Premeiotically” (MIP) or “Repeat-induced Point mutation” (RIP), respectively (17, 19). DNMT5 enzymes constitute a more recently discovered class of maintenance DNMTs in fungi (20, 21). So far, the presence of 5mC in fungi has been mainly associated with repetitive DNA suggesting a role in genome defense by silencing of transposable elements (TEs). There is little or no evidence that 5mC is also found in coding sequences to influence gene expression (22), although there are examples of promoter methylation (23, 24).
A previous study demonstrated amplification of Ztdim2, a gene encoding the homolog of N. crassa DIM-2, and inactivation by RIP in the genome of the plant pathogenic fungus Zymoseptoria tritici (25, 26). For example, the genome of the reference isolate IPO323 carries 23 complete or partial, nonfunctional copies of Ztdim2; all alleles show signatures of RIP, namely numerous C:G to T:A transition mutations. Consequently, 5mC was not detected by mass spectrometry in IPO323 (25, 26). However, the amplification of Ztdim2 must have occurred recently as two closely related sister species of Z. tritici, Zymoseptoria ardabiliae and Zymoseptoria pseudotritici were shown to carry a single intact dim2 gene and have 5mC (25).
By genome analyses of multiple Z. tritici isolates from the center of origin of the pathogen, the Middle East, we discovered several Z. tritici isolates with an intact Ztdim2 gene. This finding suggests that the loss of 5mC not only occurred very recently but is a polymorphic trait in Z. tritici. Here, we address the evolution and function of dim2 in the Zymoseptoria species complex. We show that the presence of an intact and functional Ztdim2 in some Z. tritici isolates corresponds to widespread 5mC in TEs. Integration of a wild-type, intact Ztdim2 allele into a Ztdim2-deficient background restores methylation of previously non-methylated regions. Using comparative genomics and experimental evolution we show that loss of Ztdim2 and thus 5mC affects nucleotide composition of TEs by influencing mutation rates at CA sites.
Results
The DNA methyltransferase gene Ztdim2 is functional in several Z. tritici isolates
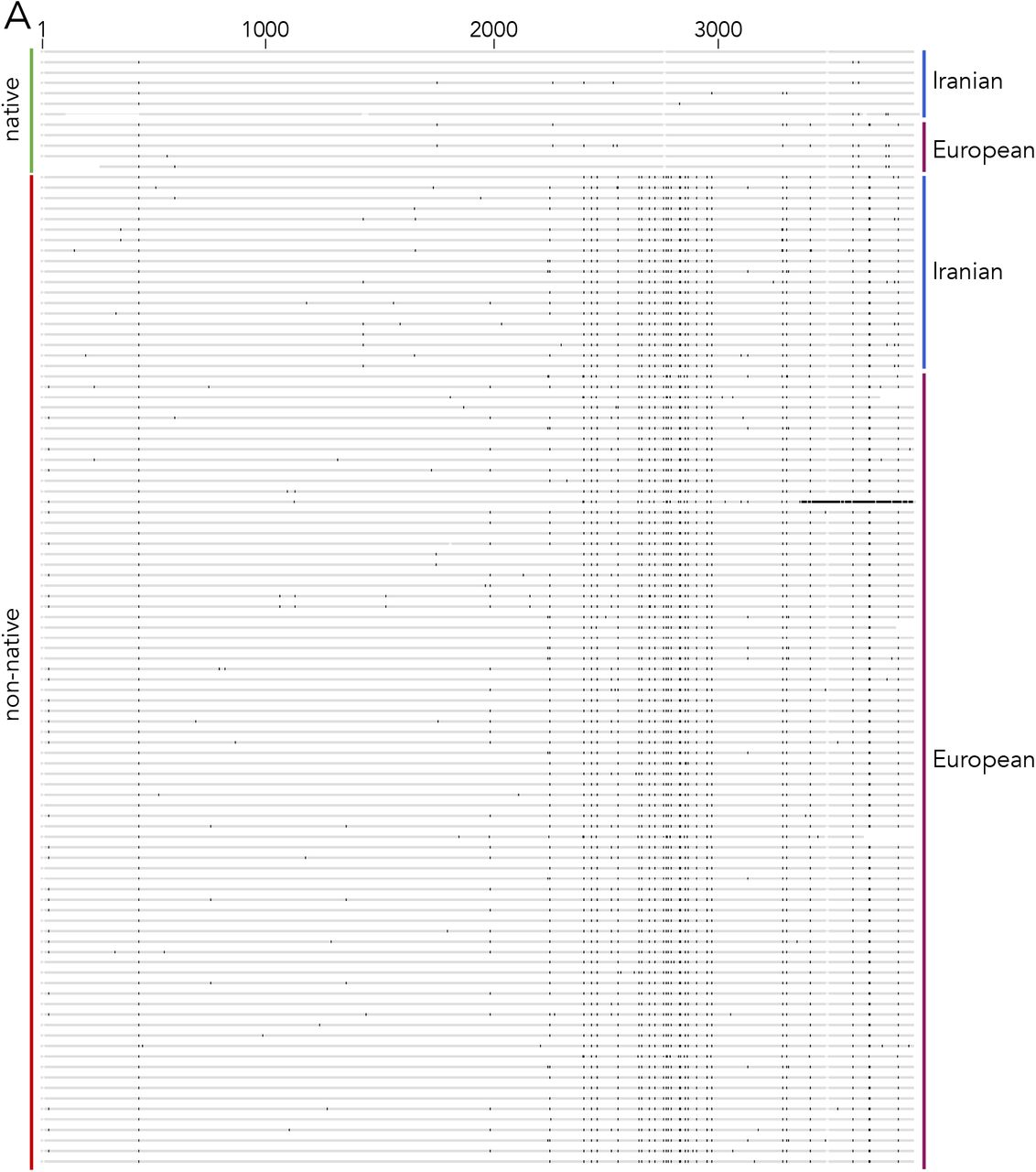
Based on our identification of a non-truncated Ztdim2 gene in an Iranian Z. tritici isolate, Zt10, we set out to investigate recent evolution of this gene in the Zymoseptoria species complex with a broader collection of genome sequences. We used BLAST to search for the sequence of Ztdim2 in 39 genomes of Z. tritici, as well as 17 genomes of Z. ardabiliae, and nine genomes of Z. brevis (Table S1), both closely related sister species of Z. tritici. We detected a single Ztdim2 homolog in each of the Z. ardabiliae and Z. brevis genomes but found multiple mutated and inactivated copies of Ztdim2 in 33 of 39 Z. tritici genomes (Fig. S1A). Six Z. tritici isolates contained an intact, non-mutated copy of Ztdim2 in addition to multiple mutated copies, except for isolate Zt469, which contained no additional copies. Zt469 was isolated from Aegilops sp., all other isolates were collected from wheat. Four of those six isolates are from Iran (Zt10, Zt11, Zt289, Zt469) (27), one is from Algeria (Zt95052, collected from durum wheat) (26), and one is from Australia (ZtWAI329) (28). A non-telomeric region of chromosome 6 of IPO323 had been previously identified as the native Ztdim2 locus; all additional non-functional copies are located in subtelomeric regions with repetitive DNA (25). Consistent with this idea, we found that all non-mutated Ztdim2 genes were located at the predicted native locus on chromosome 6 (Fig. 1A), except for that of the Australian isolate.

A) Transversion and transition mutations of Ztdim2 alleles identified by mapping to the functional allele of Zt469. Shown are six native alleles (located on chromosome 6) and two representative non-native, additional copies per isolate. Native alleles (functional and non-functional) lack transversions in the DNA methyltransferase domain (black box) that are present in all nonnative alleles. *known or putative functional allele. B) Pairwise sequence comparison of the native, non-mutated Ztdim2 allele from Zt469 with all full-length native and additional Ztdim2 sequences (see Table S2). No additional Ztdim2 copies were detected in the genome of Zt469 (from Aegilops sp.) whereas all other isolates (from Triticum aestivum) have additional copies of Ztdim2. Non-native, mutated copies (black dots) in the Iranian isolates showed higher sequence diversity compared to those of European isolates. In the European isolates, alleles at the native locus (yellow triangle) are amongst the more diverged copies, supporting the idea that this is the ancestral, now heavily mutated allele of Ztdim2.
By comparing transitions and transversions of native and additional (non-native) copies we found that all non-native copies (including the potentially functional gene in the Australian isolate) share a specific pattern of transversions in the DNA methyltransferase domain (Figs. 1A and S2). Since transversions are not a consequence of RIP, inactivation of Ztdim2 was likely not a result of amplification of the native Ztdim2 but rather the integration event of a distinct allele that was subsequently amplified. In support of this hypothesis, we find that the flanking regions of the additional, but not native, copies share homology and are annotated as class I LINE TEs (Fig. S3), suggesting that integration and amplification of non-native copies may have been TE-mediated.
We further tested whether presence of a non-mutated Ztdim2 allele is commonly found among Iranian isolates. Twelve additional isolates from Iran (Table S1) were assayed by PCR and Sanger sequencing and in all cases, we confirmed presence of an intact Ztdim2 allele.
To investigate the evolutionary history of Ztdim2 in Z. tritici, we computed the sequence identity of the non-mutated Ztdim2 gene from Zt469, as this likely represents the ancestral allele, to mutated copies of Ztdim2 using high-quality assemblies obtained by SMRT sequencing of nine Z. tritici genomes (Fig. 1B). This comparison revealed two distinct patterns: When a functional Ztdim2 is present in the genome, the additional copies show DNA sequence identities to the Zt469 allele of 84-88%. We note that this extent of sequence identity was shown to be insufficient to induce RIP in N. crassa (29). On the other hand, in isolates without a functional Ztdim2, we found overall higher levels of DNA sequence identities (85-95%) between mutated Ztdim2 copies and the functional allele from Zt469. This observation suggests an ongoing transposon- or recombination-mediated amplification of Ztdim2 pseudogenes in the genomes (Fig. 1B, Table S2). The high transition ratio in these copies may indicate that sequence amplification is followed by RIP. The two Iranian strains (Zt10 and Zt289) with functional copies at the native locus but with additional non-functional copies with sequence identities below 90% caught our attention. This is an unexpected result because when starting with identical or near-identical alleles, even after only a few sexual cycles all Ztdim2 copies should be similarly mutated. Finding alleles that are not mutated together with many alleles carrying mutations suggests either increased RIP frequency of Ztdim2 repeats by some unknown mechanism, or acquisition of functional or repeat copies in Zt10 and Zt289 via recombination. Based on the occurrence of numerous transversions in all non-native copies we favor the latter possibility. The non-mutated Ztdim2 allele in Zt289 carries a 327 bp in-frame deletion within the 5’ region, mutated copies of Zt289 lack this deletion, indicating that deletion occurred after amplification, or that this allele or the mutated copies were acquired by introgression from other isolates.
Integration of an intact Ztdim2 at the native locus restored 5mC in Zt09
To assess the role of Ztdim2 in DNA methylation we generated mutants in which we either deleted or restored Ztdim2 activity. We integrated the functional Ztdim2 gene of the Iranian Zt10 isolate into the native Ztdim2 locus of Zt09 (derived from IPO323) by selecting for hygromycin resistance, conferred by the hph gene, generating strain Zt09::Ztdim2. In Zt10, we replaced the functional Ztdim2 gene with hph, generating strain Zt10ΔZtdim2. Integration or deletion of the Ztdim2 alleles were verified by Southern analyses (Fig. S4). To test whether Ztdim2 affects growth or infection phenotypes, we compared the mutant and respective wildtype strains under different in vitro conditions as well as during host infection. We did not detect any differences between wild-type and mutant strains in vitro (Fig. S5) but integration of Ztdim2 in Zt09 significantly reduced necrosis and in planta asexual fruiting body formation (measured as number of asexual fruiting bodies, pycnidia) (Fig. S6). We therefore conclude that presence of Ztdim2 is dispensable under the tested conditions in vitro but that re-integration of Ztdim2 into the Zt09 background decreases virulence.
To validate the presence of 5mC and to compare patterns between isolates and mutants, we performed whole-genome bisulfite sequencing (WGBS). We sequenced three replicates of each wild-type isolate (Zt09 and Zt10) and mutant strain (Zt09::Ztdim2 and Zt10ΔZtdim2). As a control for the bisulfite conversion rate, we added Lambda DNA to each sample; based on this, the bisulfite conversion rate was >99.5%. To confirm the WGBS sequencing results, we performed an additional independent bisulfite conversion, amplified target regions by PCR, cloned and sequenced the fragments, and we performed Southern blots using 5mC-sensitive and -insensitive restriction enzymes (Fig. S7).
We detected high numbers of methylated cytosines in the isolates with a functional Ztdim2 gene, i.e., the wild-type Iranian isolate Zt10 and Zt09::Ztdim2, complemented with the wildtype Ztdim2 allele. Surprisingly, we also detected low levels of 5mC in the absence of Ztdim2, i.e., in Zt10ΔZtdim2 and Zt09, which had been missed by the mass spectrometry in earlier studies (25). While on average ~300,000 sites in Zt10 and ~400,000 sites in Zt09::Ztdim2 showed 5mC in the presence of Ztdim2, only ~12,000 (Zt09) and ~40,000 (Zt10ΔZtdim2) 5mC sites were found when Ztdim2 was absent (Fig. 2A). In all strains, 5mC was restricted to previously annotated TEs and adjacent non-coding regions (Fig. 2A). We did not find evidence for gene body or promoter methylation, except for genes that (partially) overlap with TEs. By correlating the 5mC data with previously published histone methylation maps (30) we found that 5mC co-localizes with H3K9me3 marks (Fig. 2B). This finding is consistent with observations in N. crassa and A. thaliana (31–33).

A) Number of 5mC methylated sites detected by whole genome bisulfite sequencing (WGBS) in TEs, intergenic regions, or genes. The vast majority of methylated sites is localized in TEs followed by intergenic regions and genes, whereby 5mC sites detected in genes overlap with TEs. B) 5mC co-localizes with H3K9me3 and TEs in Zt09. Shown are ChIP-seq tracks for H3K4me2, H3K27me3 and H3K9me3 (30), and 5mC sites detected in a representative region on chromosome 9 in the genomes of Zt09 and Zt09::Ztdim2 (three replicates). C) Genome-wide 5mC levels in the different isolates and respective mutants. 5mC levels are higher in the presence of Ztdim2, and Ztdim2 is required for all non-CG methylation. D) 5mC levels and site preferences in TEs. CG sites are preferred, followed by CA, and CC. 5mC is almost completely absent from CTs.
In strains without a functional Ztdim2, we found 5mC exclusively in CG contexts (genome wide ~0.2% in Zt09 and ~1% in Zt10ΔZtdim2; percentage based on all Cs in the genome) In presence of Ztdim2, cytosines in CG as well as CHG and CHH contexts were methylated. Genome-wide, ~5 or 6% of CGs and <1% of CHGs or CHHs were methylated in Zt10 or Zt09::Ztdim2, respectively (Fig. 2C). In sequences annotated as TEs, however, up to 60% of all CGs were methylated (Fig. 2D). Methylation of CGs occurred most frequently (~43%, ~61%), but we also detected methylation at CAs (~10%, ~22%), and to lesser extent at CCs (~3%, ~8%). At CTs, there was hardly any detectable methylation (<0.01%; Fig. 2D, Fig. S8) in Zt10 or Zt09::Ztdim2, respectively. This was surprising, as CTs are the most abundant CH dinucleotides in TEs while CAs are the least abundant sites in both Zt09 and Zt10 genomes (Table S3). This suggests a strong site-specific preference of ZtDim2 for CAs but not CTs in Z. tritici.
Upon integrating of a wild-type Ztdim2 allele in Zt09 we found that overall 5mC levels are higher when compared to Zt10 (compare Fig. 2C, left- and right-most panels), and CG methylation levels in Zt10ΔZtdim2 were higher compared to Zt09 (compare Fig. 2C, two middle panels). These findings suggest that 5mC levels can decrease over relatively short evolutionary time spans, e.g. the Ztdim2 gene amplification and RIP events in Zt09.
DNA methylation can be maintained in absence of Ztdim2
We next asked how 5mC can persist in the genome of Z. tritici in the absence of a functional Ztdim2 gene. The presence of CG methylation in isolates without Ztdim2 suggested the presence of an additional DNMT. Thus, we searched for putative DNMT coding regions in the genomes of Z. tritici, Z. ardabiliae, and Z. brevis with the conserved DNMT domain of ZtDim2 as query in a BLASTp search. In all genomes, we found two additional predicted DNMTs (Fig. S9). One is similar to Dnmt5 of Cryptococcus neoformans (20) (Ztdnmt5, Zt_chr6_00685), the other is a homolog of N. crassa RID (17) (Ztrid, Zt_chr5_00047; manually corrected gene annotation) (34). Genes for Dnmt5 and Rid are present in all Zymoseptoria spp. genomes we analyzed. The dnmt5 alleles are highly conserved and show very little inter- or intraspecies diversity (Fig. S1B). In contrast, rid shows an exceptionally high inter- and intraspecies diversity with three highly distinct alleles present in Zymoseptoria spp. (Fig. S1C). This diversity may stem from introgression events between Zymoseptoria species as recently described for highly variable regions in the genome of Z. tritici (35). However, the different Ztrid alleles do not correlate with the presence of a functional Ztdim2 gene in Z. tritici.
Masc1 and RID are involved in genome defense during pre-meiosis (17, 18). Although Masc1 is presumed to be a de novo DNMT acting during the sexual cycle of Ascobolus there are still no enzyme activity data available for either Masc1 or RID. In contrast, Dnmt5 has previously been characterized as a symmetrical CG DNMT in C. neoformans (20, 21) and is thus the most likely candidate for maintenance of 5mC at CG sites in Z. tritici that we observed here. Further studies should address the role of Ztdnmt5 in maintenance and impact on 5mC in Z. tritici.
Presence of Ztdim2 impacts nucleotide composition and activity of TEs
DNA cytosine methylation has been shown to be involved in increased C→T mutation rates (36, 37). In Z. tritici, and most other fungi (22), 5mC is enriched in TE sequences. To evaluate whether the presence of Ztdim2 impacts sequence composition of TEs, we analyzed TE sequences in isolates with (Iranian isolates) and without (European isolates) a functional Ztdim2 gene. Therefore, we extracted TE sequences, calculated dinucleotide frequencies relative to the TE content for each genome (Table S3), and visualized the difference in frequencies by correspondence analysis. Isolates lacking functional Ztdim2 have more TG/CA sites within TEs compared to isolates with functional Ztdim2 (Fig. 3 A, B). Conversely, isolates with functional Ztdim2 contain more TA sites in TEs (Fig. 3A). CA/TG sites are the least frequent sites in TEs, while TA sites are the most abundant. We observed the same pattern (i.e., increased number of TA sites), when we included the sister species Z. ardabiliae (Za17) and Z. brevis (Zb87), and the more distantly related barley pathogen Z. passerinii (Zpa63) that all contain a single functional dim2 (Fig. 3B).

A) and B) Correspondence analysis showing differences in dinucleotide (2mer) frequencies in TEs of different Z. tritici isolates (A) and other members of the Zymoseptoria species complex (B). TA and TG/CA dinucleotide frequencies have the highest impact on the observed differences between isolates and species, indicated by their relative positions on the dimension 1 versus dimension 2 plot. Isolates (even from different species) containing an intact dim2 (Zt10, Zt289, Zt469, Zb87, Za17, Zpa63) have higher TA content and cluster together, while isolates without active dim2 have higher TG/CA frequencies in TE sequences. C) Comparison of TE and GC content between different Z. tritici isolates and species with and without functional dim2. D) Number of expressed TEs during the time course of wheat infection is significantly higher in Z. tritici isolate Zt09 (inactive Ztdim2) compared to Zt10 (active Ztdim2) (Fisher’s Exact Test for Count Data, p-value <2.2 x 10−16).
Accelerated mutation rates in TEs may impact TE activity, so we next determined the TE content of the various Z. tritici isolates and compared it to that observed in other Zymoseptoria species (Fig. 3C). We tested genomes of three isolates with a functional Ztdim2 and six isolates without a functional Ztdim2 (Fig. 1) Isolates with functional Ztdim2 had slightly lower TE content than those lacking Ztdim2, average ~15.6% versus ~17.2% (Fig. 3C) but TE content in other Zymoseptoria species, all with functional dim2, was higher than in many Z. tritici strains. Thus, there is no clear and simple correlation between TE content and de novo 5mC activity. GC content of TEs, however, is considerably lower in isolates with a functional dim2 further indicating that Dim2 impacts nucleotide composition (Fig. 3C).
The TE content of a genome does not necessarily reflect the activity of TEs, as these sequences may be relics, i.e., mutated and thus inactivated TEs. We therefore assessed whether TEs in Zt09 and Zt10 contained annotated, transposon related genes as an indicator for the presence of active TEs. In Zt09, 56 genes completely overlap (>90% of the sequence) with annotated TEs. Among those, we identified 30 transposon- or virus-related genes encoding for endonucleases, transposases, ribonucleases, and reverse transcriptases, or genes encoding virus-related domains (Table S4). In contrast, we did not detect any fungal genes overlapping TEs in Zt10. We did find homologs of some of the transposon-associated genes of Zt09 in the genome of Zt10, but they all contained numerous transitions resulting in mis- and nonsense mutations.
Lastly, we compared the expression of TEs in Zt09 and Zt10 during the time course of infection by analyzing previously published RNA-seq data (38). We computed transcripts per million (TPM) originating from TEs in both the biotrophic and necrotrophic phases of infection and considered a TE expressed when TPM was >0. While ~21% of TEs produced RNA in Zt10, ~35% of TEs were transcribed in Zt09 (Fig. 3D) indicating that silencing of TE loci is less pronounced in Zt09 where Ztdim2 is inactive.
ZtDim2 promotes C→T transitions at CAs during mitosis
The difference between CA/TG and TA site abundance in genomes with and without functional Ztdim2 suggests that the mutation rate, specifically for C→T transitions, is affected by the presence of 5mC. To address whether ZtDim2 plays a role in promoting these mutations, we conducted a one-year evolution experiment that included the Iranian isolate Zt10 (functional Ztdim2) and the reference isolate IPO323 (non-functional Ztdim2). This was a mutation accumulation experiment without competition or selection on cells that were dividing by mitosis only. Each ancestral isolate was propagated as 40 independent replicates. After 52 weeks (corresponding to ~1,000 mitotic cell divisions) the genomes of the 40 evolved replicate strains per isolate and the progenitor isolates were sequenced to map SNPs. We found that the mutation rate in Zt10 was more than tenfold higher than in IPO323, on average ~170 versus ~13 mutations per replicate (Fig. 4A, Table S5). The vast majority of mutations in Zt10 (>95%) occurred in 5mC regions (Table S6) and >95% of all mutations were C→T transitions, while only ~32% of all mutations in IPO323 were C→T transitions. In Zt10, >98% of the C→T transitions occurred at CAs (versus ~ 21% in IPO323) (Fig. 4B). This was a surprise, because the majority of methylated sites are at CGs. Our findings suggest the presence of a Ztdim2-dependent mechanism that specifically targets and mutates CA/TG but not CGs in sequence repeats during mitosis. Such a mutator resembles the hitherto undescribed but much looked-for mitotic version of RIP.

A) Detected number of SNPs in the 40 replicates of each isolate. The number of SNPs in Zt10 isolates is significantly higher compared to IPO323 (Wilcoxon rank sum test, ***p-value <0.001). The vast majority of mutations are C to T transitions. B) Sequence context of the mutations in the two tested strains. The SNPs of all replicates were pooled for this analysis. C→T transitions are predominantly found in a CA context resulting in CA to TA (or GT to TA) mutations. CG sites, although preferentially methylated, are not mutated at high rates.
Discussion
Our study of the DNA methyltransferase dim2 in the fungus Z. tritici reveals an exceptional extent of polymorphism that has functional and genetic implications. The majority of Z. tritici isolates analyzed here lack a functional Ztdim2 gene, whereas dim2 was present in genomes of the sister species, Z. brevis and Z. ardabiliae. This confirms that the amplification and inactivation of Ztdim2 occurred recently and after speciation of Z. tritici (<12,000 years ago (27)). Although it has been reported that 5mC is absent in Z. tritici(25), we found evidence that the loss of most 5mC is recent, and that some isolates contain an intact and functional ZtDim2 de-novo methyltransferase. Furthermore, we find evidence that 5mC is not completely absent in Z. tritici even when there is no functional ZtDim2. The majority of isolates in which we identified non-mutated Ztdim2 genes originated in Iran, previously shown to be the center of origin of Z. tritici (27). The closely related sister species Z. ardabiliae and Z. brevis were collected in Iran as well and are so far considered to be endemic to this region (39, 40). We found only one Z. tritici isolate (Zt469) that did not carry any additional Ztdim2 copy. This isolate was collected from an Aegilops sp., also in Iran. We propose that the original state of the native dim2 locus has been maintained in the two sister species, and Z. tritici isolates collected from wild grasses. This is borne out by DNA sequence comparisons, that show that the fewest SNPs that could be transition mutations are found in Zt469. Even functional copies from two Iranian isolates, Zt10 and Zt289, show potential RIP-induced mutations or a large deletion, respectively. Thus, all available data suggest that Ztdim2 gene amplification and RIP has been specific to wheat-infecting Z. tritici isolates.
We also disproved a previous hypothesis suggesting that the mutated Ztdim2 copies in IPO323 and similar strains arose by gene duplications of the native Ztdim2 followed by transposon-mediated movement to subtelomeric regions (25). Instead, DNA sequences of additional Ztdim2 copies show patterns of increased transversion mutations in the DNMT catalytic domain (“transversion alleles”) that would not be expected as consequence of RIP, and that are a clear indication of introgression from populations with different ancestral dim2 alleles. A high extent of inter-specific hybridization among Zymoseptoria species was demonstrated by population genomic analyses suggesting that introgression can be a source of new genetic variation (35). Our data suggest recombination between isolates with Ztdim2 non-native transversion alleles, which may have induced RIP in the native Ztdim2 allele because of high sequence similarity outside of the DNMT catalytic domain, thus resulting in non-functional Ztdim2 alleles only, and therefore absence of de novo DNMT activity. Further analyses of diverse Z. tritici isolates from different hosts and geographical locations (T. aestivum, Triticum durum, or Aegilops sp.) are needed to track the evolutionary history of the dim2 gene in the Zymoseptoria species complex.
The previous study on Ztdim2 suggested absence of 5mC based on liquid chromatography coupled with electrospray ionization tandem mass spectrometry (ESI–MS/MS) (25). This finding is consistent with the low levels of methylation we discovered in strains lacking ZtDim2 activity. Based on recent results obtained in studies with Cryptococcus we hypothesize that this low level of 5mC has been maintained by the single homologue of Dnmt5 (20, 21) after the loss of Dim2. This DNMT acts as a maintenance DNMT but does not catalyze de novo DNA methylation (21). In the absence of a de novo DNMT, DNA methylation is likely to decrease over time as any loss of 5mC cannot be restored. This idea is supported by our finding that 5mC levels are higher upon deletion of Ztdim2 (Zt10ΔZtdim2) compared to Zt09, where Ztdim2 was likely inactivated thousands of generations ago.
Sequence repeats are the predominant targets of DNA methylation in fungi (15, 22, 41, 42), and 5mC enrichment is correlated with genome defense mechanisms that act to silence transposons (1). We found that nucleotide composition in TEs differs between isolates and species that have active or inactive dim2 alleles. CA sites, the preferred target sites of ZtDim2 in addition to CGs, are reduced in frequency when Dim2 is present. TA sites, however, are more frequent when Dim2 is present. This links increased C→T transition frequencies to the presence of a functional Dim2. Spontaneous deamination of 5mC yields C→T transitions (37), giving rise to accelerated mutation rates of 5mC sites, a well-known phenomenon (43). Correlation of decreased C:G to T:G transitions in absence of the DNA methyltransferase CMT3 was recently been shown in plants (44).
The observed differences in dinucleotide frequencies in Zymoseptoria and the high rate of C→T transitions in the evolution experiment suggest a role of Dim2 and 5mC in the generation of mutations. Mutations specifically affected CAs but not CGs. If mutations resulted from spontaneous deamination, we would expect a greater effect on CGs because they have higher 5mC levels. Lack of Dim2 resulted in complete loss of 5mC at CAs, however, and therefore reduced CA to TA mutation frequencies. We also did not observe a noticeable difference of CG site abundance between isolates with or without functional Dim2, suggesting that non-CG 5mC (mediated by Dim2) is the main driver of the C→T transitions we observed.
The best-known process causing C→T transitions in fungi is RIP (45, 46). DIM-2 has been suggested to be involved in RIP in N. crassa (19), and thus we propose that the absence of ZtDim2 may play a role in the efficiency and rate of RIP in Z. tritici, although mechanistic studies are still lacking because crosses with genetically-modified Z. tritici are still cumbersome. In N. crassa, RIP prefers CAs over any other context (19, 46, 47), while N. crassa DIM-2 preferentially methylates CTs (22). This suggest that, although repetitive regions are a shared target for DNMTs in different fungal species, distinct target sites of 5mC may correlate with the sequence composition of repetitive regions and the presence of other proteins involved in the 5mC pathway.
The Ztdim2-dependent mutations described here resemble a mitotic version of RIP. In several fungal species, 5mC is completely absent or methylation levels are very low (22, 48–50) raising the question about the importance of 5mC in terms of genome defense and evolution. While silencing or inactivation of TEs protects genome integrity and stability, it may limit the adaptive potential in response to changing environmental conditions. Transposon- mediated genome diversity, gene or even chromosome copy number variations are frequently found in fungal pathogens, and they form a crucial aspect of adaptability and virulence (51–54). The efficiency and abundance of DNA methylation and mutations may therefore represent an evolutionary trade-off between genome integrity and adaptive potential.
Materials and Methods
Fungal isolates and growth conditions
All Zymoseptoria spp. isolates used in this study (Table S1) were cultivated at 18°C in YMS (4 g yeast extract, 4 g malt, 4 g sucrose per 1 L, 20 g agar per L for plates) medium. Cultures for DNA extraction and plant infection experiments were inoculated directly from the −80°C glycerol stocks and grown in liquid YMS medium at 200 rpm for 5 days (DNA extractions) and in pre-cultures (3 days) and main cultures (2 days) for plant infections.
SMRT sequencing and assembly of Iranian isolates Zt289 and Zt469
High molecular weight DNA was extracted as previously described (55). Library preparation and PacBio sequencing was performed at the Max Planck Genome Center in Cologne, Germany (https://mpgc.mpipz.mpg.de/home/) with a Pacific Biosciences Sequel II. One SMRT cell was sequenced per genome. Genome assemblies were performed as described (56). Genome assembly statistics are summarized in Table S7.
Identification of TEs and analysis of TE expression
We annotated transposable elements with the REPET pipeline (https://urgi.versailles.inra.fr/Tools/REPET) (57) as described in (56). Briefly, we identified repetitive element consensus sequences in each genome using TEdenovo following the developer’s recommendations (57). We used each library of consensus sequences to annotate genomes using TEannot with default parameters. To evaluate TE activity we analyzed expression in planta using RNA-seq data of Z. tritici isolates Zt10 and Zt09 (38). First, raw sequencing reads were quality filtered and trimmed using Trimmomatic (58) and mapped to the respective genome using hisat2 (59). Reads were trimmed using the following parameters: LEADING:30 SLIDINGWINDOW:4:30 AVGQUAL:30 MINLEN:50. After mapping, the resulting aligned read files were used to assess read counts for each stage and replicate (with a total of two replicates per stage). The read count table was generated using TEcount function of the TEtranscript pipeline with the following parameter: -mode multi (60). Briefly, TEtranscript pipeline counts both uniquely and multi-mapped reads mapped to annotated transposons and genes to attribute transcript abundance. A given TE was considered transcribed only if reads mapped all along the element length, in order to avoid redundancy (60). Levels of expression were calculated using Transcript per Million (TPM) normalization which corresponds to the normalization of read counts with transposon (or gene) length per million. We considered a TE as ‘expressed’ if TPM > 0.
Sequence identification and comparison of DNA methyltransferases
All analyzed genomes and isolates used in this study are listed in Table S1 and originate from the following studies (27, 28, 35, 38–40, 56, 61–70). We identified homologs of Ztdim2 using the predicted ‘deRIPed’ protein sequence of Z. tritici IPO323 (25) as a template. To compare the sequence identity between active and inactive Ztdim2 copies in the genome we used Ztdim2 of isolate Zt469 as a reference and performed pairwise comparisons. To identify additional putative DNA methyltransferases, we used the DNA methyltransferase domain of the ZtDim2 protein as query. BLAST searches, phylogenetic trees and alignments were performed using Geneious ‘Blast’, Geneious ‘Alignment’ and Geneious ‘Map to reference’ (Geneious version 10.2.4 (http://www.geneious.com, (71)). A distance tree based on the nucleotide sequence of dim2, rid and dnmt5 was generated with the following settings: alignment type: global alignment with free end gaps, cost matrix: 65 % similarity, genetic distance: Jukes-Cantor, tree build method: neighbor joining, outgroup: Zymoseptoria passerinii (Zpa63).
Generation of Ztdim2 deletion and integration strains
We transformed the Z. tritici isolates Zt09 and Zt10 using an Agrobacterium tumefaciens-mediated transformation (ATMT) protocol as previously described (72). Briefly, we created plasmids containing the integration (pES189) or deletion (pES188) constructs using Gibson assembly (73) (Table S8). Both constructs contained the hygromycin resistance cassette as selection marker. Plasmids were amplified in E. coli TOP10 cells and sequenced to confirm correct assembly of the constructs followed by electroporation of A. tumefaciens strain AGL1. Z. tritici strains were transformed with the respective A. tumefaciens strains by co-incubation for 3-4 days at 18°C on induction medium. Following co-incubating, the strains were grown on selection medium containing hygromycin to select for integration of the constructs and cefotaxime to eliminate A. tumefaciens. Single Z. tritici colonies were selected, streaked out twice and the correct integration of the construct was verified by PCR and by Southern blot analyses (Fig. S4).
Bisulfite treatment and sequencing
We extracted high molecular weight DNA of three biological replicates of Zt10 and Zt09 and three independent transformants of Zt10ΔZtdim2 and Zt09::Ztdim2 as described previously (55). Genomic DNA was sent to the Max Planck Genome Centre in Cologne, Germany (https://mpgc.mpipz.mpg.de/home/) for bisulfite treatment and sequencing. Lambda DNA was used as spike-in (~1%) to determine conversion efficiency. Genomic DNA was fragmented with a COVARIS S2 and an Illumina-compatible library was prepared with the NEXTflex Bisulfite Library Prep Kit for Illumina Sequencing (Bioo Scientific/PerkinElmer, Austin, Texas, U.S.A). Bisulfite conversion was performed using the EZ DNA Methylation Gold Kit (Zymo Research, Irvine, CA). Illumina sequencing was performed on a HiSeq3000 machine with paired-end 150-nt read mode (Table S9).
To confirm bisulfite sequencing results, we treated genomic DNA of the fungal strains and, as a control for conversion efficiency, the Universal Methylated DNA Standard (Zymo Research, Irvine, CA) (~100 ng) with bisulfite using the EZ DNA Methylation-Lightning Kit (Zymo Research, Irvine, CA) according to manufacturer’s instructions. Two representative loci per fungal strain and the human MLH1 for the control were amplified by PCR (ZymoTaq PreMix, Zymo Research, Irvine, CA) using specifically designed primers for amplification of the bisulfite-treated DNA (designed using the ‘Bisulfite Primer Seeker’ (Zymo Research, Irvine, CA), Table S8). PCR products were cloned using the TOPO TA kit (Thermo Fisher Scientific) and sequenced (Eurofins Genomics, Ebersberg, Germany). Analysis of sequenced bisulfite-converted DNA sequences was performed using Geneious software version 10.2.4 (71).
Data analysis of whole genome bisulfite sequencing data
A list of software and input commands used in our analyses is provided in the S1 text.
Reads were quality filtered using Trimmomatic (58) and subsequently mapped using Bismark (74). Duplicate reads were removed. Methylated sites were extracted using the bismark_methylation_extractor applying the –no_overlap and CX options. We considered sites methylated, if at least 4 reads and ≥ 50 % of reads supported methylation. We then extracted these sites (see text S1) in CG, CHG or CHH contexts for further analysis. Bedtools (75) was used to correlate methylation and genomics features.
Southern blots to detect DNA methylation and confirm Ztdim2 mutant strains
To detect presence or absence of 5mC, we performed Southern blots according to standard protocols (76). Genomic DNA was extracted using a standard phenol-chloroform extraction method (77). The same amount of DNA and enzymes (BfuCI, DpnI, DpnII; 25 units, New England Biolabs, Frankfurt, Germany) was used as input for the different restriction digestions to make restriction patterns comparable between enzymes and Z. tritici strains for the detection of DNA methylation. Probes were generated with the PCR DIG labeling Mix (Roche, Mannheim, Germany) following the manufacturer’s instructions and chemiluminescent signals detected using the GelDocTM XR+ system (Bio-Rad, Munich, Germany).
Phenotypic assay in vitro
Spores were diluted in water (107 cells/mL and tenfold dilution series to 1,000 cells/mL) and three μL of the spore suspension dilutions were pipetted on plates and incubated for ten days. To test for responses to different stress conditions in vitro, YMS plates containing NaCl (0.5 M and 1 M), sorbitol (1 M and 1.5 M), Congo Red (300 μg/mL and 500 μg/mL), H2O2 (1.5 mM and 2 mM), methyl methanesulfonate (0.01% and 0.005%) and two plates containing only YMS were prepared. All plates were incubated at 18°C, except for one of the YMS plates that was incubated at 28°C.
Phenotypic assay on wheat
Seedlings of the wheat cultivar Obelisk (Wiersum Plantbreeding BV, Winschoten, The Netherlands) were pre-germinated on wet sterile Whatman paper for four days under normal growth conditions (16 h at light intensity of ~200 μmol/m−2s−1 and 8 h darkness in growth chambers at 20°C with 90% humidity) followed by potting and further growth for additional seven days. Marked areas on the second leaves (30 leaves per strain) were inoculated with a spore suspension of 107 cells/mL in H2O and 0.1 % Tween 20. Mock controls were treated with H2O and 0.1 % Tween 20 only. 23 days post inoculation treated leaves were analyzed for infection symptoms in form of necrosis and pycnidia. Evaluation was performed by assigning categories for necrosis and pycnidia coverage to each leaf (categories: 0 = 0%, 1 = 1 −20%, 2 = 21 −40%, 3 = 41 −60%, 4 = 61 −80%, 5 = 81-100%).
Analysis of dinucleotide frequencies
TE sequences were extracted from the genome using bedtools getfasta (75). k-mer frequencies in annotated TEs and masked genomes were determined using the software jellyfish (78). Correspondence analyses were carried out and visualized in R (79) using the packages “FactoMineR” and “factoextra” (80).
Mutation accumulation experiment and SNP analysis
A single colony derived directly from a plated dilution of frozen stock for Zt10 or IPO323 was resuspended in 1 mL YMS including 25% glycerol by 2 min vortexing on a VXR basic Vibrax at 2000 rpm, and 10–50 μL were re-plated onto a YMS agar plate. Forty replicates were produced. Cells were grown for 7 days at 18°C until a random colony (based on vicinity to a prefixed position on the plate) derived from a single cell was picked and transferred to a new plate as described above. The transfers were conducted for one year (52 times) before the DNA of a randomly chosen colony of each replicate was extracted and sequenced. Sequencing and library preparation were performed at the Max Planck Genome Center in Cologne, Germany (https://mpgc.mpipz.mpg.de/home/). Sequencing was performed on an Illumina HiSeq2500 machine obtaining paired-end 250-nt reads (Table S9). Paired-end reads were quality filtered (Trimmomatic), mapped (bowtie2) and SNPs were called using samtools mpileup (see text S1).
Data availability
Sequencing raw reads (FASTQ files) of all bisulfite and genomic data and the SMRT genome assemblies are available online at Sequence Read Archive (SRA) under BioProject ID PRJNA614493.
Supplementary Information
Supplementary Tables
Table S1. List of Z. tritici, Z. ardabiliae, Z. brevis and Z. passerinii isolates used in this study. Listed are collection date, origin, information about the genome assembly and presence/absence of dim2 in each isolate.
Table S2. Detected copies and pairwise identity of full-length (>3,400 bp) Ztdim2 copies in Z. tritici genomes sequenced by PacBio. The Ztdim2 gene of the Iranian isolate Zt469 was used as query for the blast search.
Table S3. Dinucleotide frequencies in transposable elements (TEs) and TE masked genomes in Z. tritici isolates and the sister species Z. brevis, Z. ardabiliae and Z. passerinii.
Table S4. Genes overlapping TEs and their functional annotation in Zt09.
Table S5. SNPs detected in Zt10 and IPO323 after 52 weeks of experimental evolution compared to the reference strain at the start of the experiment.
Table S6. Genome-wide occurrence of 5mC and SNPs in Zt10. Listed are GC and AT content, number of 5mC sites (pooled data from WGBS of all three replicates) and number of SNPs (pooled data from all 40 replicates) in 500 bp windows.
Table S7. PacBio genome assembly metrics of Iranian isolates Zt289 and Zt469.
Table S8. List of all oligos and plasmids used in this study.
Table S9. Overview of Illumina sequencing data, detected 5mC sites, and SNPs.
Supplementary text S1. Software and commands used for bisulfite and genome data analysis to detect 5mC sites and SNPs.
Supplementary Figures



Phylogenetic trees of DNA sequences encoding the dim2, dnmt5, and rid gene. A) Phylogenetic tree based on mapping of dim2 alleles to the native, functional gene of Zt469. The Z. tritici dim2 is distinct from the Z. brevis and Z. ardabiliae gene. The native, nonfunctional copies form a distinct cluster within the non-functional copies. B) Dnmt5 is present in all analyzed genomes and shows relatively little inter- or intraspecies diversity. C) The rid gene shows an exceptionally high inter- and intraspecies diversity with three highly distinct alleles present among genomes of Z. tritici, Z. ardabiliae and Z. brevis. Green background indicates Z. tritici, blue Z. ardabiliae, yellow Z. brevis. Z. passerinii is used as an outgroup. Red asterisks mark Z. tritici isolates with an intact Ztdim2.

Transversions (A) and transitions (B) in full-length Ztdim2 alleles compared to the functional allele of isolate Zt469. In addition to the isolates shown in Fig. 1, we included additional native and functional Ztdim2 alleles of Iranian isolates Zt467, Zt468, Zt471 and Zt11 (see Table S1). All native copies (functional and non-functional) lack transversions in the DNA methyltransferase domain (position ~2,300–3,500) that are present in all non-native copies suggesting that the additional copies did not emerge from amplification of the native Ztdim2. Black lines indicate differences to the reference.

Alignments of 2 kb 5’ (A) and 3’ (B) flanking regions of Ztdim2 alleles. The vast majority of flanking regions of non-native alleles (89 of 91 5’ flanks, 82 of 91 3’ flanks) are similar and annotated as RIX (LINE) class I transposons. None of the native copies shares the non-native flanking regions. In five cases, the 3’ flanks are very close to the chromosome end and contain the telomere repeats. Black lines indicate differences to the reference.

Southern blots to confirm the correct integration of A) the deletion construct for Ztdim2 in Zt10 (Zt10ΔZtdim2) and B) Ztdim2 (originating from Zt10) in Zt09 (Zt09::Ztdim2). Three positive transformants (#49, #63 and #84) were found amongst the Zt09::Ztdim2 candidates, whereas all seven candidates for Zt10ΔZtdim2 were verified (correct transformants are highlighted in bold).

Phenotypic characterization of Zt10, Zt10ΔZtdim2, Zt09 and Zt09::Ztdim2 in vitro. We compared growth phenotypes under different in vitro conditions including temperature, osmotic, oxidative, genotoxic and cell wall stress. We spotted spore dilutions of each reference isolate and three independent Ztdim2 mutant transformants on each plate. We did not detect any noticeable differences in growth between reference and mutant strains but differences between the different Z. tritici isolates Zt09 and Zt10 that were previously described (38).

Results of wheat infection experiments with Z. tritici isolates Zt09, Zt10 and the mutants Zt09::Ztdim2 and Zt10ΔZtdim2 on wheat. A) Zt09::Ztdim2 strains show significantly less necrotic lesions compared to Zt09 (*** Wilcoxon rank-sum test, p-value = 2.178 x 10−7) while there is no significant difference in the quantities of necrotic lesions caused by Zt10 and Zt10ΔZtdim2 strains. B) Coverage with pycnidia is significantly reduced between Zt09 and Zt09::Ztdim2 (** p-value = 0.003301) but not between Zt10 and Zt10ΔZtdim2 strains. Categories for necrotic lesion and pycnidia coverage: 0 = 0%, 1 = 1-20%, 2 = 21-40%, 3 = 41-60%, 4 = 61-80%, 5 = 81-100%.

Detection of cytosine methylation by enzymatic digestion and Southern blot and confirmation of bisulfite sequencing results by PCR and Sanger sequencing. Restriction enzyme analysis followed by Southern blots using the rDNA spacer (A) or a retrotransposon RIL2 (B) as probe. In addition to the cytosine methylation sensitive (BfuCI, +) or insensitive (DpnII, -) enzymes, we used DpnI (*) to test for the presence of adenine methylation. Zt09 and Zt150 contain an inactivated Ztdim2, whereas the Iranian strains Zt10, Zt11 and the Z. ardabiliae strain Za17 have an intact copy. In all strains, where Ztdim2 is not mutated, we see a clear difference between the restrictions with BfuCI and DpnII, indicating the presence of DNA methylation. In Zt09 and Zt150 this difference is not detectable except for one band that is not restricted in both strains in the rDNA blot. The genomic DNA of Zt09 treated with DpnI is not digested suggesting absence of 6mA methylation in these regions. C) Confirmation of bisulfite sequencing by an independent bisulfite treatment followed by PCR of target loci and Sanger sequencing. Two target loci (repeated region that showed 5mC signals in all isolates) were chosen per isolate, shown are the number of detected 5mC sites in these loci. N is the number of cloned PCR products that were sequenced. Based on these data we can confirm the presence of 5mC methylation in all isolates and a lower frequency in absence of Ztdim2.

5mC site preferences in CG, CHG and CHH sequence contexts. A) CGA and CGC sites are preferred in CG contexts in presence of Ztdim2, while CGA and CGG are slightly preferred targets in absence of Ztdim2. B) Among CHG sites, CAG sites are the predominant target sites of 5mC. For CHG and CHH contexts only data for Zt10 and Zt09::Ztdim2 are shown, as there is no detectable methylation outside of CG contexts in absence of Ztdim2. C) In CHH contexts, CA sites followed by CC sites have the highest methylation frequency. CT sites are almost completely devoid of 5mC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Research in the lab of EHS is supported by the State of Schleswig-Holstein, the Max Planck Society and CIFAR. Research in the lab of MF is supported by NSF grant (MCB1818006). MM is supported by the German Research Foundation (DFG, MO 3755/1-1). We thank Bruce A. McDonald for providing Iranian Z. tritici isolates, Fatemeh Salimi for sampling Iranian Z. tritici isolates, and Kathrin Happ, Anja Lach, and Maja Stralucke for assistance with experiments.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.
- 50.↵
- 51.↵
- 52.
- 53.
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵




