

Abstract
The coronavirus SARS-CoV-2 has infected more than 600,000 people and has overwhelmed hospital systems around the world. However, the factors mediating fatal SARS-CoV-2 infections are poorly understood. Here, we show that cigarette smoke causes a dose-dependent upregulation of Angiotensin Converting Enzyme 2 (ACE2), the SARS-CoV-2 receptor, in rodent and human lungs. Using single-cell sequencing data, we demonstrate that ACE2 is expressed in a subset of epithelial cells that line the respiratory tract, including goblet cells, club cells, and alveolar type 2 cells. Chronic smoke exposure triggers a protective expansion of mucus-secreting goblet cells and a concomitant increase in ACE2 expression. In contrast, quitting smoking causes a decrease in lung ACE2 levels. Taken together, these results may partially explain why smokers are particularly likely to develop severe SARS-CoV-2 infections, and they suggest that quitting smoking could lessen coronavirus susceptibility.
Introduction
In December 2019, a novel respiratory disease emerged in a seafood market in Wuhan, China1. Genomic sequencing demonstrated that the causative agent was a highly-contagious coronavirus, since named SARS-CoV-22,3. The disease, called COVID-19, rapidly spread worldwide, and as of March 2020, more than 600,000 people have been infected and more than 25,000 people have died4. No clinically-validated treatment or vaccine for COVID-19 is currently available. Thus, understanding the factors that mediate susceptibility to SARS-CoV-2 is crucial for controlling disease transmission.
Molecular analysis has begun to shed light on how SARS-CoV-2 infections occur. Like a related coronavirus that emerged in 20035, SARS-CoV-2 enters human cells by binding to the extracellular domain of Angiotensin Converting Enzyme 2 (ACE2)3,6. Importantly, ACE2 is both necessary and sufficient for infection by SARS-CoV-2: ACE2-targeting antibodies block viral uptake in permissive cells while transgenic expression of human ACE2 allows viral entry in non-human cells. ACE2 normally functions in the renin-angiotensin system (RAS) by cleaving the vasoconstrictive hormone angiotensin-II into the vasodilator angiotensin 1-77. Sequestration of ACE2 by coronavirus dysregulates the RAS pathway, contributing to morbidity8. Additionally, ACE2 levels are capable of influencing disease progression: among a cohort of mice engineered to express human ACE2, mice expressing the highest levels of ACE2 mRNA exhibited the shortest survival time following coronavirus exposure9. Thus, the regulation of ACE2 expression likely has a significant effect on SARS-CoV-2 susceptibility.
Epidemiological studies have identified several demographic features that correlate with the severity of clinical COVID-19 cases. While fewer than 5% of SARS-CoV-2 infections are fatal10, men and elderly patients are particularly at risk of developing severe disease11–14. Additionally, cigarette smokers are highly susceptible to SARS-CoV-2 and are significantly more likely to require aggressive clinical interventions: in a study of 1,099 patients with laboratory-confirmed COVID-19, 12.3% of current smokers required mechanical ventilation, were admitted to an ICU, or died, compared to only 4.7% of non-smokers12. The causes underlying these differences in clinical outcome are at present unknown.
ACE2 levels in mammalian lungs are unaffected by age or sex
In order to study factors that could potentially influence susceptibility to SARS-CoV-2 infection, we investigated the expression of the coronavirus receptor ACE2. We first assessed the expression of ACE2 in a variety of post-mortem rodent and human tissues (Figure S1 and Table S1)15–18. ACE2 was consistently expressed at high levels in mouse, rat, and human kidneys, consistent with its role as a regulator of the RAS pathway. ACE2 was also robustly expressed in rodent and human lungs, the predominant location of coronavirus infections. Interestingly, significant ACE2 expression was evident in the mouse and human small intestine. Viral RNA has been detected in stool samples from patients with COVID-1919, and gastrointestinal symptoms have been reported in a subset of affected individuals12, suggesting a potential alternate route for SARS-CoV-2 transmission. However, as SARS-CoV-2 is primarily spread through viral inhalation, we focused our study on factors that affect ACE2 expression in the lung and associated respiratory tissue.

(A) The expression of ACE2 in 17 different mouse tissues or cell types, sorted according to mean ACE2 expression.
(B) The expression of ACE2 in 11 different rat tissues, sorted according to mean ACE2 expression.
(C) The expression of ACE2 in 30 different human tissues or cell types, sorted according to mean ACE2 expression.
Age and male sex are significant risk factors for SARS-CoV-2 infections. We therefore investigated whether either feature was associated with increased ACE2 expression. Young mice (<26 weeks) and elderly mice (>78 weeks) displayed equivalent levels of ACE2 expression in the lung, as did young rats (6 weeks) and elderly rats (104 weeks)(Figure 1A-B)17,20. Similarly, ACE2 expression in rodent lungs was not significantly different between sexes (Figure 1C-D)17,21. We next assessed the expression of ACE2 in two different human cohorts: 1) lung tissue from the Genotype-Tissue Expression project (GTEx)15,16 and 2) whole-lung tissue samples from organ donors22. These datasets yielded results that were consistent with our rodent analyses: ACE2 expression was equivalent between men and women and between young individuals (<29 years) and elderly individuals (>70 years)(Figure 1E-H). In total, these findings suggest that the increased morbidity of men and older patients with COVID-19 is unlikely to result from inherent differences in ACE2 expression in the respiratory tract.

(A) Log2-normalized expression of ACE2 in the lungs of young mice (<26 weeks old) and old mice (>78 weeks old).
(B) Log2-normalized expression of ACE2 in the lungs of young rats (6 weeks old) and old rats (104 weeks old).
(C) Log2-normalized expression of ACE2 in the lungs of female mice and male mice.
(D) Long2-normalized expression of ACE2 in the lungs of female rats and male rats.
(E) Log2-normalized expression of ACE2 in lungs from the GTEx cohort by age.
(F) Log2-normalized expression of ACE2 in lungs from the GTEx cohort by sex.
(G) Log2-normalized expression of ACE2 in lungs from a cohort of organ donors by age.
(H) Log2-normalized expression of ACE2 in lungs from a cohort of organ donors by sex.
Cigarette smoke increases the expression of ACE2 in mammalian lungs
Cigarette smoking is strongly associated with adverse outcomes from COVID-1912,23. To investigate whether smoking could affect ACE2 levels, we first assessed gene expression in mouse lungs. We analyzed a cohort of mice exposed to diluted cigarette smoke for 2, 3, or 4 hours per day for five months24. Strikingly, we found a dose-dependent increase in ACE2 expression according to smoke exposure (Figure 2A). Mice exposed to the highest dose of cigarette smoke expressed ∼80% more ACE2 in their lungs compared to sham-treated mice. To determine whether this association was present in humans as well, we assessed three cohorts of current smokers and never-smokers25–27. For these analyses, lung epithelial cells were sampled from either the small or large airways by fiberoptic bronchoscopy. In each cohort, we observed that tissue collected from smokers exhibited ∼40%-50% more ACE2 compared to tissue from non-smokers (Figure 2B-D).

(A) Log2-normalized expression of ACE2 in the lungs of mice that were sham-treated or that were exposed to diluted cigarette smoke for two, three, or four hours a day (low, medium, and high smoke exposure, respectively).
(B) Log2-normalized expression of ACE2 in human small airway epithelia collected by fiberoptic brushing and analyzed according to smoking history (GSE52237).
(C) Log2-normalized expression of ACE2 in human small airway epithelia collected by fiberoptic brushing and analyzed according to smoking history (GSE3320).
(D) Log2-normalized expression of ACE2 in human large airway epithelia collected by fiberoptic brushing and analyzed according to smoking history.
(E) Log2-normalized expression of ACE2 in human lung tissue in a cohort of smokers and analyzed according to the number of pack-years smoked.
(F) Log2-normalized expression of ACE2 in human lung tissue in a cohort of lung cancer patients and analyzed according to the number of pack-years smoked.
(G) Log2-normalized expression of ACE2 in bronchial epithelia collected by fiberoptic brushing among either current smokers or former smokers. *, p < .05; **, p < .005; ***, p < .0005 (Student’s t-test).
Next, we sought to determine whether human ACE2 expression showed a dose-dependent relationship with cigarette smoke, as we had observed in mice. To investigate this, we analyzed two human datasets: 1) lung tissue from a cohort of smokers undergoing thoracic surgery for transplantation, lung volume reduction, or nodule resection28 and 2) lung tissue from a cohort of patients analyzed as part of The Cancer Genome Atlas (TCGA)29. For patients undergoing cancer-related surgeries, a sample of pathologically-normal tissue distant from the suspected tumor was used for analysis. In both cohorts, lung samples from patients who reported smoking the greatest number of pack-years also expressed the highest levels of ACE2 (Figure 2E-F). For instance, among smokers undergoing thoracic surgery, patients who had smoked more than 80 pack-years exhibited a ∼100% increase in ACE2 expression relative to patients who had smoked less than 20 pack-years (Figure 2E). Multivariate linear regression on this dataset further confirmed that smoking history was a significant predictor of ACE2 expression even when controlling for a patient’s age, sex, race, and body-mass index (Table 1). Lastly, we compared the expression level of ACE2 in lung epithelial cells from current smokers and former smokers30, and we found that quitting smoking was associated with a 30% decrease in ACE2 expression (Figure 2F). In total, our results demonstrate that exposure to cigarette smoke increases the expression of the coronavirus receptor ACE2 in rodent and human respiratory tissue, and this upregulation is potentially reversible.
Patient characteristics and ACE2 expression in the lung.
To follow up on these observations, we considered the possibility that ACE2 expression was commonly upregulated by lung diseases and/or carcinogen exposure. Indeed, we observed increased ACE2 expression in two cohorts of patients with idiopathic pulmonary fibrosis (IPF)31,32, a lung disease strongly associated with prior cigarette exposure (Figure S2A-B)33. However, ACE2 was not up-regulated in lung cells from a large cohort of patients with asthma or from patients with the lung disease sarcoidosis (Figure S2C-D)34,35. Similarly, ACE2 expression was unaffected in lung tissue from a mouse model of cystic fibrosis or in mice exposed to a variety of carcinogens, including arsenic, ionizing radiation (IR), and 1,3-butadiene (Figure S2E-G)36–39. We conclude that ACE2 upregulation in the lung is tightly associated with a history of cigarette smoking and is not a universal response to pathological insults.

(A) The expression of ACE2 in archived lung tissue from patients with idiopathic pulmonary fibrosis or from histologically-normal lung tissue from patients with lung cancer (GSE2052).
(B) The expression of ACE2 in tissue from the lungs of transplant recipients with idiopathic pulmonary fibrosis or from control donor lungs (GSE47460).
(C) The expression of ACE2 in bronchial brushing from individuals with normal lung function, mild asthma, or severe asthma.
(D) The expression of ACE2 in disease-free lung tissue resected from patients with sarcoidosis or control tissue from healthy lung donors.
(E) The expression of ACE2 in the lungs of a cystic fibrosis mouse model (Cftr-/-) or wild-type littermate controls.
(F) The expression of ACE2 in the lungs of mice provided with normal water, water with 10 parts per billion sodium arsenite, or water with 100 parts per billion sodium arsenite (low and high arsenic, respectively).
(G) The expression of ACE2 in the lungs of mice exposed to normal air or exposed to the carcinogen 1,3-butadiene.
(H) The expression of ACE2 in the lungs of mice that were sham-treated, exposed to 5 Gy of ionizing radiation to the thorax, or exposed to 17.5 Gy of ionizing radiation to the thorax (low and high IR, respectively). *, p < .05; **, p < .005 (Student’s t-test).
Coronavirus infections are facilitated by a set of host proteases that cleave and activate the viral spike (S) protein40. SARS-CoV-2 primarily relies on the serine protease TMPRSS2 but can also utilize an alternate pathway involving Cathepsin B/L in TMPRSS2-negative cells6. Interestingly, we observed that Cathepsin B expression, but not TMPRSS2 or Cathepsin L expression, was consistently increased in mice and humans exposed to cigarette smoke (Figure S3). Thus, smoking can upregulate both the coronavirus receptor as well as a protease that SARS-CoV-2 uses for viral activation.
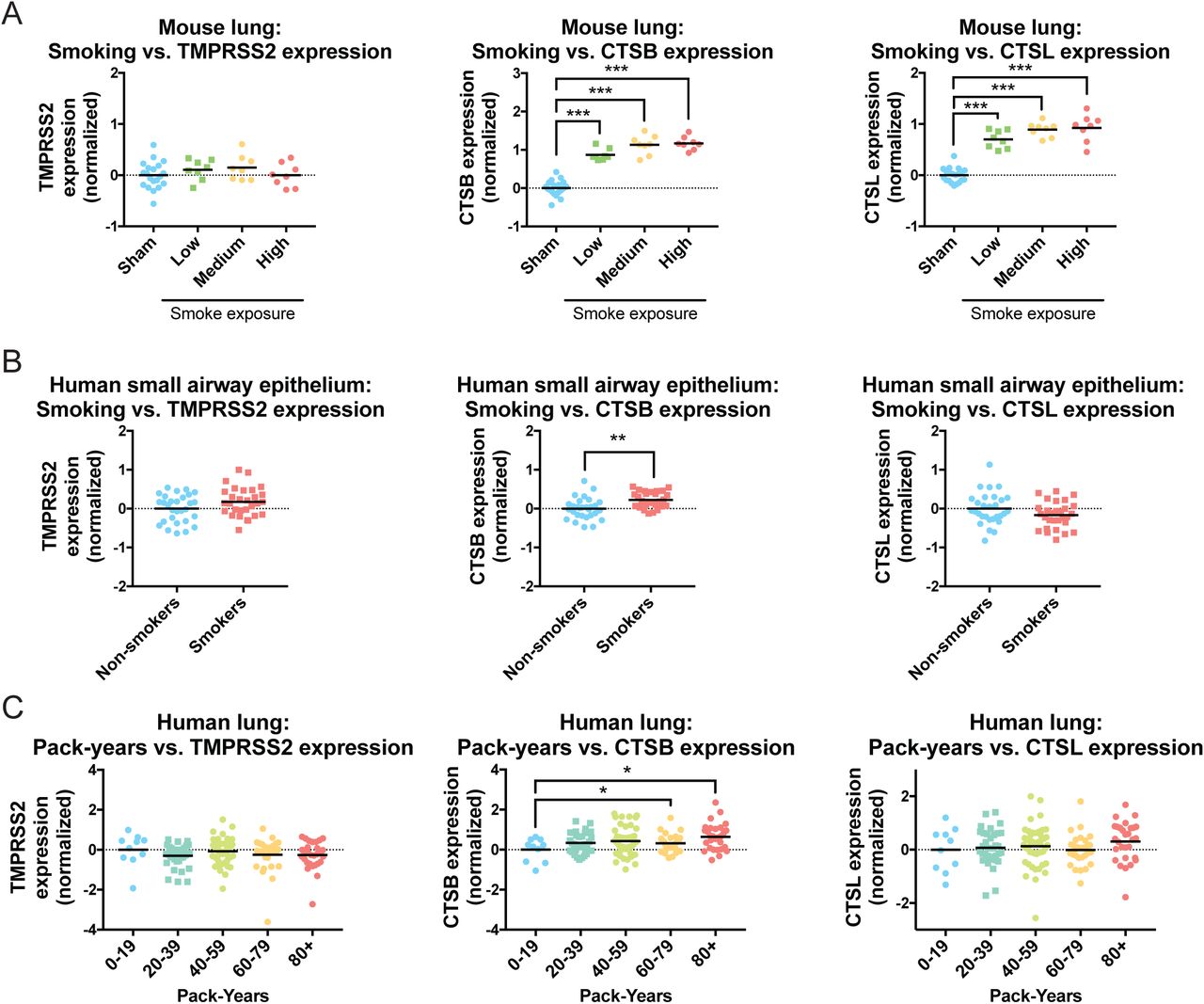
(A) The expression of TMPRSS2, Cathepsin B, and Cathepsin L in the lungs of mice that were sham-treated or that were exposed to diluted cigarette smoke for two, three, or four hours a day (low, medium, and high smoke exposure, respectively).
(B) The expression of TMPRSS2, Cathepsin B, and Cathepsin L in human small airway epithelia collected by fiberoptic brushing and analyzed according to smoking history.
(C) The expression of TMPRSS2, Cathepsin B, and Cathepsin L in human lung tissue in a cohort of smokers and analyzed according to the number of pack-years smoked. *, p < .05; **, p < .005; ***, p < .0005 (Student’s t-test).
ACE2 is expressed in secretory cells and alveolar type 2 cells in the lung
Mammalian lungs harbor more than 30 distinct cell types representing a variety of epithelial, endothelial, stromal, and immune compartments41. Of note, the upper respiratory epithelium is comprised of mucociliary cells, including goblet cells, club cells, and ciliated cells, that secrete protective fluids and remove inhaled particles from the airways42. The lower respiratory epithelium includes alveolar type 1 cells, which allow gas exchange with the blood, and alveolar type 2 cells, which regulate alveolar fluid balance and can differentiate into type 1 cells following injury43.
To gain further insight into coronavirus infections, we profiled multiple single-cell RNA-Seq experiments to identify the cell type(s) that express ACE2. We first examined a dataset containing 13,822 cells from normal mouse lungs44. We performed unsupervised Leiden clustering to separate the cells into distinct populations and then we assigned cell types to major clusters using established markers45–48. ACE2 was expressed solely in the EpCAM+ clusters that comprise the lung epithelium49, and was not detected in CD45+ immune cells50, PDGFRA+ mesenchymal cells51, or TMEM100+ endothelial cells (Figure 3A)52. We therefore focused on localizing ACE2 within the epithelial lineage. We found that ACE2 was strongly expressed in a cluster of cells that express secretory markers including MUC5AC, GABRP, and SCGB1A1 that we identified as being comprised of closely-related goblet and club cells (Figure 3B-C)48,53–55. Additionally, ACE2 expression was observed in a subset of LAMP3+ alveolar type 2 cells56, but was largely absent from RTKN2+ alveolar type 1 cells48 and FOXJ1+ ciliated cells57.

(A) T-SNE clustering of cells from the mouse lung. Cells expressing ACE2 and various lineage markers (endothelial, epithelial, mesenchymal, and immune) are highlighted.
(B) T-SNE clustering of cells from the mouse lung. Cells expressing markers for various epithelial lineages are highlighted.
(C) Violin plots display the expression of ACE2 and several lung-related genes in different cell populations obtained from Leiden clustering.
(D) Gene ontology enrichment analysis on transcripts whose expression correlates with ACE2.
We next identified the transcripts whose expression correlated with ACE2. Across all cells, ACE2 levels exhibited the strongest correlation with CLDN10 (Claudin-10), a tight-junction protein that contributes to the formation of the bronchial epithelium and that is expressed by club cells and goblet cells (Figure 3C)58,59. Gene ontology analysis revealed that ACE2-correlated transcripts were enriched for genes involved in xenobiotic metabolism, antioxidant activity, and cellular detoxification, consistent with the bronchial epithelium’s role as a barrier against toxins and foreign matter (Figure 3D and Table S2A)60.
To confirm our findings regarding ACE2’s localization, we subsequently repeated this analysis on an independent lung single-cell dataset (Figure S4). Consistent with our initial observations, we found that ACE2+ cells localized to clusters of epithelial cells that expressed the goblet and club cell markers CLDN10, GABRP, and SCGB1A1 or the alveolar type 2 marker LAMP3 (Figure S4A-C). Gene ontology analysis on ACE2-correlated transcripts revealed a similar enrichment for xenobiotic metabolism and cellular detoxification (Figure S4D and Table S2B). In total, our combined analysis demonstrates that ACE2 is expressed in secretory club and goblet cells as well as alveolar type 2 cells within the lung epithelium.

(A) T-SNE clustering of cells from the mouse lung. Cells expressing ACE2 and various lineage markers (endothelial, epithelial, mesenchymal, and immune) are highlighted.
(B) T-SNE clustering of cells from the mouse lung. Cells expressing markers for various epithelial lineages are highlighted.
(C) Violin plots display the expression of ACE2 and several lung-related genes in cell populations obtained from Leiden clustering.
(D) Gene ontology enrichment analysis on transcripts whose expression correlates with ACE2.
Cigarette smoke promotes secretory cell proliferation
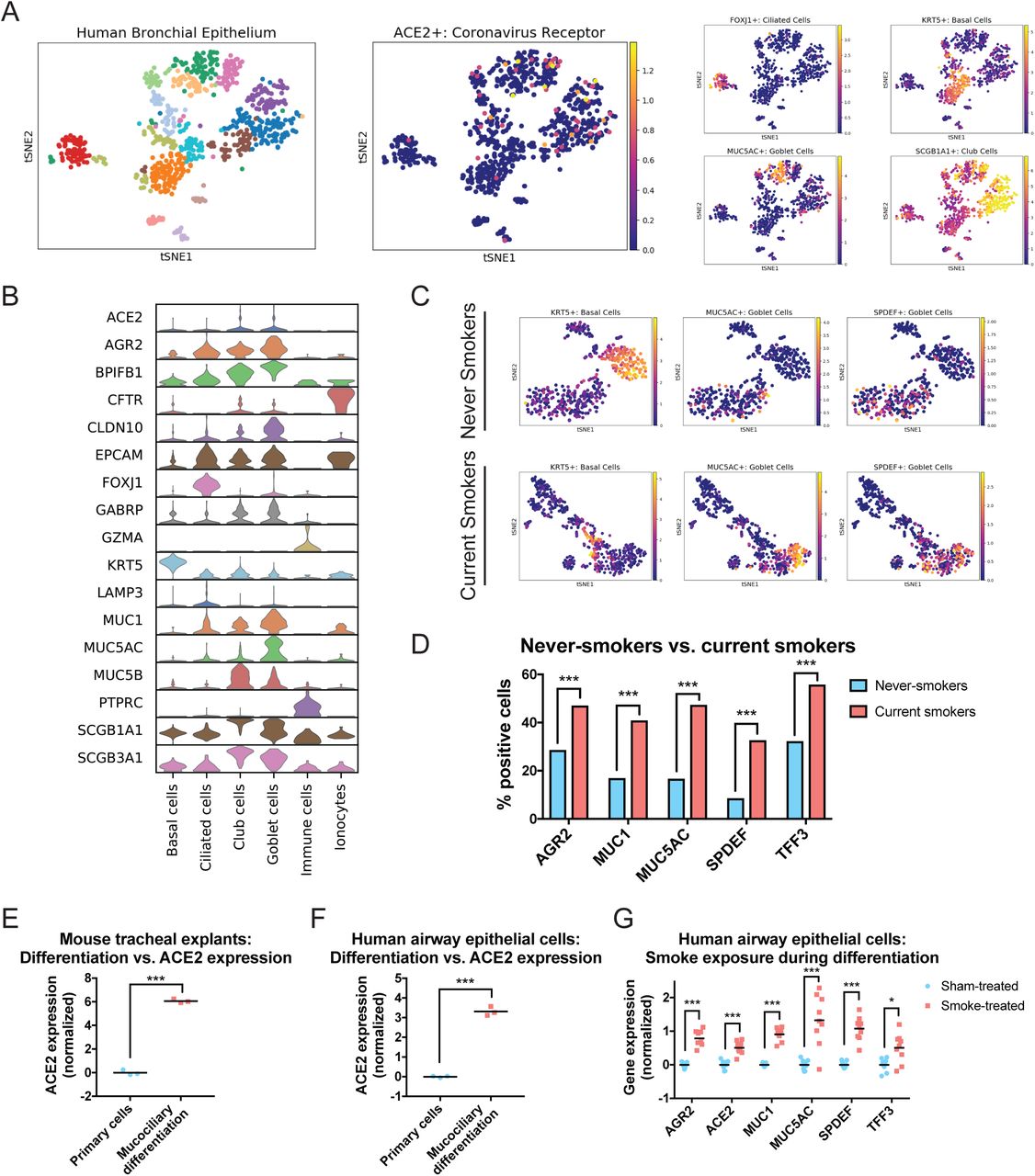
Our findings suggested a possible explanation for the upregulation of ACE2 in the lungs of cigarette smokers. Chronic exposure to cigarette smoke has been reported to trigger the expansion of secretory goblet cells, which produce mucous to protect the respiratory epithelium from inhaled irritants61–63. Thus, the increased expression of ACE2 in smokers’ lungs could be a byproduct of smoking-induced secretory cell hyperplasia. To investigate this hypothesis, we examined a dataset of single-cell transcriptomes collected by fiberoptic bronchoscopy from current smokers and never-smokers59. Notably, these cells were sorted and enriched for an epithelial cell marker (ALCAM) prior to sequencing. Additionally, they were collected from the main stem bronchus, and subsequently harbor few RTKN2+ or LAMP3+ alveolar cells59. This population was therefore highly enriched for upper-respiratory epithelial cells, and allowed us to localize ACE2 within this population with increased precision.
Using this purified dataset, we could more easily differentiate between goblet cells (marked by MUC5AC)53, club cells (marked by SCGB1A1)54, ciliated cells (marked by FOXJ1)57, ionocytes (marked by CFTR)64, and basal cells (which serve as respiratory epithelial stem cells and are marked by KRT5)65. We found that ACE2 expression was observed in both MUC5AC+ goblet cells and SCGB1A1+ club cells, but was largely absent from the basal cell, ciliated cell, and ionocyte clusters (Figure 4A-B). Next, we separated the cells harvested from current smokers and never-smokers and then analyzed each population separately. Consistent with previous reports61–63, we detected an increase in goblet cells in the lungs of current smokers: while 17% of cells from non-smokers expressed the canonical goblet marker MUC5AC53, 47% of cells collected from smokers were positive for MUC5AC (Figure 4C-D). Cells that expressed other mucus- and goblet cell-related genes, including MUC167, AGR268, and SPDEF69, were similarly over-represented in the lungs of smokers. KRT5+ basal stem cells were depleted in the smokers’ epithelia, likely reflecting their differentiation into secretory cells (Figure 4C). However, we note that in this dataset, we observed an increase in the percentage of ACE2+ cells between non-smokers and smokers that was not statistically-significant. This may reflect the small number of cells that were analyzed (∼1,000) or the small number of smokers that were profiled (six).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) T-SNE clustering of the transcriptomes from single cells derived from human bronchial cells. Cells expressing ACE2 and various epithelial markers are displayed.
(B) Violin plots display the expression of ACE2 and several lung-related genes in different cell populations obtained from Leiden clustering.
(C) Cells derived from never-smokers (top) and current smokers (bottom) were analyzed independently by t-SNE clustering. Cells expressing various epithelial markers are displayed.
(D) The percent of cells from never-smokers and current smokers expressing the indicated goblet cell-related genes are displayed. ***, p < .0005 (Fischer’s exact test).
(E) Log2-normalized expression of ACE2 in mouse cells prior to differentiation or following mucociliary differentiation at an air-liquid interface. ***, p < .0005 (Student’s t test).
(F) Log2-normalized expression of ACE2 in human cells prior to differentiation or following mucociliary differentiation at an air-liquid interface. ***, p < .0005 (Student’s t test).
(G) Log2-normalized expression of the indicated genes in human cells differentiated in the presence of diluted cigarette smoke or in the presence of clean air. *, p < .05; ***, p < .0005 (Student’s t test).
Secretory cell differentiation of lung epithelium can be modeled in vitro by culturing cells at an air-liquid interface (ALI)70,71. Under appropriate conditions, primary respiratory cells growing at an ALI will undergo mucociliary differentiation into a stratified epithelium consisting of ciliated cells, goblet cells, and club cells72. As our single-cell analysis suggested that the coronavirus receptor ACE2 is expressed in secretory goblet and club cells, we investigated whether in vitro mucociliary differentiation increases ACE2 expression. Indeed, in mouse tracheal extracts73 and primary human lung cells74, mucociliary differentiation resulted in a highly-significant upregulation of ACE2 (Figure 4E-F). Finally, to investigate the link between smoking, differentiation, and ACE2 expression, we examined data from human bronchial epithelial cells cultured at an ALI in which cells were either exposed to clean air or to diluted cigarette smoke75. Remarkably, treatment with cigarette smoke during in vitro differentiation resulted in a significant upregulation of ACE2, MUC5AC, MUC1, and other secretory cell markers, relative to cells that were differentiated in clean air (Figure 4G). ACE2 expression was increased by ∼42% in smoke-exposed cells, comparable to the increases that we observed between the lungs of non-smokers and smokers (Figure 2). In total, our results demonstrate that a subset of lung secretory cells express the coronavirus receptor ACE2, and cigarette smoke promotes the expansion of this cell population.
Discussion
While SARS-CoV-2 has infected more than 600,000 people worldwide, fewer than 5% of COVID-19 cases are fatal4,12. Here, we show that cigarette smokers harbor consistently higher levels of the SARS-CoV-2 receptor ACE2 in their lungs. This upregulation is likely mediated at least in part by the expansion of ACE2+ mucus-secreting goblet cells triggered by chronic smoke exposure. The overabundance of ACE2 in the lungs of smokers may partially explain why smokers are significantly more likely to develop severe SARS-CoV-2 infections that require aggressive medical interventions12,23. Furthermore, as quitting smoking is associated with a decrease in ACE2 expression, we speculate that giving up cigarettes may reduce susceptibility to COVID-19.
Several contrasting findings exist in the literature on ACE2 and cigarette exposure76–81. In particular, it has been reported that nicotine and/or cigarette smoke has the potential to downregulate ACE2 expression in certain tissues or cell types76–79. In this manuscript, we focused our analysis on factors affecting ACE2 expression in the mammalian lungs and associated respiratory epithelia. We observed a consistent correlation between smoking history and ACE2 expression that was dose-dependent (Figure 2E-F), that could be recapitulated in mice (Figure 2A) and in vitro (Figure 4G), and that remained significant when controlling for other demographic variables (Table 1). Thus, we propose that cigarette smoke causes an upregulation of ACE2 expression in the respiratory tract, though we recognize that smoking may have different effects on ACE2 levels in the heart, kidney, or other organs.
Our results concerning the localization of ACE2 agree with and expand on several unpublished reports82–84. Consistent with our findings, these analyses identified a population of ACE2+ cells in the alveolar type 2 compartment. However, as these previous reports predominantly analyzed tissue from the lower respiratory tract, we speculate that they were enriched for alveolar cells relative to cells from the upper respiratory epithelium. By analyzing datasets from both mice and humans, and by including cells collected from bronchial brushing, we also demonstrate the presence of ACE2 in the secretory cells that participate in mucociliary clearance42. As these cells line the upper respiratory tract, they may represent the initial site of coronavirus infections, followed by an eventual spread and migration into the alveoli. Moreover, secretory cell hyperplasia is a well-described consequence of prolonged smoke exposure61–63, potentially explaining the consistent up-regulation of ACE2 in smokers’ lungs. Interestingly, ACE2-knockout mice have been reported to be particularly vulnerable to lung failure following forced acid inhalation85. Thus, the ACE2 that we uncovered in mucociliary cells could potentially play a direct role in protecting the airways from inhaled toxins and foreign matter.
The factors that mediate susceptibility to SARS-CoV-2 infections are poorly understood. We speculate that the increased expression of ACE2 in the lungs of smokers could partially contribute to the severe cases of COVID-19 that have been observed in this population. In support of this hypothesis, mice that were engineered to express high levels of human ACE2 succumbed to infections with a related coronavirus more quickly than mice that expressed low levels of human ACE29. Nonetheless, the relevance of increased ACE2 expression as a driver of disease susceptibility in humans or for SARS-CoV-2 remains to be demonstrated. Chronic smokers may exhibit a number of co-morbidities, including emphysema, atherosclerosis, and decreased immune function86, that are also likely to affect COVID-19 progression. While the effects of smoking can last for years, smoking cessation causes an improvement in lung function and an overall decrease in disease burden86. Interestingly, quitting smoking also leads to a normalization of respiratory epithelial architecture87, a decrease in hyperplasia88, and a downregulation of ACE2 levels. Thus, for multiple reasons, smoking cessation could eventually lessen the risks associated with SARS-CoV-2 infections.
Methods
Overall analysis strategy
The analysis described in this paper was performed using Python, Excel, and Graphpad Prism. Gene expression data was acquired from the Gene Expression Omnibus (GEO)89, the GTEx portal15, the Broad Institute TCGA Firehose90, the Human Cell Atlas91, and the Single-Cell Expression Atlas92, as described below. For microarray datasets, probeset definitions were downloaded from GEO, and probes mapping to the same gene were collapsed by averaging. For each gene expression comparison, a control population was identified (e.g., young rats, sham-treated mice, non-smokers, etc.), and gene expression values were log2-transformed and normalized by subtraction so that the mean expression of a gene of interest in the control population was 0. Graphs of gene expression values were then generated using Graphpad Prism; all data points are displayed and no outliers were excluded from analysis.
Data sources
The data sources used in this paper are listed in Table S1. In general, pre-processed microarray and RNA-Seq datasets were downloaded. Additional notes on sample selection and processing are included in Table S1.
Multivariate regression and smoking history
Multivariate regression to investigate the relationship between ACE2 expression and smoking history was performed on the GSE76925 lung tissue dataset. Regressions were performed in Python using ordinary least squares from the statsmodels package93. Results reported include the standard errors (‘bse’), betas, and p-values.
Single-cell analysis
Single-cell clustering and analysis on the datasets listed in Table S1 was performed in Python using the Scanpy and Multicore-TSNE packages45,94. To filter out low-quality cells, only cells in which 500 or more genes were detected were included in this analysis. Before clustering, transcript counts were log2 transformed. Highly variable genes were selected using the Seurat approach in Scanpy, and these highly variable genes were used to produce the principal component analysis. A t-SNE projection and unsupervised Leiden clustering were then performed on each dataset using nearest neighbors, as described in the associated code.
In order to label each cluster, a gene ranking analysis was obtained using Scanpy. The 10 most highly-ranked genes from each cluster (as determined by t-test with overestimated variance) were identified. These genes were then compared against gold-standard marker lists from multiple sources to produce the cluster labels45–48.
Gene ontology analysis
In order to identify the genes whose expression correlates with ACE2, pairwise Pearson correlations coefficients (PCCs) were calculated between ACE2 and every other expressed gene. Genes whose PCC were more than four standard deviations greater than the average gene’s PCC were classified as strongly correlated with ACE2. Gene ontology terms enriched in this group were then identified with GProfiler against the background list of non-strongly correlated genes using a Benjamini-Hochberg FDR of .0595.
Code and Data Availability
All code for performing these analyses is available at github.com/joan-smith/covid19.
Declaration of Interests
J.C.S. is a co-founder of Meliora Therapeutics and is an employee of Google, Inc. This work was performed outside of her affiliation with Google and used no proprietary knowledge or materials from Google. J.M.S. has received consulting fees from Ono Pharmaceuticals, is a member of the Advisory Board of Tyra Biosciences, and is a co-founder of Meliora Therapeutics.
Footnotes
Works Cited
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.
- 63.↵
- 64.↵
- 65.↵
- 66.
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.
- 78.
- 79.↵
- 80.
- 81.↵
- 82.↵
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵




