

Abstract
G-protein coupled receptor (GPCR) 68 (GPR68, or OGR1) couples extracellular acidifications and mechanical cues to G protein signaling and plays important roles in vascular physiology, neuroplasticity and cancer progression. The mechanism of mechanosensitivity in GPR68 is currently unkonwn. Here, to study this mechanism, we designed a genetically-encoded fluorescent reporter of GPR68 by fusing a cyclic permuted green fluorescent protein to the third intracellular loop of the receptor. Stimulation with fluid shear stress, extracellular acidifications or the synthetic activator ogerin transiently and robustly increases iGlow’s baseline fluorescence up to 4-fold. Flow-induced iGlow activation was not suppressed by pharmacological uncoupling of downstream G-protein recruitment, disruption of actin filaments, inhibition of membrane stretch with the peptide toxin GsMTx4, or deletion of a C-terminal amphipathic Helix (Helix 8) proposed to mediate GPCR mechanosensitivity. These results hint that GPR68 uses a hitherto unknown, non-canonical mechanism to sense mechanical forces.
Introduction
G-protein coupled receptors (GPCRs) constitute the largest known family of membrane receptors, comprising at least 831 human homologs organized into 6 functional classes (A to F). They play essential roles in a wide range of biological functions spanning all major physiological systems such as olfaction, energy homeostasis and blood pressure regulation. They also control embryonic development and tissue remodeling in adults. The biological significance of GPCRs is underscored by the fact that ~13% of all known human GPCRs represent the primary targets of ~34% of all pharmaceutical interventions approved by the Food and Drug Administration1.
GPCRs possess a conserved structure encompassing seven transmembrane helices and switch between resting and active conformations depending on the presence of specific physico-chemical stimuli. Besides the vast repertoire of small molecules recognized by GPCRs such as odorants, hormones, cytokines and neurotransmitters, other physico-chemical cues can act as GPCR activators such as photons2, ions3, membrane depolarizations4–8 and mechanical forces9–12. An inherent challenge for the study of GPCR signaling is the fact that GPCRs may often respond to more than one stimulus, therefore acting as complex stimuli integrators.
Activated GPCRs physically interact with heterotrimeric G-proteins (Gα, Gβ and Gγ) and promote the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on the Gα subunit upon binding. This process, called G-protein engagement, enables GTP-bound Gα subunits to dissociate from the GPCR:Gβγ complex and activate downstream cellular effectors. Eighteen Gα subunit homologs have been so far identified in mammalian genomes. Gα subunits are clustered into four groups, Gαs, Gαi Gαq and Gα12, each group targeting distinct downstream signaling effectors13. Many GPCRs can engage more than one type of Gα subunits. In addition, Gα proteins are highly regulated by various regulatory proteins such as Resistance to inhibitors of cholinesterase 8 (Ric-8A/B) proteins14 and Regulator of G-protein Signaling (RGS)15. Hence, the initiation of GPCR-mediated cellular signals depends not only on the presence of specific stimuli but also on the presence and cellular availability of specific types of Gα proteins.
One example of GPCR exhibiting complex stimuli integration and pleiotropic G-protein signaling is GPR68. GPR68 is a class-A GPCR originally cloned from an ovarian cancer cell line and hence named Ovarian cancer G-protein coupled Receptor 1, or OGR116. Early studies showed that the lipid sphyngophosphorylcholine acts as an endogenous GPR68 ligand17,18, although one of these two studies has been subsequently retracted19. It is now well established that GPR68 is physiologically activated by extracellular protons20, a property shared with only three other GPCRs to date (GPR4, GPR65 and GPR132). In addition, recent evidence show that mechanical forces such as fluid shear stress and membrane tension act as an effective GPR68 positive modulator in the presence of protons9,10, enabling GPR68 to mediate flow-induced dilation of small-diameter arteries9. GPR68 can engage Gαq/11, which increases cytosolic concentration of calcium ions ([Ca2+]cyt) via phospholipase C-β (PLC-β) as well as Gαs, which increases the production of cyclic adenosine monophosphate (cAMP) via adenylate cyclase activation. Interestingly, incubation with the synthetic positive modulator ogerin increases pH-dependent cAMP production by GPR68 but reduces pH-dependent calcium signals, suggesting ogerin acts as a biased positive allosteric modulator of GPR6821. GPR68 is expressed in various tissues and is often up-regulated in many types of cancers9,22. Interestingly, ogerin suppresses recall in fear conditioning in wild-type but not GPR68−/− mice, suggesting a role of GPR68 in learning and memory21. Hence, although its contribution to vascular physiology has been established, its roles in other organs and in cancer progression remain unclear.
GPCR activation is often monitored using Fluorescence Resonance Energy Transfer (FRET) or Bioluminescence Resonance Energy Transfer (BRET)11,23,24. However, FRET necessitates complex measurements to separate donor and acceptor emissions whereas BRET often requires long integration times and sensitive detectors to capture faint signals. In contrast, recent ligand-binding reporters engineered by fusing GPCR with a cyclic permuted Green Fluorescent Protein (cpGFP) have enabled robust and rapid in vitro and in vivo detection of GPCR activation by dopamine25,26, acetylcholine27 and norepinephrine28 using simple intensity-based fluorescence measurements. Here, to facilitate future investigations of GPR68, we borrowed a similar cpGFP-based engineering approach to create a genetically-encodable reporter of GPR68-mediated signaling. We call it indicator of GPR68 signaling by mechanical forces and low pH, or iGlow.
Results
iGlow design and characterization
We sought to design iGlow by borrowing a protein engineering design from dLight1.2, a genetically-encoded dopamine sensor25. In dLight1.2, a fluorescence signal is produced by fluorescence dequenching of a cyclic permuted green fluorescent protein (cpGFP) inserted into the third intracellular loop of the DRD1 dopamine receptor. We generated iGlow by inserting cpGFP into the homologous position in the third intracellular loop of human GPR68, flanking cpGFP with the same N-terminal (LSSLI) linker and C-terminal (NHDQL) linkers as in dLight1.2 (Figure 1A, Supplementary Figure 1 and Supplementary Table 1). HEK293T cells transiently expressing iGlow were seeded into microscope-compatible flow chambers, allowing us to apply desired amount of fluid shear stress (FSS) by circulating Hank’s Balanced Salt Solution (HBSS, pH 7.3) using a computer-controlled peristaltic pump while green fluorescence emission from iGlow is continuously monitored.

(A) Top: iGlow was designed by inserting a cpGFP cassette (green) into the third intracellular loop (IL3) of GPR68 (purple). Bottom: structural model of iGlow generated using the Molecular Operating Environment (MOE) software from the crystal structure of cpGFP (PDBID: 3O77, green) and a structural model of GPR68 (purple) generated by Huang et al.21. (B) Epifluorescence images showing iGlow fluorescence in static or flow condition (bar = 10 μm). (C) Fluorescence time-course (plotted as ΔF/F0 vs. time) from a cell co-expressing iGlow (purple trace) and mCherry (black trace) in response to intermittent shear stress pulses (10 sec on, 10 sec off) of incrementally-increased amplitudes. (D) Scatter plot showing the maximal ΔF/F0 values produced by iGlow using our intermittent flow protocol. (E) Distribution of fluorescence threshold for cells stimulated using the intermittent flow protocol. (F) Time-course of iGlow fluorescence upon a 2.6 dyne cm−2 FSS pulse. (G) Time-course of dLight1.2 fluorescence upon acute perfusion with 10 μM dopamine. (H) Scatter plots comparing max ΔF/F0 values obtained between iGlow and dLight1.2 with protocols depicted in (F) and (G). (I). The Number above the scatter plots in panel (H) indicates exact p-value from a Student’s T-test. Error bars = s.e.m.
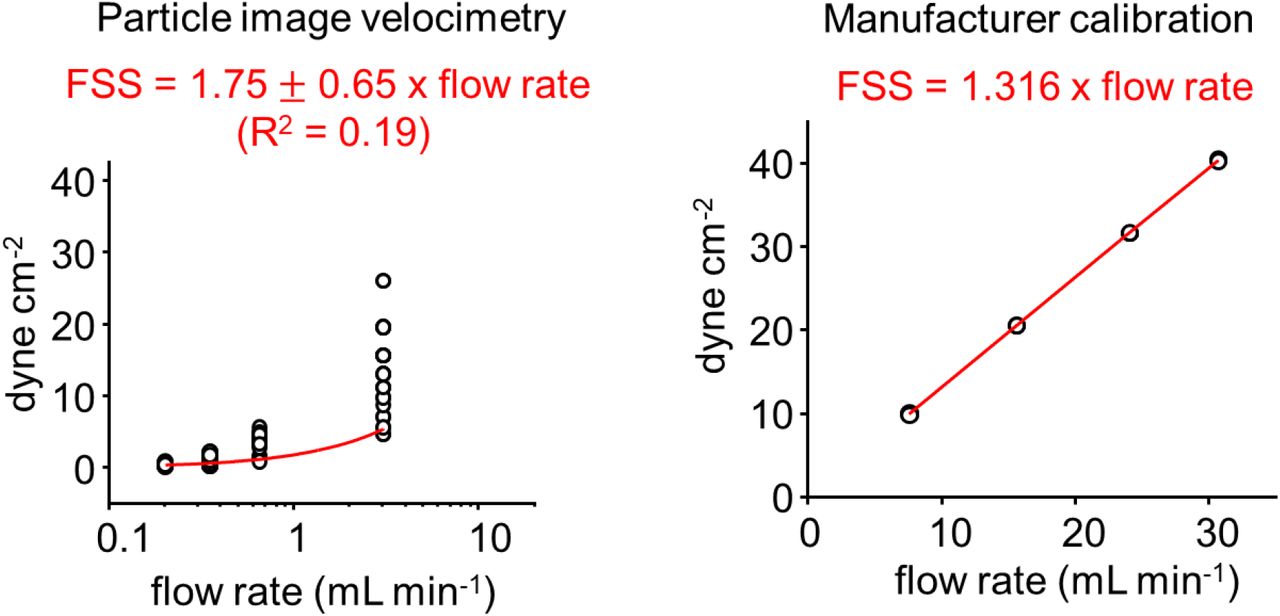
In order to determine accurately the amplitude of shear stress applied inside our commercial flow chamber, we compared shear stress values determined by multiplying the average flow-rate by a coefficient provided by the manufacturer to values calculated by particle velocimetry measurement (see Methods and Supplementary Figure 2). We observed that increasing flow rate tends to increase fluctuations of velocity measurements, yielding a poor linear fit between flow rate and shear stress (R2 = 0.19). We attribute these fluctuations to the peristaltic nature of the flow, which inevitably produces fluctuations in instantaneous flow rate. Nevertheless, the fitted slope value obtained from velocity measurements was near the manufacturer’s calibration (1.75 ± 0.65 dyne mL cm−2 min−1 vs. 1.316 dyne mL cm−2 min−1). Hence, we used the manufacturer’s calibration to determine the amplitude of shear stress in all our experiments.
When stimulated with incremental FSS pulses (10 sec on, 10 sec off), iGlow produced robust increases in green fluorescence intensity, with an average maximal response (max ΔF/F0) of +32 ± 3 % (n = 43) (Figure 1B-D). In most cells, the activation threshold of iGlow was below 1 dyne cm−2, although some cells showed higher mechanical threshold near 4 dyne cm−2 (Figures 1E). Co-transfection of iGlow with a mCherry-encoding plasmid did not yield comparable fluorescence changes in the red fluorescence channel, indicating cpGFP fluorescence changes do not result from imaging artifacts (Figure 1C). To rule out direct modulation of cpGFP fluorescence by FSS, we monitored green fluorescence from two other membrane proteins containing cpGFP and that are not anticipated to exhibit mechanosensitivity: the voltage indicator ASAP129 and Lck-cpGFP, a cpGFP we directly fused to the membrane-bound N-terminal domain of the Lymphocyte-specific kinase (Lck)30. In these cpGFP-containing proteins, no fluorescence changes other than photobleaching-induced decays occurred, even upon high flow conditions > 10 dyne cm−2, confirming FSS-induced signals in iGlow require the presence of GPR68 (Supplementary Figure 3).
We next compared fluorescence signals of iGlow and dLight1.2 when excited using a saturating physiological stimulation. We used a FSS pulse of 2.6 dyne cm−2 for iGlow and a perfusion with a 10 μM dopamine solution for dLight1.2 (Kd for dopamine in dLight1.2 = 765 nM25). In these conditions, iGlow produced signals with significantly larger maximal amplitude compared to dLight1.2 (+40 ± 7% vs. +21 ± 2%, Student’s T-test p-value = 0.0045) (Figure 1F-H).
iGlow detects several modes of GPR68 activation
We next sought to determine whether iGlow responds to all known GPR68 stimuli. Activation of iGlow using 10 μM of the small molecule GPR68 agonist ogerin produced strong fluorescence transients, with a mean maximal amplitude of +55 ± 19%. In comparison, perfusion with a vehicle solution yielded negligible fluorescence changes, with a mean max ΔF/F0 value of +6 ± 1% (Mann-Whitney U test, p-value = 0.012), likely due to unavoidable turbulent flow created during solution exchange (Figure 2A-B).

(A) Time-course of iGlow fluorescence in response to acute incubation with 10 μM ogerin (black trace) or a vehicle control (HBSS, gray trace). (B) Scatter plots showing max ΔF/F0 values obtained upon incubation with ogerin (black dots, n = 6) or a vehicle control (gray dots, n = 7). (C) Time-course of iGlow fluorescence in response to a 2.6 dyne cm−2 FSS pulse at indicated extracellular pH values. (D) Scatter plots showing max ΔF/F0 values obtained by acute application of FSS at pH 6.5 (blue dots, n = 10), pH 7.3 (black dots, n = 24) or pH 8.2 (red dots, n = 28). Numbers above plots indicate exact p-values from a Mann Whitney U-test (B), one-way ANOVA (E) or Student’s T-tests (E). Error bars = s.e.m.
Since GPR68 activates in response to extracellular acidifications in a pH range 6.5-7.5, we next stimulated transfected cells to a 2.6 dyne cm−2 FSS pulse using solutions of different pH, starting from pH 8.2 to avoid pre-stimulation and/or desensitization of the receptor that may occur at a permissive physiological (neutral) pH9. As expected, the amplitude of iGlow’s response increased as pH decreases, with mean max ΔF/F0 values ranging from +21 ± 2 % at pH 8.2, +29 ± 3 % at pH 7.3 and +44 ± 7% at pH 6.5 (Figure 2C-D). Student’s T-test pairwise statistical analyses indicate these differences in mean fluorescence changes are statistically significant, with p-values ranging from 0.044 (pH 7.3 vs. pH 6.5) to 0.0297 (pH 7.3 vs. pH 8.2). Statistically significant differences among all groups is also supported by one-way ANOVA (p-value = 0.0021).
iGlow senses GPR68 activation
How does the cpGFP insert increases its fluorescence emission upon iGlow stimulation? Two distinct scenarios are plausible. First, as seems to be the case for dLight1.2 and GRABDA, the fluorescent protein may sense stimulus-induced conformational rearrangements of the receptor25,26. Second, since cpGFP is located near the intracellular binding site for GDP-bound G proteins and regulatory kinases, we cannot exclude the possibility that iGlow could sense the interaction between the activated receptor and downstream signaling proteins. To distinguish between these two possibilities, we stimulated iGlow with a 2.6 dyne cm−2 FSS pulse in cells treated with one of several pharmacological agents preventing association or dissociation of G proteins (Figure 3A). We used GTP-γ-S, a non-hydrolyzable GTP analog which prevents Gα protein association to all GPCRs; NF449, a GDP→GTP exchange inhibitor which selectively prevents Gαs dissociation from their receptor31; and BIM-46187, a non-specific GDP→GTP exchange Gα inhibitor32. We also tested CMPD101, an inhibitor of G protein Receptor kinases 2/3 (GRK2/3). Interestingly, the GTP-γ-S treatment nearly doubles the average maximal amplitude of iGlow signals (max ΔF/F0 = +38 ± 8 % vs. +78 ± 21%, Student’s T-test p-value = 0.02), suggesting G protein binding partially quenches cpGFP fluorescence (Figure 3B-C). On the other hand, none of other pharmacological treatments induced significant change in signal amplitude (Student’s T-tests p-values between 0.19 and 0.59). We also observed that dopamine-induced dLight1.2 signals were not affected by any of these pharmacological treatments (one-way ANOVA p-value = 0.4214), as expected if dLight1.2 responds to receptor activation independently of downstream G protein signaling (Supplementary Figure 4). These results thus show iGlow specifically senses conformational rearrangements of GPR68.

(A) Expected effects of pharmacological treatments on protein-protein interactions between iGlow, Gαs/q/11 proteins and GRK2/3. (B) Time-course of iGlow fluorescence upon treatments with 0.2 mM GTP-γ-S (red trace), 20 μM NF-449 (blue trace), 20 μM BIM-46187 (green trace), 10 μM CMPD101 (purple trace) or a vehicle control (black trace) and subjected to an acute FSS pulse. (C) Scatter plots showing the max ΔF/F0 values obtained following shear stress stimulation in cells treated with GTP-γ-S (red dots, n = 33), NF-449 (blue dots, n = 20), BIM-46187 (green dots, n = 21), CMPD101 (purple dots, n = 17) or a vehicle control (black dots, n =25). Numbers above plots in panel (C) indicate exact p-values from Student’s T-tests between control and treated samples. Error bars = s.e.m.
iGlow’s mechanical activation is resilient
The ionic currents produced from mechanosensitive ion channels is often reduced or abolished by disruption of actin filaments, as cytoskeletal elements often help transmitting mechanical forces across remote cellular microdomains. We transfected HEK293T cells with LifeAct-mScarlet to monitor real-time actin disorganization upon treatment with 20 μM cytochalasin D (CD), an inhibitor of actin polymerization. Actin filaments were completely disorganized after about 20 min (Figure 4A). Since cell death could be detected after a one-hour CD treatment, we monitored iGlow’s response to FSS immediately after 20 min CD treatment. Cells produced fluorescence signals similar to untreated cells. We repeated the experiments after treating cells with 50 μM CD and found no difference in maximum amplitude of iGlow signals among all groups (one-way ANOVA p-value = 0.4980) (Figure 4B-C). These results suggest GPR68 senses flow in an actin-independent manner.

(A) Cytochalasin D (CD)-mediated disruption of actin cytoskeleton (bar = 10 μm). (B) Time-course of flow-induced iGlow fluorescence following 20 min incubation with 20 μM CD (red trace), 50 μM CD (blue trace) or untreated control (black trace). (C) Scatter plots showing max ΔF/F0 values after treatment with 20 μM CD (red dots, n = 25), 50 μM CD (blue dots, n = 16) or in untreated cells (black dots, n = 24). (D) Time-course of flow-induced Ca2+ entry in cells expressing PIEZO1 and GCaMP6f in presence (blue trace) or absence (black trace) of 2.5 μM GsMTx4. (E) Scatter plots showing max ΔF/F0 values obtained from (D) in absence (black dots, n = 31) or presence (blue dots, n = 39) of 2.5 μM GsMTx4. (F) Time-course of flow-induced iGlow fluorescence in presence (blue trace) or absence (black trace) of 2.5 μM GsMTx4. (G) Scatter plots showing max ΔF/F0 values obtained from (F) in absence (black dots, n =18) or presence (blue dots, n = 15) of 2.5 μM GsMTx4. (H) C-terminal sequences of iGlow and the deletion mutant (H8del). (I) Time-course of flow-induced fluorescence from iGlow and the H8del mutant. (J) Scatter plots showing max ΔF/F0 values from iGlow (black dots, n = 24) and H8del (grey dots, n = 6) from (I). Numbers above plots indicate exact p-values from one-way ANOVA (C), Student’s T-tests (E) and (G) or a Mann-Whitney U-test (J). Error bars = s.e.m.
Acute incubations with micromolar concentrations of the spider toxin GsMTx4 has been shown to robustly inhibit a broad range of mechanosensitive ion channels upon various mechanical stimulations, including membrane stretch, fluid shear stress and mechanical indentations33–37. We first performed a control experiments by measuring Ca2+ entry mediated by the mechanosensitive PIEZO1 channel in response to a single FSS pulse in the presence or absence of 2.5 μM GsMTx4. We monitored intracellular free Ca2+ ions by co-transfecting PIEZO1-deficient cells with a mouse PIEZO1 plasmid and a plasmid encoding the fluorescent calcium indicator GCaMP6f. Our data show that this toxin concentration was able to reduce GCaMP6f’s fluorescence response (max ΔF/F0) from +75 ± 5 % to +16 ± 2 %, a nearly 5-fold reduction (Student’s T-test p-value = 9.7×10−18) (Figure 4D-E). In contrast, the same treatment did not affect the amplitude of iGlow signals (Student’s T-test p-value = 0.2057) (Figure 4F-G).
Class-A GPCRs harbor a structurally-conserved amphipathic helical motif located immediately after the seventh transmembrane segment, called Helix 8. A recent study showed that deletion of Helix 8 abolished mechanical, but not ligand-mediated, activation in the histamine receptor H1R24. Furthermore, transplantation of H1R Helix 8 into a mechano-insensitive GPCR was sufficient to confer mechanosensitivity to the chimeric receptor24. Hence, Helix 8 seems both necessary and sufficient to confer mechanosensitivity in, at least, some GPCRs24. The online tool NetWheels indicates that GPR68 also contains an amphipathic helical motif reminiscent to Helix 8 of H1R (Supplementary Figure 5). We introduced a non-sense codon (TGA) to delete this motif and the remainder of the C-terminal region from iGlow and tested the sensitivity of the deletion mutant (H8del) to our standardized FSS protocol. H8del produced robust fluorescence signals with a mean maximal amplitude of + 29 ± 3 %, which was not statistically different than those produced by the full-length iGlow (+32 ± 3 %, Mann-Whitney U-test p-value = 0.9124) (Figure 4H-J). This result suggests that Helix 8, although present in GPR68, is dispensable for shear flow sensing by GPR68.
Discussion
In this study, we introduce iGlow, the first cpGFP-based fluorescence reporter for a polymodal GPCR. To work as a useful investigational tool, cpGFP-based reporters need to exhibit a large fluorescence intensity change between the bright and the dark states. Achieving such as large dynamic range often requires iterative rounds of mutagenesis and positive selection, for example through directed evolution. However, the first version of iGlow produced robust signals without needing additional optimization steps. Many studies have shown that a critical molecular determinant for modulating the dynamic range of cpGFP-based intensiometric reporters are the linkers connecting the N- and C-termini of cpGFP to the host protein(s) or protein domain(s). The linker sequences in iGlow are identical to the linker sequence of dLight1.2, a dopamine sensor where the linkers have been optimized through high-throughput mutational screening25. Hence, perhaps these specific linker sequences could confer a high fluorescence dynamic range to a broad range of host proteins. If true, these sequences could be used as a starting point to jump-start the design of new cpGFP-based indicators, in particular those using the sensory machinery of GPCRs.
To date, mechanosensitive GPCRs have been reported in at least one class-B GPCR (parathyroid hormone type 1 receptor)38 and many class-A subfamilies including A3 (bradykinin receptor B2, Apelin receptor and angiotensin II type 1 receptor)11,39,40, A6 (vasopressin receptor 1A)40, A13 (sphingosine receptor 1)41, A15 (GPR68)9,10, A17 (dopamine receptor DRD5)42 and A18 (muscarinic receptor M5R and histamine receptor H1R)24,40. While a physiological role in flow-induced vasodilation has been clearly demonstrated for GPR68 and H1R, the physiological importance of GPCR-mediated mechanotransduction signaling in non-vascular tissues remains unclear9,24. Helix 8 has been shown both necessary and sufficient to confer mechanosensitivity in certain class-A GPCRs such as H1R24. However, while H1R’s mechanosensitivity is fully uncoupled from histamine-induced activation24, GPR68’s mechanosensitivity is coupled to the concentration of extracellular protons. Indeed, mechanical stimuli are not sufficient to activate GPR68 at high extracellular pH9. This suggests mechanical stimuli acts as positive modulators rather than independent activators of GPR6810,22. These observations hint that mechanosensation by class-A GPCRs is not mediated by a unique molecular mechanism. The existence of distinct mechanosensory mechanisms among GPCRs is further supported by our data showing Helix 8 is not necessary for GPR68 mechanosensitivity. How does mechanical stress differentially activate GPCRs? Some GPCRs like H1R may use the amphipathic Helix 8 as a gauge to sense outward lipid displacement upon membrane stretch. In GPR68, in contrast, mechanical forces may modulate pH-dependent activation, perhaps by acting on the spatial orientation of pH-sensitive histidine residues, modulating their pKa20.
It is still unclear whether fluid shear stress is directly sensed by GPR68, via a physical effect of solvent molecules and/or membrane lipids, or indirectly via the action of other mechanosensitive cellular components. Certain G proteins that are physiologically recruited by GPR68, such as Gαq/11, have been reported to exhibit GPCR-independent mechanosensitivity43. However, broad range pharmacological disruption of G protein signaling did not attenuate mechanical activation of iGlow, suggesting G proteins are not involved in mediating mechanosensitivity of GPR68. In addition, our data show that neither the spider toxin GsMTx4 nor disruption of actin filaments were effective to attenuate flow-induced iGlow activation, in contrast to numerous mechanosensitive ion channels which show at least partial reduction of mechanosensitivity in response to these treatments33,37,44–50. Future studies will be needed to determine whether GPR68 acts as a bonda fide mechanotransducer or rely on other mechanotransducers to sense mechanical forces. To conclude, iGlow represents the first intensiometric indicator for a multimodal mechanosensitive GPCR. We anticipate this reporter will be compatible with in vivo studies to probe the biological roles of GPR68 in vascular and non-vascular physiology such as hippocampal plasticity.
Methods
Molecular cloning
A fragment containing the human GPR68 cDNA was obtained by digesting a pBFRT-GPR68 plasmid (a gift from Drs. Mikhail Shapiro, Caltech and Ardèm Patapoutian, Scripps Research) by NdeI and BamHI. The insert was ligated into an in-house pCDNA3.1-Lck-GCaMP6f plasmid linearized by the same enzymes, creating the plasmid pCDNA3.1-GPR68. A cpGFP cassette was amplified by PCR from a pCDNA3.1 plasmid encoding ASAP1 (a gift from Dr. Michael Lin, Stanford, available as Addgene #52519) and inserted into pCDNA3.1-GPR68 using the NEBuilder HiFi DNA Assembly kit (New England Biolabs). The pCNDA3.1-jRGECO1a plasmid was cloned by assembling PCR-amplified fragments from pGP-CMV-NES-jRGECO1a (Addgene # 61563, a gift from Dr. Douglas Kim51) and pCDNA3.1. All constructs were confirmed by Sanger sequencing (GENEWIZ). The pLifeAct-mScarlet-N1 plasmid was obtained from Addgene (#85054, a gift from Dr. Dorus Gadella52). All molecular biology reagents were purchased from New England Biolabs.
Cell culture, transfection and drug treatment
HEK293T cells were obtained from the American Tissue Culture Collection and ΔPZ1 cells were a gift from Ardèm Patapoutian (Scripps Research). Cells were cultured in standard conditions (37 °C, 5 % CO2) in a Dulbecco’s Modified Eagle’s Medium supplemented with Penicillin (100 U mL−1), streptomycin (0.1 mg mL−1), 10 % sterile Fetal Bovine Serum, 1X Minimum Essential Medium non-essential amino-acids and without L-glutamine. All cell culture products were purchased from Sigma-Aldrich. Plasmids were transfected in cells (passage number < 35) seeded in 96-well plates at ~50 % confluence 2-4 days before the experiment with FuGene6 (Promega) or Lipofectamine 2000 (Thermo Fisher Scientific) and following the manufacturer’s instructions. 1-2 days before experiments, cells were gently detached by 5 min incubation with Phosphate Buffer Saline and re-seeded onto 18 mm round glass coverslips (Warner Instruments) or onto disposable flow chambers (Ibidi μ-slides 0.8 or 0.4mm height), both coated with Matrigel (Corning). CMPD101 (#5642) and NF-449 (#1391) were purchased from R&D Systems (Biotechne), GTP-gamma-S was purchased from Cytoskeleton, Inc (#BS01), Dopamine (#H8502) and Gαq inhibitor BIM-46187 (#5332990001) were purchased from Sigma-Aldrich.
Fluorescence imaging
Excitation light of desired wavelengths were produced by a Light Emitting Diode light engine (Spectra X, Lumencor), cleaned through individual single-band excitation filters (Semrock) and sent to the illumination port of an inverted fluorescence microscope (IX73, Olympus) by a liquid guide light. Excitation light was reflected towards a plan super apochromatic 100X oil-immersion objective with 1.4 numerical aperture 1.4 (Olympus) using a triple-band dichroic mirror (FF403/497/574, Semrock). Emission light from the sample was filtered through a triple-band emission filter (FF01-433/517/613, Semrock) and sent through beam-splitting optics (W-View Gemini, Hamamatsu). Split and unsplit fluorescence images were collected by a sCMOS camera (Zyla 4.2, ANDOR, Oxford Instruments). Spectral separation by the Gemini was done using flat imaging dichroic mirrors and appropriate emission filters (Semrock). Images were collected by the Solis software (ANDOR, Oxford Instruments) at a rate of 1 frame s−1 through a 10-tap camera link computer interface. Image acquisition and sample illumination were synchronized using TTL triggers digitally generated by the Clampex software (Molecular Devices). To reduce light-induced bleaching, samples were only illuminated during frame acquisition (200 ms exposure). To reduce auto-fluorescence, the cell culture medium was replaced with phenol red-free HBSS approximately 20 min prior experiments.
Fluid shear stress stimulation and calculations
Fluid shear-stress stimulation was done by circulating extracellular physiological solutions at various speeds into a μ-slide channel (Ibidi) using a Clampex-controlled peristaltic pump (Golander). The average amplitude of wall shear stress τ applied at the cell surface was estimated using the manufacturer’s empirical equation relating τ with flow rate Φ for μ-slide channel with 0.4 mm height:

We independently measured τ using particle image velocimetry measurements. Briefly, we dispensed 6 μm diameter polystyrene beads (Polybead microspheres, Polysciences) diluted in HBSS into recording μ-slide channels and let them settle to the bottom of the μ-slide. Beads were imaged using a 100X immersion objective (Olympus) focused at the fluid-wall boundary. Bead velocity was estimated using high-speed imaging (300-500 frames s−1) for various flow rates (Supplementary Table 1). τ depends on the distance between the fluid and the boundary y, the dynamic viscosity of the fluid μ and the flow velocity u according to:

Since the beads are located very close to the boundary, we can assume they are within the viscous sublayer53. Hence, in these conditions, the fluid velocity profile is linear with the distance from the boundary:

Shear stress values were calculated using the experimentally measured u values (in m s−1) and using an averaged bead radius of 2.5 × 10−6 m. For μ, we measured the dynamic viscosity of HBSS at room temperature using a rotary viscometer (USS-DVT4, U.S. SOLID) and obtained a value of 1.04 ± 0.04 × 10−3 Pa s−1 (n = 3).
Statistical analyses
The number n represents the number of independent experiments. To evaluate pairwise differences of mean data sets, we performed Mann-Whitney U-tests when n ≤ 10 and Student’s T-tests when n > 10 in both data set. To compare difference of mean values between different treatments, we performed one-way ANOVA. All error bars are standard errors of the mean. All statistical tests were performed on OriginPro 2018.
Author contributions
J.J.L. conceived the project. A.D.O., T.G., A.T. and W.P. performed experiments. A.D.O., T.G. and J.J.L. analyzed data. J.J.L. wrote the manuscript with inputs from A.D.O. and T.G.
Supplementary Materials

Black: GPR68; bold purple: linkers and green: cpGFP.

Shear stress applied through our flow chamber was calculated using particle image velocimetry (see methods) or using the manufacturer’s calibration.
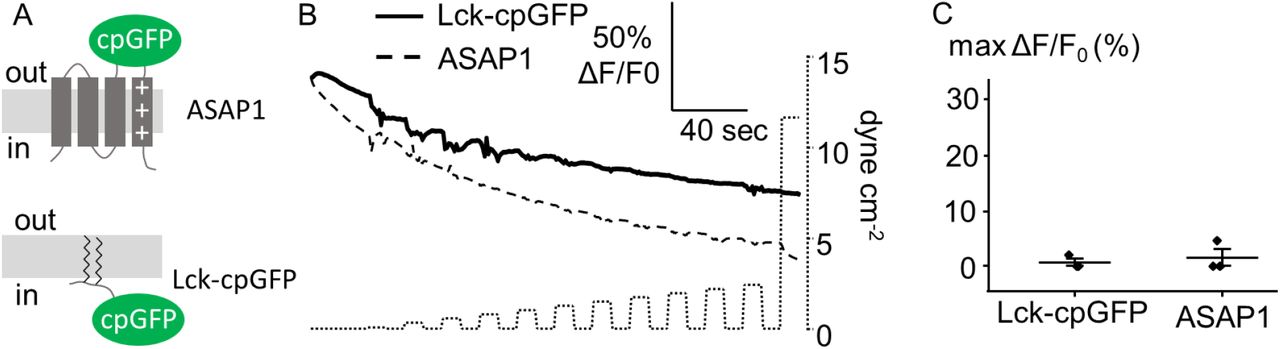
(A) Cartoons illustrating the position of cpGFP in the voltage indicator ASAP1 and Lck-cpGFP. (B) Example of fluorescence time-course (shown as ΔF/F0 vs. time) from a single cell expressing ASAP1 (dashed line) or Lck-cpGFP (solid line) in response to FSS pulses of incrementally increased amplitudes (dotted line). (C) Scatter plot showing the maximal ΔF/F0 values obtained with ASAP1 (n = 3) and Lck-cpGFP (n = 3) using the FSS protocol shown in (B).

The scatter plots show max ΔF/F0 values obtained upon acute perfusion with 10 μM dopamine and CMPD101 (purple dots, n = 13), BIM-46187 (BIM, green dots, n = 8), NF-449 (blue dots, n = 5), GTP-γ-S (red dots, n = 6), or a vehicle control (black dots, n = 20). The number above the graph indicates the exact p-value from a one-way ANOVA. Error bars = s.e.m.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Helical wheel plot of Helix 8 in the long isoform of the swine gonadotropin-releasing hormone receptor (ssGnRHR2), the guinea pig histamine H1 receptor (gpH1R) and human GPR68 (hGPR68). The dotted line indicates the separation between the polar vs. apolar interfaces of the Helix.
Acknowledgments
We thank Ardèm Patapoutian for the gift of the human GPR68 cDNA. This work was supported by intramural funds from Western University of Health Sciences (to J.J.L), federal work-study (to L.G) and NIH grants GM130834 and NS101384 (to J.J.L.).
References




