

Abstract
Cytotoxic necrotizing factors (CNFs) are single-chain exotoxins. They are secreted by several bacterial pathogens to modulate cytokinetic/oncogenic and inflammatory processes through activation host cell Rho-GTPases, but their secretion-translocation mechanism still remains an enigma. Here, we determined the crystal structure of full-length Yersinia pseudotuberculosis CNFY, revealing five separate domains (D1-D5) of which D1-D3 act as translocation module for the catalytic unit (D4-5) and for other fused reporter proteins. By integrating structural and functional data, we suggest a model in which the α-helical D1 domain constitutes a membrane-spanning translocation unit. This unit promotes bacterial export and exposes the host cell recognition sites of D2. Receptor binding then triggers endosomal uptake, release and structural reorientation of the catalytic unit implicating D3. Sequence comparison also suggests that this translocation mechanism is used by many other bacterial proteins and could be employed as universal drug delivery tool.
Introduction
Amongst the plethora of traits developed by pathogenic bacteria to establish infections, toxins play the most prominent role, since they are responsible for the majority of clinical symptoms including extensive tissue lesions during disease (Popoff, 2005). Many bacterial exotoxins are key virulence factors which target different functions of host cells to break barriers, improve excess to nutrients, defeat immune responses and promote bacterial dissemination to colonize, proliferate, persist and spread within and among hosts. Some of these toxins, including the family of cytotoxic necrotizing factors (CNFs), were shown to modulate the expression of inflammatory mediators that orchestrate innate immune responses and promote tissue damage, leading to the development of acute disease symptoms (Knust & Schmidt, 2010; Schweer et al, 2013; Diabate et al, 2015; Cavaillon, 2018; Heine et al, 2018).
The CNFs belong to a class of bacterial exotoxins which enter host cells via receptor-mediated endocytosis and deaminate a glutamine (Q61 or Q63) in the active domain (switch II region) of proteins belonging to the small Rho GTPase family, i.e. RhoA, Rac1 and Cdc42 (Flatau et al, 1997; Schmidt et al, 1997; Knust & Schmidt, 2010). This locks these key regulators in their active states, causing a plethora of downstream effects that are most readily observed as alterations of the actin cytoskeleton or perturbations of other cellular processes including phagocytosis, cell migration/proliferation (multinucleation), reactive oxygen species production, and the release of pro-inflammatory and anti-apoptotic factors (Fabbri et al, 2013; Hodge & Ridley, 2016; Ho et al, 2018). CNFs are found in several pathogenic bacteria, predominantly in pathogenic Escherichia coli, but also in Y. pseudotuberculosis, Shigella species, Salmonella enterica, as well as in Moritella viscosa and Photobacterium damselae, pathogens of economically important fish (Appendix Fig S1) (Morgan et al, 2019). In addition, CNF-related toxins such as the dermonecrotizing toxin DNT of Bordetella pertussis and the PMT toxin of Pasteurella multocida have been identified (Sugai et al, 1999; Boquet, 2001).
CNF1, the most thoroughly investigated representative of the CNF family, is a major virulence factor in uropathogenic E. coli (UPEC) strains, which live in the intestine and enter the urinary tract via the uretha (Boquet, 2001; Knust & Schmidt, 2010; Ho et al, 2018). CNF1-containing strains exhibit a higher viability, have a higher potential to colonize the urinary tract, affect the function of immune cells and increase the inflammation rate (Falzano et al, 1993; Fournout et al, 2000; Rippere-Lampe et al, 2001). CNF1 was also identified in some intestinal and extraintestinal E. coli (ExPEC) and found to increase invasion into endothelial cells (Khan et al, 2002). Moreover, CNF1 was recently shown to favor malignant tumor conversion and invasiveness by inducing epithelial to mesenchymal transition in intestinal epithelial cells as well as to provoke reversible senescence of human colon cancer cells (Zhang et al, 2018; Fabbri et al, 2019). Similarly, the homologous toxin CNFY, which shares 65% identity with E. coli CNF1, is crucial for the pathogenicity of Yersinia pseudotuberculosis, which causes food-borne and zoonotic enteric infections that manifest themselves as enteritis, mesenterial lymphadenitis and more rarely, in sequelae such as reactive arthritis (Koornhof et al, 1999a; Smego et al, 1999; Heine et al, 2018). The importance of CNFY is emphasized by the fact that a knock-out mutation of the cnfY gene leads to avirulence, allowing bacteria to become persistent in mice (Heine et al, 2018). Recent studies demonstrated that Rho-GTPase activation by CNFY enhances the translocation of Yersinia outer proteins (Yops) into neutrophils and macrophages via a type III secretion system (T3SS). This blocks phagocytosis, triggers immune cell death and contributes to massive tissue damage by induction of pro-inflammatory responses and necrosis (Schweer et al, 2013; Wolters et al, 2013).
CNFs may also hold promise for human treatment (Maroccia et al, 2018). For example, it has been demonstrated that injection of CNF1 into the brains of mice can enhance neurotransmission and synaptic plasticity, leading to improved learning and memory function (Diana et al, 2007). Moreover, CNF1 is able to rescue wildtype-like mitochondrial morphology in fibroblasts derived from patients with myoclonic epilepsy accompanied by ragged-red fibers, and it reduced tumor growth and spared neuron structure as well as function (Vannini et al, 2016; Fabbri et al, 2018). CNFs may therefore be of use in the treatment of neurological disorders and cancer. As they are efficiently delivered into a broad range of host cells, transport modules of the toxin may also be useful for drug delivery (Haywood et al, 2018). To exploit and further develop this tool, detailed knowledge of the molecular mechanisms underlying CNF secretion, translocation and activity is required. However, little is known about the global structure and the individual functional units of CNFs and so far, only the structure of the catalytic domain of CNF1 has been determined (Buetow et al, 2002).
At the sequence level, CNF-type toxins of different species share at least 55% overall identity (Appendix Fig S1), indicating similar structures and conserved modes-of-action, although they show differential preferences with respect to the targeted Rho-GTPase and interact with different host cell receptors (Hoffmann et al, 2004; Blumenthal et al, 2007). CNF1 uses two cellular receptors to enter host cells, the 37-kDa laminin receptor precursor p37LRP, which is recognized by sequences located within the N-terminus of the toxin, and the Lutheran adhesion glycoprotein/basal cell adhesion molecule (Lu/BCAM), which interacts with motifs in the C-terminal half (Fabbri et al, 1999; Chung et al, 2003; Kim et al, 2005; McNichol et al, 2007; Piteau et al, 2014; Reppin et al, 2017). The receptor(s) of CNFY are still unknown, but it has been shown that binding of CNF1 to host cells has no effect on CNFY uptake (Blumenthal et al, 2007). The CNFs are taken up into endosomes and their release into the host cytoplasm requires two predicted α-helices in their N-terminal half. These helices are separated by a loop containing an acidic patch of four conserved acidic amino acids, and they are believed to insert into the endosomal membrane upon charge neutralization in the course of endosome acidification. An unidentified protease then cleaves CNF (i.e. CNF1 between residues 532 and 544), and the C-terminal fragment including the catalytic domain (residues 720-1014) is released into the cytosol of the host cell to mediate the cellular effects of the toxin (Pei et al, 2001; Knust et al, 2009).
In this study, we resolved the crystal structure of the full-length Y. pseudotuberculosis CNFY protein, necessary to achieve an understanding of its transport and functional mechanisms and its potential therapeutic use. The CNFY structure revealed a complex set-up of individual functional building blocks and allowed us to obtain detailed information about the minimal bacterial secretion and translocation domain required to transport the catalytic domain or other fusion partners into the host cell cytosol, which could be exploited for drug delivery.
Results
CNFY contains five structural building blocks and displays novel protein folds
Recombinant full-length CNFY was produced in E. coli and crystallized in space group I212121. These crystals diffracted to 2.7 Å and contained one CNFY molecule in the asymmetric unit. Since no suitable search model for molecular replacement was available and crystallization of full-length seleno-L-methionine-labelled protein failed, we also crystallized different fragments of CNFY: (i) one containing the deamidase domain (residues 720-1014), (ii) another consisting of the subunit which is likely released into the cytosol (residues 526-1014) based on the homology to E. coli CNF1 (Hoffmann et al, 2004; Blumenthal et al, 2007), and (iii) a third fragment including the complete N-terminal portion with parts of the released subunit (residues 1-704). A detailed description of structure determination by Se-SAD and molecular replacement is given in the Materials and Methods section and an overview over data collection and refinement statistics as well as the respective Protein Data Bank (Berman et al, 2000) deposition codes is provided in Appendix Tab S1.
CNFY adopts a compact, modular structure of five structural building blocks (D1-D5) with approximate dimensions of 115*73*65 Å (Fig 1A-C). All residues of the protein could be traced in the structure of the holo-protein with the exception of residues N430-K431, S550-L553 and P701-L717. The unresolved amino acids resided in surface loops, indicating intrinsic flexibility. The fragment comprising residues 1-704 (D1-D4) is fully superimposable with the respective residues of the holo-protein, whereas the structure of the isolated D4-D5 fragment showed a different orientation of the two domains with respect to the full-length protein, which likely is linked to the postulated flexibility of the P701-L717 region (Fig 2). The domain organization of CNFY is also corroborated by computational analysis with PiSQRD (Aleksiev et al, 2009) which assigns domain boundaries to residues 1-22/135-424 (D1), 23-134 (D2), 425-529 (D3), 530-700 (D4) and 718-1014 (D5, deamidase domain).

A Domain boundaries and sequence motifs mapped to the sequence of CNFY.
B Cartoon representation of CNFY, colored according to domain boundaries determined with PiSQRD (Aleksiev et al, 2009). Dark blue: domain D1, cyan: domain D2, dark green: domain D3, yellow: ADP-ribosyltransferase-like domain D4, pink: deamidase domain D5. Other colors indicate the position of sequence motifs that have been identified in E. coli CNF1, namely light blue: p37LRP/67LR receptor-binding motif, red: hydrophobic stretches predicted to form membrane-inserting α-helices, orange: cleavage site, magenta: main Lu/BCAM receptor-binding motif. The positions of N- and C-terminus are indicated by N and C, respectively.
C Surface representation of CNFY as seen from two different orientations with respect to B. Note that the cleavage site between D3 and D4 (orange) as well as the deamidase active site in D5 are partially blocked in the structure of full-length CNFY. The C-terminal domain D5 interacts mainly with D3 (607 Å2), which partially hides the catalytic site of D5, but it interacts only weakly with D4 (382 Å2), which itself establishes an extensive interface with D1 (1376 Å2) by mainly hydrophilic interactions (17 hydrogen bonds and 6 salt bridges).

A Crystal structure of the free D4-5 subunit. Note the different relative orientations of domains D4 and D5 with respect to the structure of full-length CNFY (thin grey lines). The domain D4 forms a large interface area (1097 Å2) with the catalytic domain D5 involving several polar interactions (8 hydrogen bonds and 8 salt bridges), whereby the active crevice is extended and fully solvent-exposed.
B Surface representation of the free D4-5 subunit as seen from two different orientations. Note that the deamidase active site of domain D5, unlike in the full-length structure (Fig 1), is fully accessible and that its extended shape is also determined by domain D4.
Domain D1 (residues 1-22/135-424) forms a bundle of α-helices flanked by a four-stranded anti-parallel β-sheet that is covered with three α-helices from the other side (Fig 1B). It contains elements that are required for translocation of the catalytic fragment of E. coli CNF1, suggesting that it is a major component of the translocation machinery of CNFs. For E. coli CNF1, two hydrophobic α-helices have been predicted in residues 350-372 and 387-412, which are believed to insert into the endosomal membrane after charge neutralization of a conserved acidic patch in the connecting loop (D373, D379, E382 and E383) (Pei et al, 2001). However, the respective segments do not fold into the predicted α-helices in CNFY but adopt mostly loop-like structures with a helical part at their C-terminus (Fig 1B).
Although the D1 domain of CNFY is, due to its overall α-helical character, reminiscent of the translocation domain of other toxins such as diphtheria toxin (DT), searches with DALI (Holm & Rosenström, 2010) detected no significant structural homology to these proteins. Instead, it identified only the segment containing the four-stranded anti-parallel β-sheet (residues 152-343) as being somewhat similar to a fragment of the translocation domain of nigritoxin, a toxin of crustaceans and insects (PDB entry 5M41; 177 residues aligned, rmsd 3.8 Å, 11% sequence identity) (Fig 3A) (Labreuche et al, 2017). However, the translocation domain of nigritoxin is significantly smaller than the D1 domain of CNFY and does not contain hydrophobic sequence motifs, hinting at distinct translocation mechanisms.

A Side-by-side comparison of CNFy and nigritoxin. Nigritoxin is a toxin of crustaceans and insects. The translocation domain of nigritoxin (PDB entry 5M41, (Labreuche et al, 2017)) and domain D1 of CNFY show partial structural similarity (highlighted areas). This similarity was identified with DALI (Holm & Rosenström, 2010) which was also used to align both structures.
B The ART-like domain D4 of CNFY. Essential residues of canonical ARTs are not conserved in CNFY (RSE-ARTs exemplified by C. perfringens iota toxin, PDB entry 4H03 (Tsurumura et al, 2013); HYE-ARTs exemplified by P. aeruginosa ExoA, PDB entry 2ZIT (Jørgensen et al, 2008); carbon atoms of NAD+ shown in black).
C The released fragment of Pasteurella multocida toxin PMT contains three domains of which C1 is required for membrane binding, the C2 domain has an unknown function and the C3-domain activates heterotrimeric G-proteins by deamidation. The two Pseudomonas syringiae proteins A0A0P9UH04 and A0A0N8SZE6 represent two uncharacterized toxins that encode catalytic domains of the indicated type. While sequence alignments unequivocally reveal a CNF-like imperfect β-barrel in PMT, the presence of this domain in the P. syringiae toxins is less obvious.
Unlike the N-terminal α-helix (residues 5-18), which is an integral part of the helical bundle that dominates domain D1, residues 23-134 seem to establish a separate structural building block (domain D2) that protrudes from the mostly α-helical subunit, potentially suggesting its insertion during evolution (Fig 1B). It consists of a three-stranded anti-parallel β-sheet flanked by α-helices at the side facing D1 and by several surface-exposed loops at the other. One loop contains residues 53-75, a segment that has previously been implicated in host cell binding of E. coli CNF1 to receptor p37LRP/LR67 (Fig 1B) (Fabbri et al, 1999; Chung et al, 2003; Kim et al, 2005). It is hence conceivable that this domain represents a receptor binding domain of CNFs. There are five amino acid changes between CNF1 and CNFY within this loop region, which may account for the distinct receptor specificity observed for CNFs (Blumenthal et al, 2007).
The third domain, D3 (residues 425-529), is reminiscent of an incomplete β-barrel, containing six anti-parallel strands in CNFY. No homologous structures could be discovered with DALI. The imperfect β-barrel domain D3 and the following domain D4 are connected via a linker that is partially shielded by the C-terminal deamidase domain D5 in the structure of full-length CNFY (Fig 1C). In CNF1, this linker is cleaved between residues 532-544 to release the D4-5 subunit into the cytosol of the host cell (Knust et al, 2009), suggesting that the respective segment must become solvent-exposed in the course of host cell intoxication to become accessible to proteases.
Sequence analysis places the fourth domain D4 (residues 530-700) into the DUF4765 family, a building block that is also found in a number of other uncharacterized bacterial proteins. Surprisingly, structure similarity searches reveal distant but significant homology to ADP-ribosyl transferase (ART) domains (Appendix Tab S2), which are widespread in protein toxins (Fieldhouse & Merrill, 2008). This similarity is exemplified by two examples shown in Fig 3B and will be discussed below.
The C-terminal catalytic deamidase domain D5 is linked to D4 via the unstructured residues P701-L717 (Fig 1B). These belong to the postulated binding epitope for the Lu/-BCAM host receptor of CNF1 (Piteau et al, 2014), and their flexibility may be a requirement for receptor binding by the toxin. The D5 domain is very similar to the respective domain of E. coli CNF1 (PDB entry 1HQ0; (Buetow et al, 2001); 1.8 Å rmsd over 295 residues, 59% sequence identity), featuring a central β-sandwich with shielding α-helices on both sides (Figs 1B and 2). The active site employs a conserved cysteine/histidine couple (C866/H881; (Hoffmann et al, 2004)) that lies in a crevice on the surface of the domain.
Rearrangements of subunits D4-5 after cleavage from full-length CNFY
The compact arrangement of D1-D5 in the full-length structure of CNFY prompted us to investigate the interactions between the five individual domains of CNFY in more detail. Analysis with PISA (Krissinel & Henrick, 2007) reveals large hydrophobic interfaces between D1 and D2 (interface area 872 Å2) as well as between D1 and D3 (744 Å2) (Appendix Tab S3). The C-terminal domain D5 interacts mainly with D3 (607 Å2) partially hiding the catalytic site, but it interacts only weakly with D4 (382 Å2), which itself establishes an extensive interface with D1 (1376 Å2) (Fig 1C).
In contrast to the full-length protein, domain D4 forms a large interface (1097 Å2) with the catalytic domain D5 in the isolated D4-5 fragment, whereby the active site crevice of D5 is extended by D4 and becomes fully solvent-exposed (Fig 2). To reach this position, domain D4 has to rotate over 140°, which can probably only be achieved after cleavage from D1-3 through the flexibility of the linker connecting both domains. The contact area between both domains in the free D4-5 subunit overlaps largely with that of D3-5 in the full-length structures such that both conformations are mutually exclusive, i.e. the D4-5 subunit cannot adopt the conformation observed in the free state when it is bound to D1-3.
Structure-guided mutagenesis provides insights into the function of the structural building blocks of CNFY
In order to gain insights into the biological function of the individual building blocks, we constructed truncated or mutated marker-tagged versions of CNFY and investigated their secretion, translocation and enzymatic activity in human epithelial cells. The ability of the CNFY variants to activate GTPases was tested by (i) deamination of RhoA, resulting in a slower gel migration behavior, (ii) induction of actin rearrangements and the inhibition of cell division (formation of multinuclear cells), and (iii) the ability of CNFY to translocate into the cytosol of host cells measured by translocation of a CNFY-β-lactamase (TEM) fusion constructs using the FRET substrate CCF4-AM.
We first investigated C-terminally truncated 3xFlag-tagged or TEM-tagged forms of CNFY missing one or more domains of the toxin. These were tested either using cleared bacterial lysates of Y. pseudotuberculosis or as purified recombinant proteins produced in E. coli. The truncated forms of CNFY were all detected at similar levels by western blotting, showing that the results were not due to protein instability or lack of detection (Fig 4A). All C-terminally deleted CNFY derivatives were efficiently secreted and bound to host cells (Figs 4E and F). Moreover, all truncated toxin variants harboring N-terminal domains D1-3 were able to translocate and transport cargo proteins such as the β-lactamase TEM into the host cell cytosol (Fig 4D), demonstrating that D1-3 forms the secretion and membrane translocation unit. Strikingly, only the full length CNFY protein was able to activate Rho-GTPase RhoA (Fig 4B), resulting in the induction of polynucleation in living cells (Fig 4C), whereas all protein variants with deletions of the C-terminal 720-1014 aa domain D5 or the replacement of the cysteine residue C866 with serine in the active site eliminated toxicity (Figs 4B and C). This is consistent with studies showing that the C-terminal 300 amino acids (709-1014) of the related E. coli CNF1 protein, including the catalytically active residues C866 and H881, are important for its activity (Koornhof et al, 1999b; Zhang et al, 2018; Fabbri et al, 2019). Neither the deletion nor site-directed mutagenesis of the catalytic domain affected secretion, host-cell binding, or protein translocation (Figs 4D-F), indicating that the sole role of the C-terminal domain is the targeting and modification of Rho-GTPases.

A C-terminally 3xFlag-tagged CNFY and different C-terminally deleted toxin variants were expressed in Y. pseudotuberculosis YP147 (ΔcnfY) from plasmids under control of their own promoter and were detected in whole cell extracts using an anti-Flag antibody.
B HEp-2 cells remained untreated or were incubated with full-length CNFY or the C-terminally deleted toxin variants for 4 h. Cells were lysed and the deamidation of RhoA was analyzed by the mobility shift of the modified Rho GTPase on SDS PAGE.
C HEp-2 cells were incubated with 500 nM full-length CNFY or the C-terminal deleted toxin variants for 24 h. The cell nuclei were strained with DAPI (blue) and the actin cytoskeleton was stained using FITC-phalloidin (green). The formation of large, multinuclear cells was observed by fluorescence microscopy and the formation of actin stress fibers and membrane actin folding were only observed with full-length-CNFY-treated cells. The white scale bar is 40 µm. Cells incubated with extracts of YP147 (ΔcnfY) harboring the empty expression vector were used as negative controls.
D HEp-2 cells were incubated with 500 nM full-length CNFY or the C-terminally deleted toxin variants fused to beta-lactamase (TEM) for 4 h. Cleavage of the reporter dye CCF4-AM was used to visualize toxin delivery. After cell entry CCF4-AM is rapidly converted into the negatively charged form CCF4, which is retained in the cytosol and emits a green fluorescence signal (520 nm). In the presence of translocated beta-lactamase fusion proteins, CCF4-AM is cleaved, and disruption of FRET results in blue fluorescence (447 nm). White bar: 20 µm.
E 3xFlag-tagged CNF1, CNFY and C-terminally deleted toxin variants were added to HEp-2 cells for 4 h. The cells were thoroughly washed, pelleted, lysed and the toxin variants bound to the cells were identified by western blotting using an anti-Flag antibody.
F To test secretion of the CNFY variants, CNFY and different C-terminally deleted variants fused to beta-lactamase (TEM) were expressed in Y. pseudotuberculosis YP147 (ΔcnfY). Beta-lactamase activity in the culture supernatant was subsequently measured using nitrocefin as substrate.
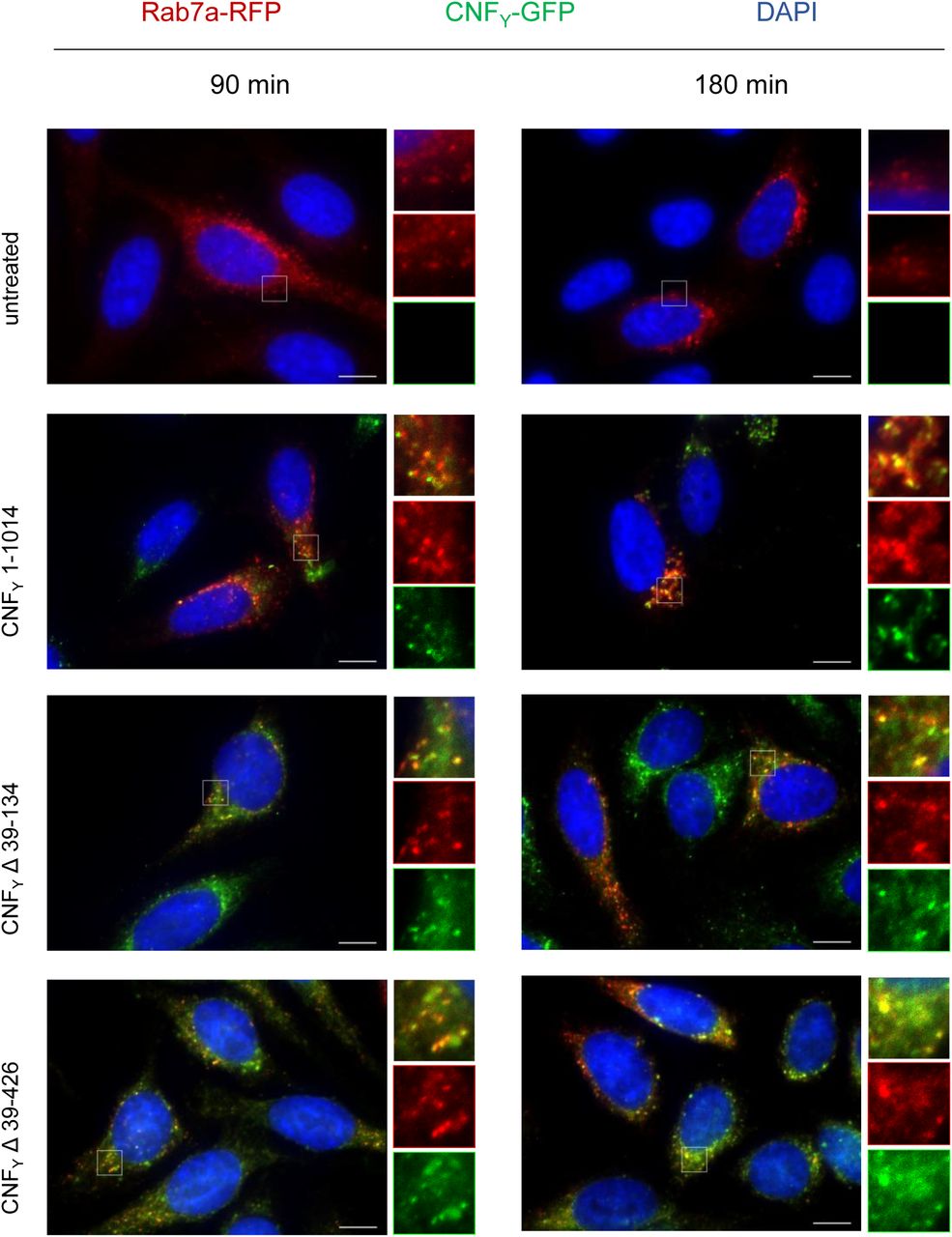
We next investigated Y. pseudotuberculosis strains expressing deletions of the N-terminal domains D1 and/or D2 (Δ39-134, Δ134-426, Δ39-426) of CNFY (Figs 5A and B). CNFY Δ39-134 and CNFY Δ39-426 were efficiently expressed and enzymatically active, whereas the obtained amount of CNFY Δ134-426 was low. This derivative was unable to deamidate RhoA in host cell extracts (Fig 5D), indicating that it is improperly folded and less stable. All N-terminally deleted proteins failed to be secreted (Fig 5C) and were thus unable to trigger RhoA activation and multinucleaction in host cells (Figs 5D and F). The C-terminally truncated CNFY protein, containing only the first two sub-domains D1-2 (1-443), on the other hand, was efficiently secreted (Fig 4F), corroborating that both domains are necessary for CNFY to exit the bacterial cell. Binding and colocalization studies further showed that the D1-2 (1-443) fragment was able to bind host cells (Fig 4E) and reached the early and late endosomes (Fig 6, Appendix Fig S2), indicating that this truncated version of CNFY includes a cell receptor binding site in agreement with data obtained with CNF1 (Fabbri et al, 1999; Chung et al, 2003; Kim et al, 2005). However, this also demon-strates that CNFY, unlike previous work with CNF1 suggests (Piteau et al, 2014; Reppin et al, 2017), does not require a second recognition site in the C-terminal region to enter host cells.

A Schematic overview of the N-terminal CNFY deletion variants.
B C-terminally 3xFlag-tagged CNFY and different N-terminally deleted toxin variants were expressed in Y. pseudotuberculosis YP147 (ΔcnfY) from plasmids under the control of their own promoter and were detected in whole cell extracts using an anti-Flag antibody.
C To test secretion of the CNFY variants, CNFY and different N-terminally deleted variants fused to β-lactamase (TEM) were expressed in Y. pseudotuberculosis YP147 (ΔcnfY). Beta-lactamase activity in the culture supernatant was subsequently measured using nitrocefin as substrate.
D Upper panel: HEp-2 cells remained untreated or were incubated with full-length CNFY or the N-terminally deleted toxin variants for 4 h. Cells were lysed and the deamidation of RhoA was analyzed by the shift of the modified Rho GTPase band in SDS PAGE gels; lower panel: HEp-2 cells were lysed and the cell extracts were incubated with full-length CNFY or the N-terminally deleted toxin variants for 4 h. The deamidation of RhoA in the cell extracts was analyzed by the mobility shift of the modified Rho GTPase on SDS PAGE.
E 3xFlag-tagged CNFY and N-terminally deleted toxin variants were added to HEp-2 cells for 4 h. The cells were intensively washed, pelleted, lysed and the toxin variants bound to the cells were identified by western blotting using an anti-Flag antibody.
F HEp-2 cells were incubated with 500 nM full-length CNFY or the N-terminally deleted toxin variants for 24 h. The cell nuclei were strained with DAPI (blue) and the actin cytoskeleton was stained using FITC-phalloidin (green). The formation of large, multinuclear cells was observed by fluorescence microscopy and the formation of thick actin stress fibers and membrane actin folding were only observed with CNFY-treated cells. The white scale bar is 40 µm.

HEp-2 cells were intoxicated with 500 nM CNFY 1-1014-GFP, CNFY 1-719-GFP, CNFY 1-526-GFP or CNFY 1-443-GFP (green) for 90 or 180 min. Cells were fixed and processed for fluorescence microscopy. The red fluorescent signal represents early endosomes (CellLight Late Endosomes-RFP (Rab7a)). Nuclei were stained with DAPI (blue). A merged image of the different channels is shown, and insets are magnified views of boxed areas. White scale bar is 10 µm.
Site-directed mutagenesis of the two acidic residues E382 and E383 in the acidic patch between the hydrophobic stretches of the D2 domain (Appendix Fig S3A), which is important for CNF1 host cell binding (McNichol et al, 2007) still allowed secretion and host cell entry of this CNFY derivative, but it failed to induce multinucleation (Appendix Figs S3B, C and F). The E382/383K exchange also abolished RhoA activation in living HEp-2 host cells, but not when added to cell lysates (Appendix Figs S3D and E). This indicated that these residues are important for the association with endosomes.
Notably, translocation and delivery of the β-lactamase TEM into the host cell cytoplasm was achieved when the D1-2 fragment was extended by the third domain (D3) comprising the imperfect β-barrel domain (Fig 4D), demonstrating that this subunit is an essential part of the translocation apparatus. Interestingly, the N-terminus of Pasteurella multocida exotoxin PMT possesses high homology to the first three domains of CNFY (Fig 3C) and a hybrid toxin consisting of this N-terminal fragment (residues 1-505) of PMT and the ADP-ribosylating domain of DT was able to intoxicate cells (Bergmann et al, 2013), indicating a conserved translocation module consisting of domains D1-3.
In order to identify amino acids that are important for the toxin cleavage upon trans-location, we introduced mutations within the linker region including the potential cleavage site between D3 and D4. All recombinant mutant proteins were able to bind to host cells and deaminate RhoA in cell lysates, indicating proper folding and full activity (Appendix Fig S4). Interestingly, the CNFY I535L/P536A/V537G mutant promoted translocation into the host cell cytoplasm and was able to deaminate RhoA when added to living cells (Appendix Fig S4). However, this was not the case for the CNFY variant I535L/-P536A/V537G/F539L/D541A/K542A (Appendix Fig S4), suggesting that this variant is unable to escape the endosome because it might not be cleaved.
Identical assays were used to characterize the properties of the recombinant D4-5 domains (residues 526-1014) constituting the C-terminal fragment that is translocated into the host cell cytoplasm after cleavage of CNFY. Although catalytically active when added to host cell extracts (Appendix Fig S5E), D4-5 is unable to deamidate RhoA when given onto intact host cells (Appendix Fig S5D). This suggested that it was either not taken up into the host cell or unable to escape the endosome to reach the cytoplasm. Interestingly, cell binding assays demonstrated that the D4-5 fragment specifically interacts with host cells (Appendix Fig S5C), indicating a second host cell binding site in the C-terminal region. This assumption is strongly supported by the fact that N-terminal deletions missing parts of D1-2 are still able to promote cell binding and endosomal uptake as indicated by colocalization studies (Figs 5E, 7 and S6). In E. coli CNF1, the binding site for the Lu/BCAM receptor was shown to include amino acids 720-730 of the catalytic domain (Reppin et al, 2017). Thus, although the host cell receptors for the toxin are still unknown, CNFY, similar to CNF1, seems to contain two distinct host cell binding sites, one each at the N- and C-terminus. However, unlike CNF1 (Piteau et al, 2014; Reppin et al, 2017), they are not both required to enter host cells. Presence of two receptor binding sites might broaden the range of targeted cells or may increase host cell binding affinity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HEp-2 cells were intoxicated with 500 nM CNFY 1-1014-GFP, CNFY Δ39-134-GFP or CNFY Δ39-426-GFP green) for 90 or 180 min. Cells were fixed and processed for fluorescence microscopy. The red fluorescent signal represents late endosomes (CellLight Late Endosomes-RFP (Rab7a)). Nuclei were stained with DAPI (blue). A merged image of the different channels is shown, and insets are magnified views of boxed areas. White scale bar is 10 µm.
In order to further characterize the function of D4, which is released together with the catalytic domain, we analyzed two mutant variants harboring internal deletions of amino acids 527-719 or 527-699. However, both protein variants did not deamidate RhoA in vitro, indicating that these mutant proteins failed to fold properly (Appendix Fig S7). Since structure similarity searches revealed distant homology to the ADP-ribosyltransferase (ART) domains (Fig 3B), we hypothesized that CNFs may possess a second, previously unrecognized enzymatic function encoded in D4. The active sites of ARTs fall into two groups, the RSE-ARTs, containing a conserved arginine-serine-glutamate active site motif, and the HYE-ARTs, using a histidine-tyrosine-glutamate triad (Cohen & Chang, 2018). The ART-like domain D4 contains arginine, glutamate and histidine at the respective positions instead (R599, E639, H676), which could, in principle, support similar chemistry (Fig 3B). However, changes of CNFY E639, which is in the comparable position of the conserved glutamate of bacterial ARTs, to alanine or glutamine had no effect on CNFY function (Appendix Fig S8).
Discussion
Here we show that CNFY and related CNFs consist of five individual structural building blocks that enable the different steps of the intoxication process, namely secretion, cell attachment, entry, translocation and enzymatic activity. The three N-terminal domains D1-3 all possess novel folds and constitute the secretion and membrane translocation apparatus, whereas the two C-terminal domains D4-5 form the toxicity-mediating unit of the toxin. We further show that the D1-3 unit is sufficient to transport cargo proteins such as β-lactamase into the cytosol of host cells. Strikingly, both the Rho deamidation and β-lactamase activity were preserved when the reporter was fused to the C-terminal end of the full-length protein (Fig 4D), indicating that the secretion and transport module of the CNFY protein is very robust and insensitive to C-terminal extensions, making it an attractive tool for drug delivery.
The molecular mechanisms by which domains D1-3 promote the delivery of the cargo from the bacterial to the host cell cytosol are still unknown. However, considering the compact arrangements of the modules with large hydrophobic interfaces between several domains, it is likely that the full-length toxin is secreted from the bacterial cell and endocytosed by the host cells as monolithic compact structure. In fact, the CNFY toxin has recently been identified on the surface of outer membrane vesicles (OMVs) isolated from Y. pseudotuberculosis culture supernatants (Monappa et al. 2018). While this could indicate that the toxin might be predominantly delivered into endosomes by OMVs, data obtained in this study further show that also the purified CNFY toxin interacts with and is efficiently internalized into host cells on its own. This suggests that the toxin is also directly secreted by the bacterial cell and/or exposed on the OMVs to promote contact with target cells.
The data presented here further show that the cellular toxin uptake process not only requires segments identified for receptor binding in D2 and for translocation in D1, but also the imperfect β-barrel domain D3. It is interesting to note that D1 is, due to its mostly α-helical character, reminiscent of the translocation machinery of other toxins including that of the diphtheria toxin DT. DT, CNFs and several other AB-type toxins contain two hydrophobic stretches that are believed to fold into α-helices and insert into the endosomal membrane after charge neutralization of surrounding acidic residues (Pei et al, 2001; Orrell et al, 2017). In CNFY, the respective residues 350 – 372 and 387 – 412 were not found in the predicted α-helical structure (Fig 1B). However, since the crystal structures presented here have been obtained at neutral pH, it is conceivable that this region undergoes refolding during endosomal acidification. Work with DT suggests that the catalytic subunit of this toxin is unfolded in the translocation process (Murphy, 2011). The fact that (i) translocation in CNFs also involves two hydrophobic motifs interrupted by acidic residues and (ii) the observation that a sequence that gets cleaved to release the catalytic unit (D4-5) of CNFY (residues 532 to 544) is not accessible in the full-length structure (Fig 1C) may hint at a similar unfolding in the CNFs. In this respect, the similarity of parts of the CNFY’s D1 domain to the putative translocation domain of nigritroxin (Fig 3A) (Labreuche et al, 2017) is interesting, because the translocation domain of this toxin does not contain hydrophobic α-helices. This could indicate that the translocation process occurs through several steps that involve different parts of the translocation machinery, most of which are not shared between the CNFs and nigritoxin.
Sequence searches in the UniRef50 database (Suzek et al, 2015) revealed that large sections of the D1-3 domain of CNFY are also found in a number of un- or less characterized bacterial proteins, suggesting that these proteins are toxins that apparently utilize a similar secretion and translocation device for their catalytic domains (Fig 3C). For example, about 530 amino acids of the N-terminus of CNFY share between 30-50% sequence identity to the N-terminus of Pasteurella multocida toxin PMT (Bergmann et al, 2013). Moreover, members of a group consisting of 372 proteins with approximately 900 residues each were found to possess a canonical RSE-type ART-domain at their C-terminus (represented by UniProt entry A0A0P9UH04 from Pseudomonas syringae pv. maculicola) in addition to a CNF-like translocation apparatus. A second group of 206 proteins with more than 1000 residues contains a predicted C-terminal glycosyltransferase (represented by UniProt entry A0A0N8SZE6 from Pseudomonas syringae pv. syringae). Since the C-termini of these proteins differ from CNFY, it is likely that the toxins consist of individual modules that have been shuffled in the course of evolution. This aligns with a recent analysis of the distribution of CNF-like deamidase domains, which are also found at different positions within the sequence of other toxins or as stand-alone proteins (Cruz-Migoni et al, 2011; Ho et al, 2018).
The finding that the DUF4765 domain D4 shows similarity to ADP-ribosyltransferases was surprising and led us to investigate if the released D4-5 may possess an additional and previously unrecognized enzyme activity that may contribute to the toxicity of CNFs. However, the observation that CNFY toxicity strictly depended on the activity of the deamidase D5 domain, whereas mutations of the potential/suggested NAD+ binding domain within D4 had no effect (Appendix Fig S8), speaks against such an additional activity. Alternatively, the grossly different and mutually exclusive relative orientations of the D4 and D5 domains in the free D4-5 subunit with respect to the full-length CNFY structure (Fig 2) could suggest that D4 may have a regulatory role. On the one hand, the finding that the active site of D5 becomes solvent-accessible and that the crevice leading to this site becomes extended by parts of D4 (Fig 2B) could fine-tune the deamidase function of D5 with respect to general activity levels or substrate specificity towards RhoA, Rac1 or Cdc42. On the other hand, D4 could contribute to localizing the catalytical unit within the host cell by promoting access to membrane-associated Rho GTPases. In fact, CNFs act predominantly on Rho GTPases bound to GTP, a form essentially found at the cytoplasmic face of the host cell membrane (Boquet, 2001). Clearly, the importance of D4 merits future studies.
In summary, the data presented here provide insight into the full-length and released active D4-5 structure, and they illustrate the importance of the individual building blocks of CNFs and related exotoxins. This not only forms the basis for the detailed analysis of the molecular secretion and transport mechanism, but also enables the rational design of the transport module as a toxin-based cargo delivery tool for cytosolic drug/therapeutics delivery and the structure-guided development of inhibitors of CNF-like virulence factors.
Materials and Methods
Bacterial strains, cell lines, plasmids and growth conditions
All bacterial strains and plasmids used in this study are listed in Appendix Tab S4. All oligonucleotide primers used for cloning are listed in Appendix Tab S5. E. coli strains were grown in Luria-Bertani (LB; Becton Dickinson) broth at 37°C. Yersinia strains were aerobically grown in LB at 25°C or 37°C. Other media used for bacterial growth were brain-heart infusion broth (BHI) (Gibco) and Double Yeast Tryptone medium (DYT) (Gibco). Cultures were supplemented with 30 μg/ml kanamycin (Kan) or chloramphenicol (Cm) where necessary. HEp-2 cells (ATCC CCL-23) were grown at 37°C, 5% CO2 in RPMI (Gibco) supplemented with 7.5% newborn calf serum (NCS; Sigma).
Cloning, expression and purification of CNFY constructs for crystallographic experiments
For crystallography purposes, truncated constructs were generated comprising a fragment lacking the catalytically active C-terminal domain (CNFY1-704), a construct comprising both C-terminal domains D4-5 (CNFY526-1014) and another containing only the catalytic domain D5 (CNFY720-1014). For crystallization of the full-length protein, a construct containing the inactive C866S variant of CNFY was produced.
The coding sequences of D4-5 (CNFY1-704) and D5 (CNFY720-1014) were both cloned into pET28 containing sequences coding for an N-terminal hexa-histidine tag and a thrombin protease cleavage site. The constructs were transformed into E. coli BL21 (DE3) (CNFY1-704) or Rosetta II (DE3) (CNFY720-1014). Native protein was expressed in lysogenic broth (LB) medium at 20°C after induction with 0.5 mM isopropyl-β-D-thiogalactopyranosid (IPTG) for 16-18 h (D1-4; CNFY1-704) or 4 h (D5; CNFY720-1024) (5), respectively. Seleno-L-methionine (Se-Met) labeled protein of CNFY1-704 (D1-4) was expressed using M9 minimal medium.
After harvesting, the cell pellets were resuspended in lysis-buffer (for D1-4/CNFY1-704: 1 x PBS, 400 mM NaCl, 5 mM β-mercaptoethanol, 5 mM MgSO4, 10 mM imidazole; for D5/CNFY720-1014: 50 mM Tris/HCl pH 8.0, 400 mM NaCl, 5 mM imidazole) and lysed by sonification. The supernatant after centrifugation was mixed with 1 ml Ni-NTA resin preequilibrated with wash I buffer (D1-4/CNFY1-704: 1 x PBS, 400 mM NaCl, 10 mM imidazole, 5 mM MgSO4, 5 mM β-mercaptoethanol; D5/CNFY720-1014: 50 mM Tris/HCl pH 8, 400 mM NaCl, 5 mM imidazole) and incubated for 1 h on an overhead-shaker at 4°C. After washing with wash I buffer and wash II buffer (D1-4/CNFY1-704: 1 x PBS, 400 mM NaCl, 20 mM imidazole, 5 mM MgSO4, 5 mM β-mercaptoethanol; D5/CNFY720-1014): 50 mM Tris/HCl pH 8, 400 mM NaCl, 20 mM imidazole), elution of the protein was carried out with 12 × 1 ml of elution buffer (D1-4/CNFY1-704: 1 x PBS, 400 mM NaCl, 250 mM imidazole, 5 mM MgSO4, 5 mM β-mercaptoethanol; D5/CNFY720-1014: 50 mM Tris/HCl, 250 mM NaCl, 250 mM imidazole). Buffer exchange and tag cleavage with thrombin (1:50 mg/mg) were achieved over night by dialysis at 4°C in wash I buffer. To remove cleaved His-Tag, uncleaved protein and the thrombin protease, 1 ml of Ni-NTA resin and 5 ml of benzamidine-sepharose resin, respectively were mixed with the dialyzed protein solution. The collected flow-through predominantly contained pure protein. Further purification was achieved by size-exclusion chromatography. D1-4 (CNFY1-704) was purified using a HiLoad 16/600 Superdex 200 pg (GE Healthcare) pre-equilibrated in buffer containing 20 mM Tris pH 8.0, 150 mM NaCl, 5 mM DTT. D5 (CNFY720-1014) was purified using a HiLoad 16/600 Superdex 75 pg (GE Healthcare) pre-equilibrated in buffer containing 25 mM Tris pH 8.0, 100 mM NaCl. The proteins were then concentrated to 20 mg/ml, flash-frozen in liquid nitrogen and stored or directly used for crystallographic screens.
The gene encoding for the full-length protein of the CNFY C866S variant was cloned into a modified pCOLA Duet-1 vector (Novagen) encoding for an N-terminal Strep-tag II and TEV-protease recognition site (construct: CNFY C866S). In the case of D4-5 (CNFY 526-1014), the insert was amplified from pCNFY3xFlag as template so that the three C-terminal FLAG-epitopes were included in the insert and cloned into the same modified pCOLA Duet-1 vector that was also used for the full-length toxin (construct: pVP-CNFY526-1014-3xFlag). Both proteins were heterologously expressed in E. coli BL21 (DE3) in ZYM-5052 auto-inducing medium (Studier, 2005) at 20°C for 20-24 h.
In the case of D4-5 (CNFY526-1014), the cell pellet was resuspended in a buffer containing 20 mM HEPES/NaOH pH 7.5, 300 mM NaCl, 2 mM TCEP, one tablet of complete EDTA-free protease inhibitor cocktail (Roche) and lysed by sonication. The protein was isolated from the supernatant after centrifugation for 1 h at 100.000 x g using a self-packed 10 ml column with Strep-Tactin Superflow High Capacity resin (IBA) and eluted from the column with a single step of 5 mM d-desthiobiotin. The affinity tag was cleaved off with TEV protease (1:50 mg/mg) at 4°C overnight. Gel filtration was carried out using a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) in 20 mM HEPES/NaOH pH 7.5, 300 mM NaCl, 2 mM TCEP. The peak fractions were concentrated to 5 mg/ml and flash-frozen in liquid nitrogen for crystallization screening.
For the full-length protein, the cell pellet was resuspended in a buffer containing 20 mM HEPES/NaOH pH 7.5, 100 mM NaCl, 1 mM TCEP, one tablet of complete EDTA-free protease inhibitor cocktail (Roche) and lysed by sonication. The protein was isolated from the supernatant after centrifugation for 1 h at 100.000 x g using a self-packed 10 ml column with Strep-Tactin Superflow High Capacity resin (IBA) and eluted from the column with a single step of 5 mM d-desthiobiotin. The affinity tag was cleaved off with TEV protease (1:50 mg/mg) at 4°C overnight. Gel filtration was carried out using a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) in 20 mM HEPES/NaOH pH 7.5, 100 mM NaCl, 1 mM TCEP. The fractions corresponding to the second peak in the chromatogram (elution volume 70-75 ml) were pooled and subjected to further size exclusion chromatography on a Superdex 200 Increase 10/300 GL column (GE Healthcare) in the same buffer. The peak fractions were concentrated to 27.5 mg/ml and flash-frozen in liquid nitrogen for crystallization screening. All chromatographic steps were carried out using an Äkta Purifier system (GE Healthcare). The samples were analyzed by SDS-PAGE (12%), and protein concentrations were determined from the absorbances at 280 nM with the extinction coefficients as calculated by Protparam (Gasteiger et al, 2003).
Crystallization
Crystallization trials were set up at room temperature with a HoneyBee 961 crystallization robot (Digilab Genomic Solutions) in Intelli 96-3 plates (Art Robbins Instruments) with 200 nl protein solution at different concentrations and 200 nl reservoir solution. Native D1-4 (CNFY1-704) was crystallized in 0.1 M Tris pH 7.3-7.9, 0.2 M ammonium sulfate and 19-21% (w/v) PEG 5000 MME. The Se-Met derivative of D1-4 (CNFY1-704) was crystallized in 0.1 M tri-sodium citrate pH 5.9-6.2, 0.2 M ammonium acetate and 28-32% (w/v) PEG 4000. Macro-seeding was applied in order to obtain well-diffracting crystals. As all tested compounds for cryoprotection were not tolerated by the samples, the crystals were flash-cooled without any additional cryoprotection. The catalytic domain D5 (CNFY720-1014) yielded crystals in several PEG or ammonium sulfate containing conditions and the best diffracting crystals were obtained in 0.2 M ammonium fluoride with 20% (w/v) PEG 3350. Crystals were cryo-protected with either 25% glycerol or 100% Type A oil (Hampton Research) prior to flash freezing in liquid nitrogen. A single well-diffracting crystal of D4-5 (CNFY526-1014) was obtained in the presence of 1 mM ATP in a condition containing 0.24 M magnesium chloride, 22.5% (w/v) PEG 2000 monomethyl ether. The crystal was harvested after 130 days of growth and cryo-protected by addition of 10% (v/v) (2R,3R)-2,3-butanediol. A single crystal of sufficient diffraction quality of full-length CNFYC866S was obtained in 1.4 M ammonium sulfate, 0.13 M lithium acetate, 0.1 M HEPES/NaOH Ph 7.1. The crystal was harvested after 21 days of growth and after removal of satellite crystals cryo-protected by addition of 10% (v/v) (2R,3R)-2,3-butanediol.
Data collection and processing
Data collection of native and Se-Met-derivatized D1-4 (CNFY1-704) was performed on beamline X06DA (PXIII) of the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland) and BESSY BL14.1 (Helmholtz Zentrum Berlin, Germany) (Mueller et al, 2015). High-resolution data of D5 (CNFY720-1014) were recorded at beamline BL 14.2 of the BESSY II (Helmholtz-Zentrum Berlin, Germany). Datasets of domain D4-5 (CNFY526-1014) and full-length CNFYC866S were measured at beamline X06DA (PXIII) at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland). Data processing was achieved either manually via the XDS software package (Kabsch, 2010) or by using the AutoPROC (Vonrhein et al, 2011) toolbox (Global Phasing) executing XDS (Kabsch, 2010), Pointless (Evans, 2006), and Aimless (Evans & Murshudov, 2013). All datasets were recorded at a temperature of 100 K.
Structure determination, refinement and model building
The structure of domain D1-3 (CNFY1-704) was solved by single anomalous dispersion (SAD) using data collected at the selenium absorption edge. The initial phases were calculated using AutoSol (Terwilliger et al, 2009) and a partial model was generated running AutoBuild (Terwilliger et al, 2008), both components of the Phenix software package (Adams et al, 2010). The output model was analyzed in Coot (Emsley et al, 2010) and misplaced main chains were removed or corrected manually in order to obtain a reliable search-model for the following molecular replacement procedures against the dataset of native D1-4 (CNFY1-704) and the full-length C866S variant. The structure of D5 (CNFY720-1014) was determined by molecular replacement using the structure of the catalytic C-terminal domain of CNF1 from E. coli (PDB: 1HQ0, (Buetow et al, 2001)) as search-model. The structure of domain D4-5 (CNFY526-1014) was determined by molecular replacement using the structure of domain D5 (CNFY720-1014) and the region comprising residues 526-704 from domain D1-4 (CNFY1-704). Phases for full-length CNFYC866S were obtained by using both domain D1-4 (CNFY1-704) and D5 (CNFY720-1014) as search-models in molecular replacement. The molecular replacement procedures were carried out using Phaser (McCoy et al, 2007) from the Phenix suite (Adams et al, 2010). The structural models were built using Coot (Emsley et al, 2010) and crystallographic refinement was performed with Phenix.refine (Afonine et al, 2012) including the addition of hydrogens in riding positions and TLS-refinement. 5% of random reflections were flagged for the calculation of Rfree. The model of domain D1-4 (CNFY1-704) was at 3.0 Å resolution and refined to R/Rfree of 24/26% in space group P21. The structure of domain D5 (CNFY720-1014) was at 1.1 Å resolution and refined to R/Rfree of 17/19% in space group P21. The structure of domain D4-5 (CNFY526-1014) was at 1.8 Å resolution and refined to R/Rfree of 16/19% in space group P212121. The structural model of the full-length C866S variant of CNFY was at 2.7 Å resolution and refined to R/Rfree of 21/24% in space group I212121. Data collection and refinement statistics are summarized in Appendix Tab S1. Figures of crystal structures were prepared using the PyMOL Molecular Graphics System version 2.0.0 (Schrödinger, LLC).
Construction of fusion plasmids and C-terminal cnfY deletions
To construct the plasmids for CNFY fusion proteins, the blaM gene and the 3xFlag tag were amplified using primers listed in Appendix Tab S5. pFU189 was used as a backbone from which the luxCDABE operon was removed by digestion with PstI and NotI after which blaM or the 3xFlag tag were ligated into the vector, resulting in pTEM and p3xFLAG, respectively.
The PCR fragments of C-terminal cnfY deletions containing the cnfY promoter region were cloned into the BamHI and PstI sites of pTEM and p3xFLAG using the Quick-Fusion cloning kit (Biotool) with primers listed in Appendix Tab S5. For the construction of pCNFY-GFP, gfp was excised from pFU31 and ligated into the PstI and NotI sites of digested pCNFY-TEM. All clones were transformed into E. coli DH10β and confirmed by sequencing. The plasmids were electroporated into Y. pseudotuberculosis YP147 (ΔcnfY) and selected for on LB agar plates containing the appropriate antibiotics.
Site-directed mutagenesis of cnfY
Single residue mutants were generated by site-directed mutagenesis with primers listed in Appendix Tab S5. pCNFY-TEM, pCNFY-3xFLAG were used as templates. Clones were selected on LB containing the appropriate antibiotics. Mutations were verified by DNA sequencing.
Detection of fusion proteins by immunoblotting
For protein expression, strain YP147 harboring the overexpression plasmids encoding full-length CNFY or the deletion variants was grown in BHI at 25°C overnight. Cells were harvested by centrifugation at 6,500 rpm and 4°C for 5 min. Cell pellets were washed with PBS and resuspended with lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.3% Triton X-100, 3 mg/ml lysozyme and protease inhibitor cocktail). After incubation at room temperature for 1 h, protein samples were centrifuged for 10 min and supernatants were sterilized with a 0.2 μm filter. To detect proteins, Western blot analysis was performed. The proteins were separated on a 10% SDS-polyacrylamide gel and transferred onto an Immobilon PVDF membrane (Millipore). Membranes were blocked in 5% BSA/TBST at 4°C overnight. Subsequently, the membrane was washed and incubated with primary antibody diluted in 5% BSA/TBST (1:10,000 anti-Flag (Sigma-Aldrich) or anti-Beta lactamase (Abcam)) at room temperature for 1 h. After washing, the secondary antibody diluted in 5% skim milk/TBST (1:5,000 anti-mouse IgG HRP (Cell Signaling Technology)) was added for 30 min at room temperature. After washing the membrane, proteins were visualized using the Western Lightning ECL II Kit (Perkin Elmer) and exposed on X-Ray film (GE Healthcare Amersham Hyperfilm ECL, Fisher Scientific).
Nitrocefin secretion assay
Bacteria were grown overnight at 25°C in BHI containing the appropriate antibiotics. Subsequently, equal amounts of bacteria were pelleted by centrifugation at 13,000 rpm for 10 min. 95 μl of each supernatant was transferred to a 96-well plate in triplicate. 5 μl nitrocefin (2 mM) were added to each well and the plate was incubated at room temperature for 30 min. Beta-lactamase activity was determined at 390 nm (yellow) and 486 nm (red) using a VarioSkan plate reader (Thermo Scientific).
Microbial viability assay
The microbial viability was assessed in equalized bacterial cultures using the BacTiter-Glo™ Microbial Cell Viability Assay kit (Promega) according to the manufacturer’s recommendations and luminescence was measured using a VarioSkan plate reader (Thermo Scientific).
Fluorescent actin staining
HEp-2 cells were seeded onto coverslips at a concentration of 5×104 cells/well and allowed to attach overnight. The next day, cells were washed and incubated with an equal amount of cleared bacterial cell lysates for 24 h at 37°C, 5% CO2. After washing with PBS, cells were fixed in 4% paraformaldehyde for 15 min at room temperature. Subsequently, washed cells were permeabilized with 0.1% Triton X-100 in PBS for 1 min. The actin cytoskeleton was stained with FITC- or TRITC-Phalloidin (0.5 μg/ml in PBS; Sigma-Aldrich) and mounted on slides using ProLong® Gold Antifade mounting medium containing DAPI (Thermo Scientific). Cells were visualized by fluorescence microscopy using an Axiovert II inverted fluorescence microscope (Carl Zeiss) with Axiocam HR and the AxioVision program (Carl Zeiss).
CNFY translocation assay
In order to study the CNFY translocation into the host cells, a β-lactamase reporter assay was performed using the LiveBLAzer-FRET B/G Loading Kit (Life Technologies). HEp-2 cells were seeded in 8-well μ-slides (Ibidi) at a concentration of 1.7×104 cells/well and allowed to attach overnight. The next day, cells were washed and incubated with equal amounts of cleared bacterial cell lysates for 24 h at 37°C, 5% CO2. Cells were washed with PBS, followed by the addition of fresh media containing 20 mM HEPES. Cells were then stained with loading dye according to the manufacturer’s protocol. After staining for 1 h at room temperature, translocation was visualized by fluorescence microscopy using an Axiovert II with Axiocam HR and the AxioVision program (Carl Zeiss).
Fluorescence microscopy to visualize endocytosis
To test whether the CNFY deletion constructs are able to enter the cells through the endocytotic pathway, CellLight® Early or late Endosomes-RFP, BacMam 2.0 (Thermo Scientific) were used to investigate toxin entry to the host cells. HEp-2 cells were seeded 5×104 cells/ml onto coverslips in 24 well plates and allowed to attach overnight. The next day, CellLight® reagent was added to HEp-2 cells around 20 particles per cells for 16 h and the cells were then incubated with CNFY toxin on ice for 30 min. Subsequently, cells were washed and transferred to 37°C for 30, 90 or 180 min. To investigate colocalization of CNFY with early or late endosome, cells were then fixed and visualized by using a fluorescence microscope (Axiovert II with Axiocam HR, Carl Zeiss) and the AxioVision program (Carl Zeiss).
Biochemical analysis of RhoA deamidation
Cells were seeded in 10 cm cell culture dishes at the concentration of 2.2 × 106 cells/dish and allowed to attach overnight. The next day, cells were washed and incubated with 500 nM of cleared bacterial cell lysates for 4 h at 37°C, 5% CO2. Cells were washed with cold PBS and lysed in 150 μl lysis buffer containing 50 mM Tris-HCl (pH7.4), 100 mM NaCl, 2 mM MgCl2, 10% NP-40 and 0.5 mM phenyl-methyl-sulfonyl fluoride (PMSF). Cells were then scraped off and centrifuged for 30 min (13,000 rpm, 4°C). Sodium dodecyl sulfate (SDS) sample buffer was added to the clear lysates and samples were separated on 12% SDS-gel. After blotting onto a PVDF membrane, RhoA was detected using mouse anti-RhoA IgG (Millipore) (1:1000) as a primary antibody and followed by secondary antibody goat anti-mouse IgG-HRP (Cell signaling). Membranes were visualized using the Western Lightning ECL II Kit (Perkin Elmer) and exposed on X-Ray film.
In vitro RhoA shift assay
In vitro Rho-shift assays were performed in order to check for proper folding and catalytic activity of CNFY deletion constructs. Cells were seeded on 150 mm dish at the concentration of 5 × 106 cells/dish and allowed to attach overnight. The next day, cells were washed with PBS and lysed in 300 μl lysis buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM EDTA, 10% NP-40 and 1 mM dithiothreitol (DTT)). Cells were then scraped off and centrifuged 13,000 rpm at 4°C for 30 min. 20 μl of cytosolic extracts were incubated with 1 μM of CNFY lysates at 37°C for 4 h. The reactions were stopped by adding SDS-sample buffer and heated at 95°C for 10 min. Samples were then subjected to 12% SDS-PAGE. After blotting onto a PVDF membrane, blots were developed as mentioned above.
Author contributions
PD and WB conceived the project. PL, EMG and TVH produced recombinant proteins and performed crystallization and structure determination. PC, SM, JNGS and TEBS conducted work with Y. pseudotuberculosis and analyzed microscopy experiments with eukaryotic cells. SM, PL, PD and WB wrote the manuscript.
Conflict of interest
The authors declare no competing interests.
Data availability
Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank (Berman et al, 2000) with accession codes 6YHK (full-length CNFY C866S), 6YHL (CNFY 1-704), 6YHM (CNFY 720-1014) and 6YHN (CNFY 526-1014).
Acknowledgements
We thank the beamline staff at the Helmholtz Centre Berlin (Germany) and the Paul Scherrer Institute (Villigen, Switzerland) for providing access to beamlines BL14.1 and BL14.2 at the BESSYII electron storage ring and to beamline X06DA at the SLS synchrotron. Experiments at the SLS have received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement n.° 730872, project CALIPSOplus. Andrea Berger and Ute Widow are acknowledged for excellent technical assistance. T.H. and P.C. were supported by the HZI Graduate School for Infection Research.
References




