

Abstract
COVID-19 virus is the cause of a debilitating and life-threatening infectious pulmonary disease that is now responsible for a global pandemic. Currently, there are no specific drugs or vaccines to contain this virus. The main protease (Mpro) of COVID-19 virus is a key enzyme, which plays an essential role in viral replication and transcription, making it an ideal drug target. An FDA-approved antineoplastic drug, carmofur, has been identified as an inhibitor that targets COVID-19 virus Mpro. However, its inhibitory mechanism is unknown. Here, we report the 1.6-Å crystal structure of COVID-19 virus Mpro in complex with carmofur. The crystal structure shows that carmofur contains an electrophilic carbonyl reactive group, which covalently binds to C145, a member of the catalytic dyad. As a result, its fatty acid tail occupies the hydrophobic S2 subsite of Mpro whilst its 5-fluorouracil head is cleaved as product of the new covalent bond that has formed. Carmofur is active in a cell based antiviral assay with an EC50 of 24.87 μM. It is therefore a promising lead compound for the development of new antivirals to target COVID-19.
Introduction
Starting in December 2019, a highly infectious viral disease has now spread and reached over 200 countries leading to a global public health emergency and pandemic. The etiological agent of the disease is a coronavirus (identified as COVID-19). According to the WHO COVID-2019 Situation Report-77, there were 1,210,956 confirmed cases and 67,594 deaths, with a mortality rate at 5.58%. The number of confirmed cases worldwide continues to grow at a rapid rate and is far from peaking. However, there are no specific drugs or vaccines available to control symptoms or the spread of this disease.
The COVID-19 virus has a ~30,000 nt RNA genome encoded with two translation products, polyproteins 1a and 1ab (pp1a and pp1ab) which are crucial for replication and transcription1,2. These polyproteins become matured non-structural and structural proteins through auto-cleavage by the main protease (Mpro) and by a papain-like protease3. Because of this, Mpro is an excellent target for anti-coronavirus (CoV) drug development4–6. In order to rapidly discover new drug leads that target COVID-19 virus Mpro, our group screened over 10,000 compounds from a library that consisted of approved drugs, drug candidates in clinical trials, and other pharmacologically active compounds. Amongst these we identified carmofur as compound that can inhibit Mpro with an IC50 of 1.82 μM 7.
Carmofur is an FDA-approved antineoplastic drug, and a derivative of 5-fluorouracil (5-FU) a widely drug used against solid cancers. 5-FU is especially efficient for controlling head, neck, and gastrointestinal tumors8. Carmofur (Figure 1A) is a derivative of this compound and has been used in colon cancer therapy since 19819. Clinical research has also shown that carmofur has a curative effect on breast, gastric, bladder, and colorectal cancers10–12. The target for carmofur is believed to be thymidylate synthase an enzyme that converts deoxy uridine monophosphate (dUMP) to deoxythymidine monophosphate13,14. Carmofur has also been shown to target human acid ceramidase (AC)15, a potential drug target for the treatment of melanoma and glioblastoma tumors16,17. Carmofur inhibits human AC through the covalent modification of its catalytic cysteine18. Our previous data shows that carmofur inhibits Mpro activity7, but the molecular details as to how it inhibits this target are unresolved. Here, we have determined the 1.6-Å crystal structure of COVID-19 virus Mpro in complex with this compound. The structure shows that the fatty acid tail from carmofur covalently binds to C145 at the catalytic center of the viral protease (Figure 1B). Furthermore, cell-based assays show that carmofur has an EC50 value of ~25 μM for the COVID-19 virus. This high-resolution crystal structure thus provides the structural basis for the design of new carmofur analogs with clinical potential to treat COVID-2019.
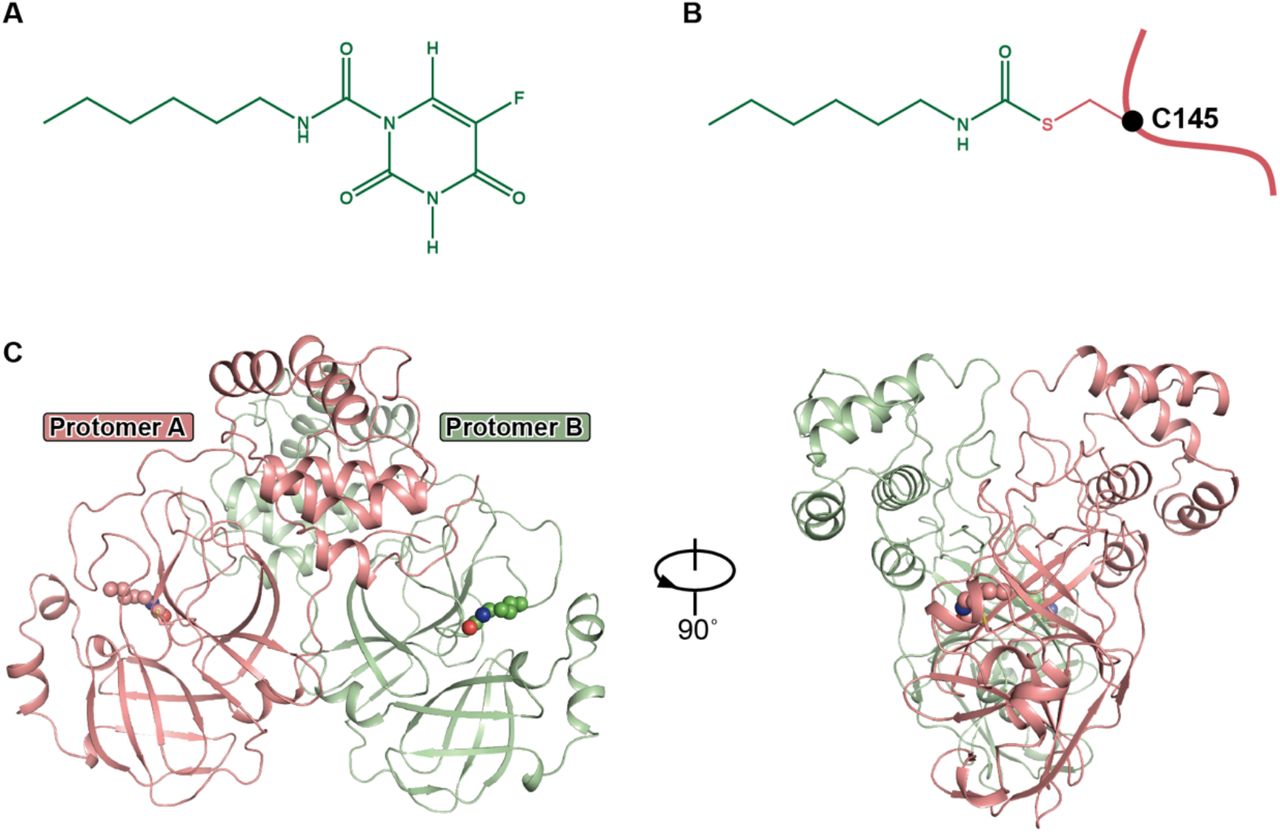
A. The chemical structure of carmofur.
B. The binding mode of carmofur to COVID-19 virus Mpro. The red curve represents COVID-19 virus Mpro polypeptide with the sidechain of Cys145 protruding.
C. The overall structure of COVID-19 virus Mpro in complex with carmofur. The salmon and green represent the different protomers. The carmofur atoms are shown as solid spheres.
Results
Overall structure of Mpro-carmofur complex
The structure of COVID-19 virus Mpro in complex with the fatty acid tail of carmofur has been solved at a resolution of 1.6 Å (Extended Data Table 1). In accord with our previous studies4,5,19–21, this Mpro complex also forms a homodimer. In the crystal structure, this is formed by two polypeptides (protomer A and B) related by crystallographic symmetry (Figure 1C). All of the residues, 1-306, in the polypeptide could be traced in the electron density map. Each protomer is composed of three domains, similar to those found in other Mpro structures (Figure 1A). Domain I (residues 10-99) and domain II (residues 100-184) are two β-sheet rich domains. Domain III (residues 201-303) is linked to domain II by a long loop region (residues 185-200). The buried surface area at the dimer interface is ~1409 Å2. This is mainly due to interactions by domain II, domain III and the N-termini of each polypeptide.
There are twelve cysteine residues across the protein with six buried in the core (Figure S3). The other six cysteines are exposed to the surface, with one of these (C145) located in the catalytic center, which lies in a cleft between domain I and domain II. The long loop connecting domain II and domain III also participates in the formation of the substrate binding pocket. S1 of one protomer interacts with E166 of the neighbor protomer to stabilize the S1 subsite of the substrate-binding pocket. This structural feature is essential for catalysis.

A. Putative inhibition mechanism. Red curve represents Mpro polypeptide and the black sphere represent the Cα of C145.
B. Schematic diagram of Mpro-carmofur interactions. Black spheres represent Cα atoms.

A. Overall structural comparison between the Mpro-carmofur and Mpro-N3 complexes. The salmon cartoon represents the carmofur bound structure and the light cyan represents the N3 bound structure. Carmofur, N3 and DMSO are represented by the purple, yellow and green balls and sticks, respectively.
B. The binding pocket of Mpro. Carmofur and N3 are represent in the same way as in Figure S2A.
C. Schematic diagram of carmofur and N3.

Each cysteine side chain is represented by a colored sphere.
Carmofur is covalently linked to the catalytic cysteine
The substrate-binding pocket lies in the cleft between domain I and domain II and is characterized by the presence of the catalytic dyad residues, C145 and H41 (Figure 2A, 2B). The electron density map unambiguously shows that the fatty acid moiety (C7H14NO) of carmofur is linked to the Sγ atom of C145 through a 1.8-Å covalent bond, and that the fatty acid tail is inserted into the S2 subsite (Figure 2B, 2C). It suggests that the sulfhydryl group of catalytic C145 attacks the electrophilic carbonyl group of carmofur, resulting in the covalent modification of the cysteine residue by the fatty acid moiety (C7H14NO) and the release of the 5-fluorouracil head (Figure S1A). This result is also consistent with our previous tandem MS/MS studies7.

A. The structure of a single protomer. The three domains are shown in three different colors. The catalytic center is located within the dashed square.
B. Zoom in of the catalytic center. The residues that participate in carmofur binding are shown as stick models. Carmofur is show as a ball and stick model with the carbons in magenta. Water is presented as a red sphere.
C. A rotated view of the binding site, but with the surface removed.
In addition to the C-S covalent bond, the inhibitor is stabilized by numerous hydrogen bonds and hydrophobic interactions (Figure 2C, S1B). The carbonyl oxygen of the inhibitor occupies the oxyanion hole and forms hydrogen bonds (3.0-Å) with the backbone amides of C143 and C145, mimicking the tetrahedral oxyanion intermediate formed during protease cleavage (Figure 2C). The fatty acid tail, which presents in an extended conformation, inserts into the bulky hydrophobic S2 subsite (composed of the side chains of H41, M49, Y54, M165, and the alkyl portion of the side chain of D187) (Figure 2B, 2C). The hydrophobic interactions are mainly contributed by the side chains of H41, M49 and M165, all of which run parallel with the alkyl part of the fatty acid tail of the inhibitor (Figure 2C, S1B).
Compared with that for the Michael acceptor, inhibitor N37,22, the covalent modification mechanism for carmofur is different. In this case, the catalytic C145 attacks the electrophilic carbonyl group of carmofur and results in the covalent modification of the cysteine residue while in the case of N3, covalent modification occurred through Michael addition of the vinyl group.
The overall structures of the Mpro-carmofur complex and the MproN3 complex are similar with an RMSD of 0.286 Å for all Cα atoms. Though slight, the largest conformational differences occur in the substrate binding pocket (Figure S2A). Compared with the Mpro-N3 complex structure, the backbone surrounding the inhibitor binding site of carmofur complex structure moves in a slightly outward direction (Figure S2A). A major difference between N3 and carmofur binding is that N3 occupies four subsites (S1, S2, S4 and S1′), (Figure S2B) whereas carmofur only occupies the S2 subsite. Thus, there is good scope for structural elaboration of this lead. Interestingly, a molecule of DMSO fills the S1 subsite, a region corresponding to the location where the lactam ring in the N3 bound complex is located (Figure S2B). This observation should also assist in the design of more potent carmofur analogs as inhibitors of COVID-19 Mpro.
Antiviral activity assay
In our preliminary antiviral studies reported recently7, at a concentration of 10 μM treatment in COVID-19 virus infected Vero cells, ebselen showed potent antiviral effect while carmofur did not demonstrate strong antiviral activity at this concentration. It indicates that carmofur have a larger EC50 value than that for Ebselen (EC50=4.67 μM). We then set out to determine the accurate EC50 value for carmofur. As described in our previous study23, Vero E6 cells were treated with a series of concentrations of carmofur, and were then were infected with COVID-19 virus. As a result, the EC50 for carmofur was determined to be 24.87 μM (Figure 3B). To verify this result, infected cells were fixed and subjected to immunofluorescence assay (IFA) using anti-sera against viral nucleocapsid protein (NP). Figure 3A shows NP expression levels decreased after the treatment of carmofur, in accordance with the quantitative RT-PCR study. A cytotoxicity assay was also performed in Vero E6 cells with a CC50 value of 132.7 μM (Figure 3C). Thus, the compound has a favorable selectivity index (SI) of 5.34, but optimization is needed to make it an effective drug.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Vero E6 cells infected with SARS-CoV-2 at a MOI of 0.05 were treated with carmofur at a range of different concentrations.
A. At 24 hours p.i., cells were fixed, and intracellular NP levels were monitored by immunofluorescence. Chloroquine (CQ, 10 μM) was used as a positive control. Bars: 400 μm
B. Cell viability was measured using a CCK8 assay. The Y-axis represents mean % of cell viability. The experiments were performed in triplicate, and data shown are for the mean values ± SE.
C. Supernatant was collected and viral copy number in the supernatant was measured with quantitative RT-PCR. The Y-axis of the graph indicates mean % inhibition of virus, the experiments were performed in triplicate, and data shown are mean values ± SE.
Discussion
In conclusion, we have solved the 1.6-Å crystal structure of Mpro in complex with an FDA approved antineoplastic, carmofur. This study shows that carmofur directly modifies the catalytic cysteine of COVID-19 virus Mpro leading to its inhibition. The study also provides a basis for rational design of carmofur analogs to treat COVID-19 infections. Since Mpro is highly conserved among all CoV Mpros, carmofur and carmofur analogs could also be effective against other CoV Mpros.
We also showed that carmofur can inhibitor COVID-19 virus at a lower concentration than its cellular cytotoxicity (SI > 5), further suggesting that carmofur is a good starting point to develop inhibitors with clinical potential to treat COVID-19. In addition, it has been reported that carmofur might be a novel therapeutic agent for acute lung injury (ALI)24 which can also benefit COVID-19 patients since lung injury is an outcome that can occur as the result of this infection.
Methods
Cloning, protein expression and purification of COVID-19 virus Mpro
The cell cultures were grown and the protein expressed according to a previous report7. The cell pellets were resuspended in lysis buffer (20mM Tris-HCl pH 8.0, 150 mM NaCl, 5% Glycerol), lysed by high-pressure homogenization, and then centrifuged at 25,000g for 30 min. The supernatant was loaded onto Ni-NTA affinity column (Qiagen, Germany), and washed by the lysis buffer containing 20 mM imidazole. The His tagged Mpro was eluted by lysis buffer that included 300 mM imidazole. The imidazole was then removed through desalting. Human rhinovirus 3C protease was added to remove the C-terminal His tag. COVID-19 virus Mpro was further purified by ion exchange chromatography. The purified Mpro was transferred to 10 mM Tris-HCl pH 8.0 through desalting and stored at −80 degrees until needed.
Crystallization, data collection and structure determination
COVID-19 virus Mpro was concentrated to 5 mg/ml incubated with 0.3 mM carmofur for 1 hour and the complex was crystallized by hanging drop vapor diffusion method at 20 °C. The best crystals were grown using a well buffer containing 0.1 M MES pH 6.0, 5% polyethylene glycol (PEG) 6000, and 3% DMSO. The cryo-protectant solution was the reservoir but with 20% glycerol added.
X-ray data were collected on beamline BL17U1 at Shanghai Synchrotron Radiation Facility (SSRF) at 100 K and at a wavelength of 0.97918 Å using an Eiger X 16M image plate detector. Data integration and scaling were performed using the program XDS25. The structure was determined by molecular replacement (MR) with the PHASER26 and Phenix 1.17.1 27 using the COVID-19 virus Mpro (PDB ID: 6LU7) as a search template. The model from MR was subsequently subjected to iterative cycles of manual model adjustment with Coot 0.8 28 and refinement was completed with Phenix REFINE29. The inhibitor, carmofur, was built according to the omit map. The phasing and refinement statistics are summarized in Extended Data Table 1. Coordinates and structure factors have been deposited in Protein Data Bank (PDB) with accession number 7BUY.
Antiviral and cytotoxicity assays for carmofur
A clinical isolate COVID-19 virus (nCoV-2019BetaCoV/Wuhan/WIV04/2019) was propagated in Vero E6 cells, and viral titer was determined as described previously23. For the antiviral assay, pre-seeded Vero E6 cells (5×104 cells/well) were pre-treated with the different concentration of carmofur for 1 h and the virus was subsequently added (MOI of 0.05) to allow infection for 1 h. Next, the virus-drug mixture was removed, and cells were further cultured with fresh drug containing medium. At 24 h p.i. (post infection), the cell supernatant was collected and vRNA in supernatant was subjected to qRT-PCR analysis, while cells were fixed and subjected to immunofluorescence to monitor intracellular NP level as described previously23. For cytotoxicity assays, Vero E6 cells were suspended in growth medium in 96-well plates. The next day, appropriate concentrations of carmofur were added to the medium. After 24 h, the relative numbers of surviving cells were measured by the CCK8 (Beyotime, China) assay in accordance with the manufacturer’s instructions. All experiments were performed in triplicate, and all the infection experiments were performed at biosafety level-3 (BSL-3).
Author contributions
Z.R. and H.Y. conceived the project; Z.J., Y.Z., Z.R., and H.Y. designed the experiments; Y.Z., Z.J., H.W., Y. Zhu., C.Z., X.D. J.Y. and X.Y. cloned, expressed, purified and crystallized proteins; Y.Z., B.Z., Z.J. and T.H. collected the diffraction data; Y.Z, B.Z. and Xiang Liu solved the crystal structure; Y.S., and Y.W. performed cell-based antiviral assay; Y.D. and L. Z performed qRT-PCR and cytotoxicity assay analysis; Y.Z., J.Z., L.Z., Y.D., X. L., L.G., G.X., Z.R. and H.Y. analyzed and discussed the data; Y.Z., Z.J., L.Z., L.G. H.Y, and Z.R. wrote the manuscript.
Competing interests
The authors declare no competing interests.
Data and materials availability
The PDB accession No. for the coordinates of COVID-19 virus Mpro in complex with carmofur is 7BUY.
Acknowledgement
We are grateful to the staff at the BL17U beamline of the Shanghai Synchrotron Radiation Facility (SSRF), where data were collected. This work was supported by grants from National Key R&D Program of China (grant No. 2017YFC0840300 to Z.R.), National Key R&D Program of China (grant No. 2020YFA0707500 to Z.R.), Project of International Cooperation and Exchanges NSFC (grant No. 81520108019 to Z.R.), Science and Technology Commission of Shanghai Municipality (grant No. 20431900200), Department of Science and Technology of Guangxi Zhuang Autonomous Region (grant No. 560 2020AB40007), and the Natural Science Foundation of China (grant No. 31970165 to Leike Zhang.
Footnotes
↵† e-mail: zhangleike{at}wh.iov.cn
References




