

ABSTRACT
The rapid, sensitive and specific detection of SARS-CoV-2 is critical in responding to the current COVID-19 outbreak. Here, we explore the potential of targeted mass spectrometry based proteomics for the detection of SARS-CoV-2 proteins in both research and clinical samples. First, we assessed the limit of detection for several SARS-CoV-2 proteins by parallel reaction monitoring (PRM) mass spectrometry. For Nucleocapsid the limit of detection was found to be in the mid-attomole range (0.9 x 10−12 g). Next, we apply this PRM assay to the detection of viral proteins in in vitro mucus substitutes, as well as in various clinical specimens such as nasopharyngeal swabs and sputum. In this proof-of-concept study SARS-CoV-2 proteins could unambiguously be detected in clinical samples, suggesting that the sensitivity of this technology may be sufficiently high to further explore its potential role in diagnostics.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 2019 (COVID-19), which is a severe respiratory disease 1. The World Health Organization (WHO) has designated the ongoing pandemic of COVID-19 a Public Health Emergency of International Concern 2. As of now, over one million deaths have been reported worldwide and this is probably an underestimation because of lack of testing capacity in large parts of the world.
SARS-CoV-2 is a positive-sense single-stranded RNA virus, which encodes several non-structural proteins such as spike, envelope, membrane and nucleocapsid protein 3. Rapid, sensitive and specific diagnosis of SARS-CoV-2 is widely recognized to be critical in responding to this outbreak, but also for long-term improvements in patient care. Importantly, the reduction of time required to identify SARS-CoV-2 infections will significantly contribute to limiting the enormous social and economic consequences of this large global society paralyzing outbreak. Conventional methods for diagnostic testing of viral infections, which are also widely used for SARS-CoV-2 testing, are based on polymerase chain reaction (PCR) or other (multiplexed) nucleic-acid based technologies and antigen detection. Since its emergence late 2019 it has become clear that additional diagnostic tools that target SARS-CoV-2 should be developed to complement existing tools in a “proactive approach” proposed by the Coronaviridae Study Group of the International Committee on Taxonomy of Viruses 1. Alternative and/or complementary SARS-CoV-2-specific diagnostic tests are desperately needed since the current testing capacity is insufficient, amongst others because of shortages of supplies such as RNA extraction kits, PCR reagents and delivery issues for primers and probes.
Besides PCR based approaches, immunoassays have been employed in the detection of other viruses. In addition, mass spectrometry (MS) based techniques have been applied previously, for instance to detect influenza virus proteins 4 and human metapneumovirus (HMPV) in clinical samples 5. Recent developments in targeted proteomics methods and Orbitrap mass spectrometry such as parallel reaction monitoring (PRM) have shown a substantial sensitivity increase. Although mass spectrometry based approaches have been used in a few SARS-CoV-2 studies recently 6–9 is not yet clear whether state-of-the-art proteomics technologies could provide the sensitivity and specificity needed in diagnostics.
Here, we explore the use of targeted mass spectrometry based proteomics for SARS-CoV-2 detection in research and clinical samples. For this, we first assessed the limit of detection by parallel reaction monitoring (PRM) on an Orbitrap mass spectrometer for specific tryptic peptides of SARS-CoV-2 proteins. The sensitivity was found to be in the mid-attomole range (~0.9 x 10−12 g) for Nucleocapsid (NP). Next, we sought whether this sensitivity is sufficient for the detection of SARS-CoV-2 in clinical samples such as nasopharyngeal swabs, mucus and sputum. This largely depends on the absolute amounts of viral proteins as well as on the complexity and abundance of the proteinaceous matrix background present in such samples. Using PRM, we could indeed detect various proteolytic peptides of several SARS-CoV-2 proteins in sputum and swab samples.
The data presented here suggest that the sensitivity of targeted proteomics is sufficiently high for the detection of viral material in patient samples such as swabs, sputum, mucus and possibly also other types of body fluids. Subsequent steps should be focused on sample preparation protocols that are in agreement with validated virus inactivation procedures, improvements in sample throughput and increase in sensitivity of detection.
Finally, providing novel mass spectrometry based diagnostic tools that complement genomic approaches is also the major goal of the recently formed COVID-19 mass spectrometry coalition (www.covid19-msc.org). The aim of this ‘proof-of-concept’ study is to highlight the potential of mass spectrometry in identifying SARS-CoV-2 proteins for diagnostics and research.
METHODS
Virus and cells
Vero E6 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10 % fetal calf serum (FCS), HEPES, sodium bicabonate, penicillin (final concentration 100 IU/mL) and streptomycin (final concentration 100 IU/mL) at 37 °C in a humidified CO2 incubator. SARS-CoV-2 (isolate BetaCoV/Munich/BavPat1/2020; European Virus Archive Global #026V-03883; kindly provided by Dr. C. Drosten) was propagated on Vero E6 cells in Opti-MEM I (1X) + GlutaMAX (Gibco), supplemented with penicillin (final concentration 100 IU/mL) and streptomycin (final concentration 100 IU/mL) at 37 °C in a humidified CO2 incubator. Stocks were produced by infecting cells at a multiplicity of infection (MOI) of 0.01 and incubating the cells for 72 hours. The culture supernatant was cleared by centrifugation and stored in aliquots at −80 °C. Stock titers were determined by preparing 10-fold serial dilutions in Opti-MEM I (1X) + GlutaMAX. Aliquots of each dilution were added to monolayers of 2 × 104 VeroE6 cells in the same medium in a 96-well plate. Plates were incubated at 37 °C for 5 days and then examined for cytopathic effect. The TCID50 was calculated according to the method of Spearman & Kärber. All work with infectious SARS-CoV and SARS-CoV-2 was performed in a Class II Biosafety Cabinet under BSL-3 conditions at Erasmus University Medical Center.
Organoid-derived human airway culture secretions
Organoid-derived human airway culture secretions were harvested from cultures that had been differentiated at air-liquid interphase for 3 weeks as described by Lamers et al. 10. Secretions could be harvested by pipetting using a P1000 tip and were not diluted. Secretions were stored at −80 °C until use. Ten-fold dilutions of virus stock containing 1.21 × 106 TCID50/ml were made in Opti-MEM I (1X) + GlutaMAX. Next, 25 μl of each virus dilution was mixed with 25 μl of airway culture secretions. Virus was inactivated by adding 50 μl of 2X Laemmli buffer (BioRad) and incubating at 95 °C for 10 minutes.
Collection and treatment of patient material samples
Nasopharyngeal swabs from COVID-19 patients were stored in universal transport medium (UTM; contains bovine serum albumin) after collection. Next, they were centrifuged at 15,000 g for 3 min to pellet down cell debris (termed ‘swab pellet’). The swabs were then washed twice with PBS to remove excessive albumin and fixed in 80% acetone (termed ‘swab supernatant’). Sputum from COVID-19 patients was collected and diluted in UTM. Alternatively, sputum was diluted in medium after collection and a few droplets were pipetted on glass slides, dried and fixed in 80% acetone.
The nasopharyngeal and throat swabs and sputum samples were obtained from different patients. Samples of sputum deposited on glass slides were obtained from one single patient.
Proteins present in patient nasopharyngeal and throat swabs or sputum samples in transport medium were first precipitated with acetone-TCA to remove excessive albumin according to 11. Briefly, 40 μl of the sample was mixed with 400 μl acetone and 1 % TCA and left overnight at −20 °C. Proteins were pelleted, washed once with ice-cold acetone and left to dry for 5 min. The protein pellet was then resuspended in 40 μl 50 mM Tris/HCl, 4 M urea (pH 8.2) and diluted with 160 μl 50 mM Tris/HCl (pH 8.2).
Cellular human and viral material in sputum deposited on glass slides was lysed in 50 μl 2% SDS dissolved in 50 mM Tris/HCl (pH 8.2) followed by sonication in a Bioruptor Pico (Diagenode). Proteins were digested using the SP3 protocol as described below.
Sample preparation for MS
A 90 % confluent T75 flask of VeroE6 was infected at a MOI of 0.3 and incubated for 24 hours at 37 °C in a humidified CO2 incubator. Next, cells were collected by scraping and the medium was removed after centrifuging at 400 g for 5 min. Cells were lysed in 2X Laemmli buffer (final concentration; Bio-Rad) and boiled at 95 °C for 20 min to inactivate the virus. Proteins were reduced and alkylated with DTT (Sigma) and IAA (Sigma) and precipitated using chloroform/methanol 12. The protein pellet was then dissolved in 100 μl of a 50 mM Tris/HCl buffer (pH 8.0) with 2 M urea. Proteins were quantified using the BCA protein kit (ThermoFisher Scientific / Pierce, #23225); peptides were quantified with a quantitative colorimetric peptide assay (ThermoFisher Scientific / Pierce, #23275). Fifty μg of protein was digested with 1 μg trypsin (Thermo) overnight at room temperature. The peptide digest was cleaned on a 50 mg tC18 Sep-Pak cartridge (Waters) and the peptides were eluted with 2 ml acetonitrile/water (1:1) with 0.05 % TFA.
Alternatively, proteins were digested with trypsin using the SP3 protocol 13, with minor modifications. Briefly, proteins in 30 μl Laemmli buffer were reduced for 30 min at 50 °C with 5 mM DTT and alkylated with 10 mM IAA. A slurry of 10 μg of Sera-Mag speedbeads (GE Healtcare) in 20 μl milliQ/ethanol (1:1, vol/vol) was added to the solution and mixed for 10 min at RT. Using a magnetic rack, the beads were immobilized and washed three times with 100 μl 80 % ethanol. 1 μg trypsin and 0.5 μg Lys-C in 100 μl 50 mM Tris/HCl pH 8.3 were added to the beads and the sample was incubated overnight at 37 °C. The tryptic digest was then acidified with TFA and desalted using a StageTip. Peptides were eluted with 100 μl 40 % acetonitrile and 0.1 % formic acid and dried using a Speedvac. Before analysis by LC-MS peptides were dissolved in 20 μl 2 % acetonitrile / 0.1% formic acid.
For PRM measurements, peptide samples with concentrations ranging from 0 to 25 ng/μl were prepared. For global proteomics, peptides were fractionated off-line using high pH reversed-phase (ThermoFisher / Pierce, #84868) into four fractions.
Synthetic AQUA peptide analogs containing a heavy stable isotope labeled C-terminal Arginine (R10) residue were purchased from Thermo.
LC-MS
Peptide mixtures were trapped on a 2 cm x 100 μm Pepmap C18 column (ThermoFisher Scientific, #164564) and separated on an in-house packed 50 cm x 75 μm capillary column with 1.9 μm Reprosil-Pur C18 beads (Dr. Maisch) at a flow rate of 250 nL/min on an EASY-nLC 1200 (ThermoFisher Scientific), using a linear gradient of 0–32% acetonitrile (in 0.1 % formic acid) during 60 or 90 min. The eluate was directly sprayed into the mass spectrometer by means of electrospray ionization (ESI).
For targeted proteomics, a parallel reaction monitoring regime (PRM) was used to select for a set of previously selected peptides on an Orbitrap Eclipse Tribrid mass spectrometer (ThermoFisher Scientific) operating in positive mode and running Tune version 3.3. Precursors were selected in the quadrupole with an isolation width of 0.7 m/z and fragmented with HCD using 30 % collision energy (CE). See Supplementary Table 2 for the isolation list. For global DDA proteomics, data were recorded on an Orbitrap Fusion Lumos Tribrid mass spectrometer (ThermoFisher Scientific) in data dependent acquisition (DDA) mode. All MS1 and MS2 spectra were recorded in the orbitrap at 30,000 resolution in profile mode and with standard AGC target settings. The injection time mode was set to dynamic with a minimum of 9 points across the peak. The sequence of sampling was blanks first and then in order of increasing peptide input amounts to avoid any contamination of previous samples.
Data analysis
Mass spectrometry data were analyzed using Mascot v 2.6.2 within the Proteome Discoverer v 2.3 (PD, ThermoFisher Scientific) framework or with MaxQuant v 1.6.10.43 (www.maxquant.org), all with standard settings (note: fragment tolerance set to 20 ppm). Raw data recorded on the Orbitrap Eclipse with the FAIMS option were first converted into mzXML format using the FAIMS MzXML Generator software tool (Coon Lab) before MaxQuant analysis. PRM data were analyzed with Skyline (skyline.ms). Spectra and chromatograms were visualized in PD 2.3, Skyline or the PDV proteomics viewer (pdv.zhang-lab.org). For global proteome analyses the UniprotKB SARS2 database (https://covid-19.uniprot.org/; 14 entries; May 2020) was concatenated with the UniprotKB database, taxonomy Chlorocebus (African green monkey) or taxonomy Homo sapiens (version Oct 2019).
RESULTS
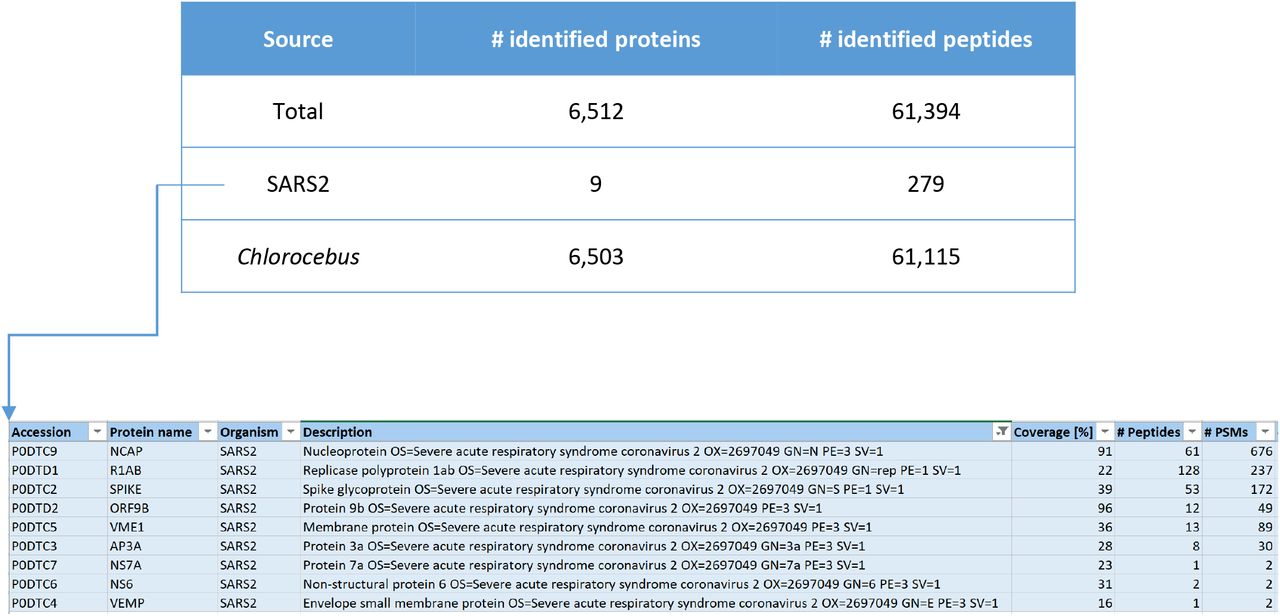
We set off by analyzing the global proteome of Vero E6 cells infected with SARS-CoV-2 using standard bottom-up proteomics. Upon off-line high pH reversed-phase (RP) peptide fractionation, LC-MS was performed on an Orbitrap Lumos and RAW files were combined during data analysis. SARS-CoV-2 proteins were measured with high sequence coverage as exemplified in Figure 1 and Supplementary Figures 1 and 2. Based on a label free semi-quantitative (LFQ) analysis of MaxQuant output data, we estimate that 4-5% of the total proteome of this sample (composed of Vero cells, viral proteins inside cells and viral particles outside of cells in the supernatant) is made up of viral proteins. Of all SARS-CoV-2 proteins covered NP is the most abundant one, making up > 88 % of all signal intensity as calculated from MaxQuant intensity values. Therefore, if intensity values can be used as a proxy for total protein abundance, almost 90 % of the SARS-CoV-2 proteome would consist of NP. Abundance of the NP protein in the samples is due to the high level production of this protein in cells as a result of the nested set of mRNAs produced during replication and the resulting overproduction of this protein. Moreover, the high number of identified Chlorocebus proteins (>6,000; see Supplementary Table 1) suggests that it is possible to not only study SARS-CoV-2 proteins, but to also investigate the effects of viral infection on the host cell proteome in great detail.

Numbers of identified proteins in SARS-CoV-2 infected Vero cells (PD2.3/Mascot search engine, offline high pH RP fractionation into four fractions, total input material … μg, 90 min LC gradients on an Orbitrap Lumos).
Based on the extensive sequence coverage for NP and several other SARS-CoV-2 proteins we established a list of peptide targets that can be used for PRM targeting. These molecular finger prints are used to program the mass spectrometer in such a way that it acts as a filter to let only those specific SARS-CoV-2 proteolytic fragments pass. This way, a specific set of target peptides/proteins can be searched for in basically any sample from which proteins can be isolated (e.g., in vitro cell cultures, patient derived samples, etc.).
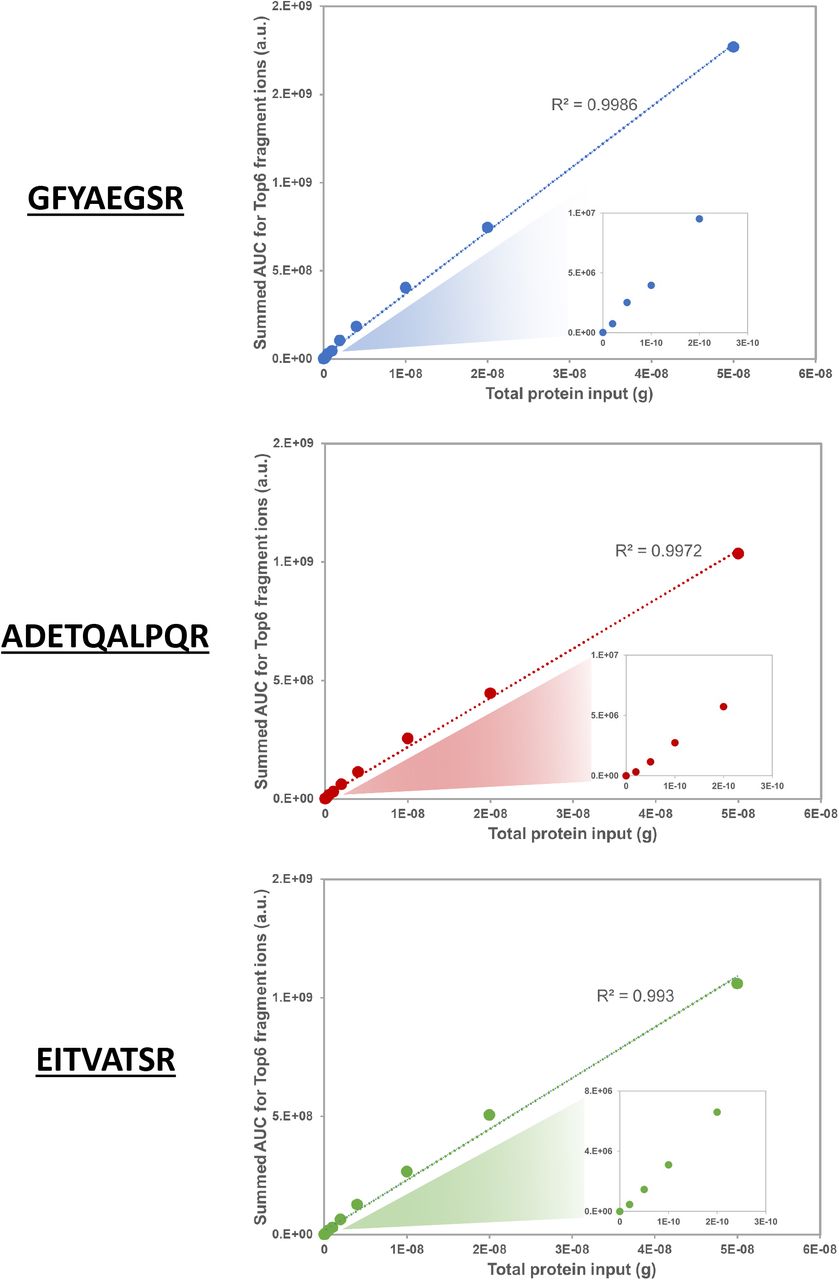
Three highly mass spectrometric responsive tryptic peptides were selected from the global proteome data set as targets for PRM, i.e. GFYAEGSR (NACP_SARS2), ADETQALPQR (NACP_SARS2) and EITVATSR (VME1_SARS2). Importantly, there are potentially a few dozens of specific SARS-CoV-2 peptides that could be used for targeting, although some of these may show slightly lower mass spectrometric responsiveness.
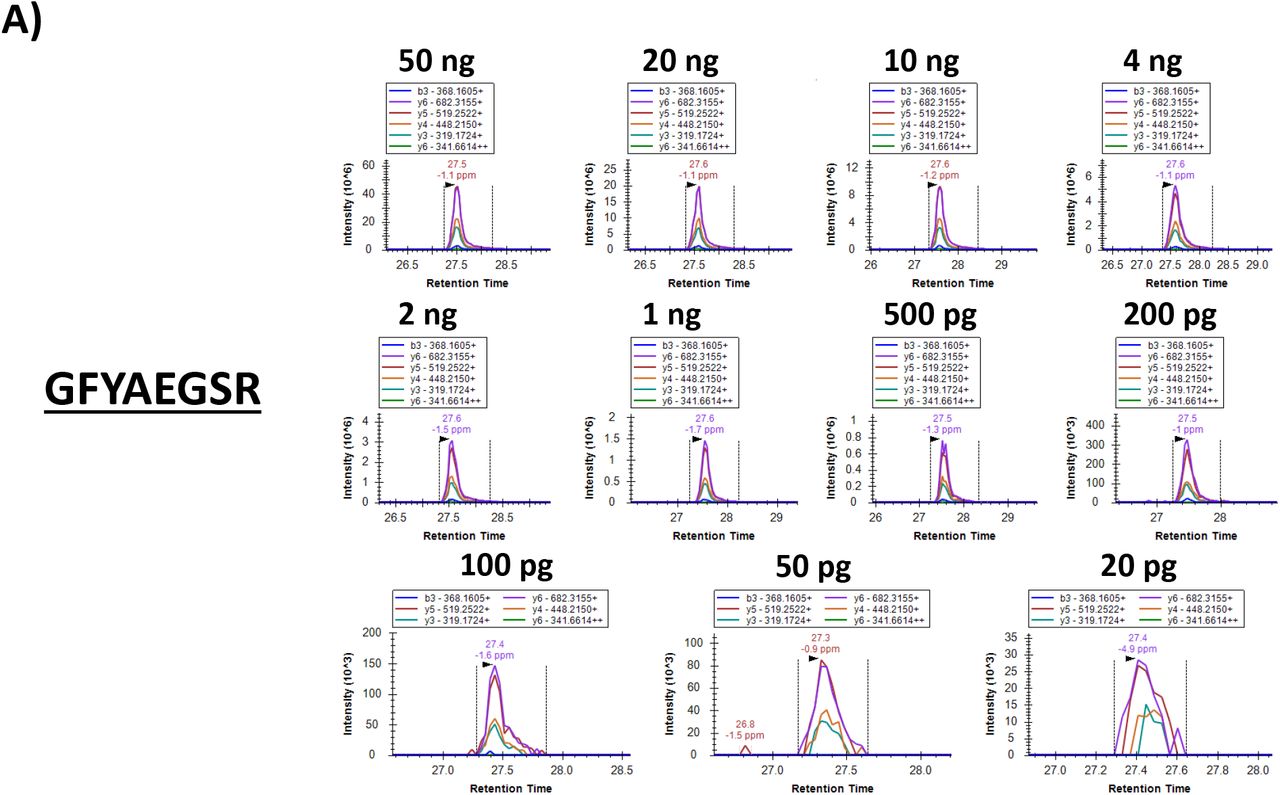
Our test sample (i.e., Vero E6 cells infected with SARS-CoV-2, referred to as ‘cells+virus’) contained 2.0 mg/ml protein based on a BCA assay. The results of the colorimetric peptide quantification after digestion were in agreement with this concentration. A dilution series was prepared from this sample and the injected total peptide quantities ranged from 50 ng down to 20 pg. These extensively diluted samples were then subjected to PRM on an Orbitrap Eclipse. Figures 2 and 3 show the results of this PRM assay. The six most intense (Top6) fragment ion peaks are shown in different colors as overlapping (in terms of retention time) peaks. The chromatogram excerpts are shown from top to bottom and left to right for decreasing total protein input concentrations. The lower right chromatogram in each panel shows the Top6 fragment ions in the sample corresponding to 20 pg total protein input, which could thus be regarded as the limit of detection (LOD). It should be noted that all PRM assays are performed on peptide targets that are present in a complex matrix, i.e. a Vero cell lysate.



PRM results visualized in Skyline (skyline.ms). Chromatograms for each of the Top6 fragment ions are shown in different colors in a dilution series for tryptic peptides A) GFYAEGSR (NP_SARS2), B) ADETQALPQR (NP_SARS2) and C) EITVATSR (VME1_SARS2). The lower right chromatogram represents the lowest sample input, i.e. 20 pg. The MS/MS spectrum on the right is the library spectrum.

Calibration curves based on PRM data for three target peptides recorded on an Orbitrap Eclipse. The summed AUC values for the Top6 fragment ions of each peptide were taken for relative quantitation. ‘Input’ is total protein input from the ‘cells+virus’ sample; inserts are zoom-ins of the input range 0 – 300 pg.
Next, we tried to detect viral proteins diluted in a complex and viscous background matrix composed of organoid derived airway cell secretions, which served as a substitute for mucus (‘in vitro mucus’). For this, we used secretions from organoid-derived human airway cultures that had been differentiated at air-liquid interphase for three weeks 10. A SARS-CoV-2 stock was added to these excretions in a ten-fold dilution series format. Using PRM mass spectrometry, several target peptides could still be detected in a 100X dilution (Figure 4).

A) PRM results from the mucus + SARS-CoV-2 spike in visualized in Skyline (skyline.ms). Chromatograms for each of the Top6 fragment ions of the tryptic peptide AYNVTQAFGR are shown for a dilution series of SARS-CoV-2 spiked into a stable background of surrogate mucus. B) bar graph visualization of PRM data.
As a proof-of-concept experiment we then applied this targeted proteomics technology to detect SARS-CoV-2 proteins in samples from COVID-19 patients. Several different types of patient samples were collected and provided to us by the Erasmus MC diagnostic department. Since all viral infectivity in these clinical specimens needs to be abolished according to established protocols in an BSL-3 facility before they can be further processed, the condition of the starting material was not optimized for subsequent proteomics. Notably, some clinical samples contained high amounts of contaminants such as detergents, albumin, etc. Sputum diluted in viral transport medium deposited on glass slides and then simply fixed in 80 % acetone turned out to be the sample type that was most compatible with the subsequent proteomics workflow. Apparently, the relatively simple background matrix composition combined with a sample preparation protocol that does not involve the addition of detergents or albumin offers a substantial advantage for proteomics workflows.
For PRM, we focused on the SARS-CoV-2 NP tryptic peptide AYNVTQAFGR, since this peptide was found to be one of the most prominent and responsive peptides in the ‘cells+virus’ sample. Also, this amino acid sequence is unique to SARS-CoV-2, even in comparison to SARS-CoV. Figure 5 shows the Skyline PRM chromatograms of various patient derived samples such as swabs and sputum compared to a pure virus positive control. Clearly, the fragmentation ion chromatogram patterns match those in the positive control and the sputum samples show the highest intensities.


A) PRM chromatograms of SARS-CoV-2 NP tryptic peptide AYNVTQAFGR in positive control (pure virus) and various patient derived samples (pellet, supernatant, sputum). B) & C) PRM chromatograms of heavy labeled synthetic AQUA peptide AYNVTQAFG[R] (m/z 568.79) and the corresponding co-eluting endogenous peptide (m/z 563.79) in various patient derived samples in which the AQUA peptide was spiked in.
In order to unambiguously confirm the presence of NP peptides we compared the chromatogram patterns of AYNVTQAFGR with those of a variant of this peptide that contains a heavy isotope labeled C-terminal Arginine. This synthetic AQUA peptide was spiked in all patient samples and co-elutes with the corresponding (non-labeled) endogenous peptide in LC-MS because of its similar biophysical properties. For several clinical samples the chromatograms of the AQUA peptides are shown in Figure 5B, while the corresponding endogenous peptides are shown in panel C. The similarities in both fragment ion chromatogram pattern and elution time confirm the presence of SARS-CoV-2 proteins in all sputum samples and sample ‘swab supernatant 4’.
CONLUSIONS & PERSPECTIVES
We show that proteolytic peptides of SARS-CoV-2 proteins can be detected down to the mid-attomole range by targeted mass spectrometry. Our rough calculations indicate that the level of sensitivity should be sufficient to detect protein amounts corresponding to 1.2E7 copies. In addition, we have shown that the current sensitivity of PRM targeted mass spectrometry is sufficiently high for the detection of virus proteins, in particular NP, present in patient material such as nasopharyngeal swabs and sputum. The identification of SARS-CoV-2 tryptic peptides was confirmed in an assay using AQUA synthesized heavy isotope labeled peptides spiked in as a positive control. Since we did not detect all SARS-CoV-2 tryptic peptides in every clinical sample that was positively tested for COVID-19 by PCR, the success of mass spectrometry based methods may depend on both the total absolute amount of viral proteins present in such samples as well as on the specific type of clinical specimens and the preparation thereof. Larger sample cohorts need to be included in future studies to further look into this.
PRM sensitivity in terms of numbers of detected virus particles is – as expected – not as high as that of RT-qPCR, which has been reported to be able to detect viral RNA in copy numbers as low as several 100s per reaction 14. A major difference compared to conventional methods of viral diagnostics is that in this study proteins are analyzed as opposed to RNA in case of PCR based methods. This makes it an orthogonal detection method that could serve as a complementary tool for diagnosing SARS-CoV-2 infection.
The excellent label free quantitation capacity of targeted mass spectrometry over a wide concentration range makes this method particularly useful for e.g. the study of infection courses over time. By using spiked in AQUA peptides it should be possible to absolutely quantitate viral proteins, which would allow for the accurate monitoring of SARS-CoV-2 protein abundances in e.g. time series. This could be useful in studies to the course of infection and for solving open questions on the importance of viral load in COVID-19 spreading.
In conclusion, the current level of sensitivity of PRM proteomics methodology and the successful detection of SARS-CoV-2 proteins in patient material opens up ways to explore the use of mass spectrometry as a technology for clinical and diagnostics labs to detect viral infection in clinical specimens. Subsequent steps should now be focused on the optimization of fast sample preparation procedures and LC-MS throughput.
DATA AVAILABILITY
All raw mass spectrometry data were uploaded to the PRIDE repository (www.ebi.ac.uk/pride/) under accession number PXD018760.

MS/MS spectra of tryptic peptides A) GFYAEGSR, B) ADETQALPQR and C) EITVATSR. Data visualization in PDV proteomics viewer (pdv.zhang-lab.org).

Tryptic peptide coverage in light green of A) Nucleocapsid (NP_SARS2) and B) Membrane protein (VME1_SARS2). Data visualization in PD2.3.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
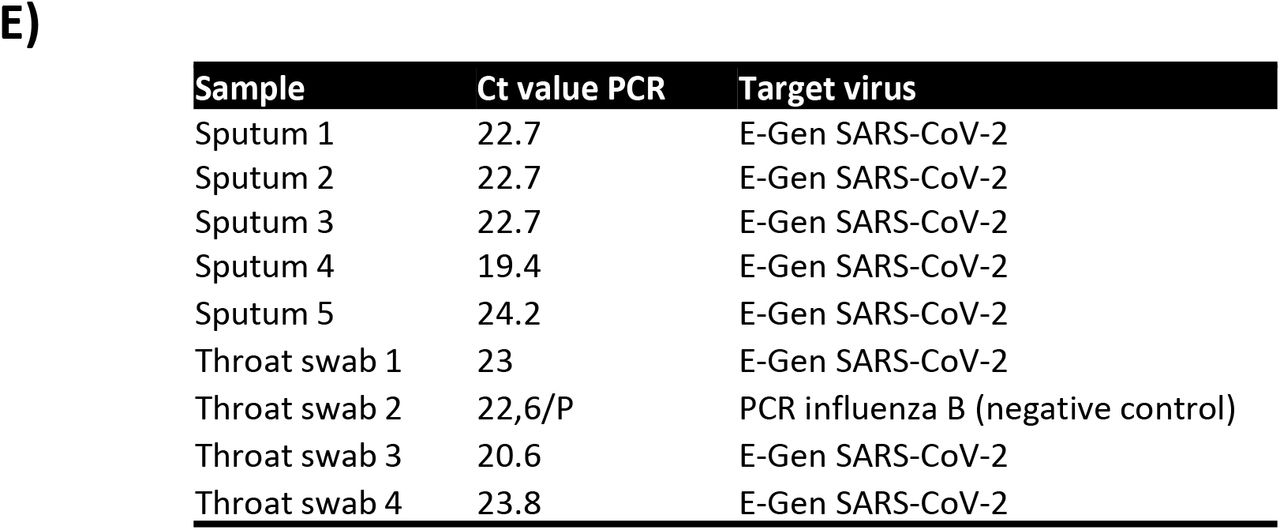
PRM chromatograms of SARS-CoV-2 NP tryptic peptide A) GFAYEGSR, B) RGPEQTQGNFGDQELIR, C) KADETQALPQR and D) ADETQALPQR in positive control and several patient derived samples, i.e. sputum deposited on glass slides (sputum 1-3), sputum collected in a tube (4,5) and throat swabs. E) The corresponding Ct values for the sputum and throat swab samples from PCR assays.
MaxQuant results of ‘cells+virus’ sample. Intensity values were taken directly from the ProteinGroups.txt output file.
SUPPLEMENTARY TABLE 1 | Mascot/PD2.3 results of fractionated ‘cells+virus’ sample.
SUPPLEMENTARY TABLE 2 | PRM isolation m/z list.
SUPPLEMENTARY TABLE 3 | Skyline export files for all PRM assays.
Footnotes
Manuscript now includes data from COVID-19 patient samples.




