

Abstract
Early detection of infection with SARS-CoV-2 is key to managing the current global pandemic, as evidence shows the virus is most contagious on or before symptom onset1,2. Here, we introduce a low-cost, high-throughput method for diagnosis of SARS-CoV-2 infection, dubbed Pathogen-Oriented Low-Cost Assembly & Re-Sequencing (POLAR), that enhances sensitivity by aiming to amplify the entire SARS-CoV-2 genome rather than targeting particular viral loci, as in typical RT-PCR assays. To achieve this goal, we combine a SARS-CoV-2 enrichment method developed by the ARTIC Network (https://artic.network/) with short-read DNA sequencing and de novo genome assembly. We are able to reliably (>95% accuracy) detect SARS-CoV-2 at concentrations of 84 genome equivalents per milliliter, better than the reported limits of detection of almost all diagnostic methods currently approved by the US Food and Drug Administration. At higher concentrations, we are able to reliably assemble the SARS-CoV-2 genome in the sample, often with no gaps and perfect accuracy. Such genome assemblies enable the spread of the disease to be analyzed much more effectively than would be possible with an ordinary yes/no diagnostic, and can help identify vaccine and drug targets. Using POLAR, a single person can process 192 samples over the course of an 8-hour experiment, at a cost of ~$30/patient, enabling a 24-hour turnaround with sequencing and data analysis time included. Further testing and refinement will likely enable greater enhancements in the sensitivity of the above approach.
Introduction
There have been over 2.8 million cases of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection to date, claiming over 200,000 lives worldwide3.
Knowing who is infected is a key first step towards pandemic containment. When a virus has a relatively high basic reproductive ratio (R0) and evidence of asymptomatic transmission, early identification of infected individuals is critical1,2. High sensitivity (i.e. a low limit of detection, or LoD) could facilitate detection of early infections.
Most SARS-CoV-2 diagnostic assays approved by the US Food and Drug Administration (FDA) are based on viral nucleic acid detection via amplification of a small number of specific viral target loci via Real-Time Polymerase Chain Reaction (RT-PCR). Although RT-PCR reactions can be extraordinarily specific, they suffer from key limitations. First, since RT-PCR assays amplify specific target loci, the assays will report a negative result if the particular target locus is not present in the sample. Consequently, RT-PCR will often produce an incorrect result when the sample is positive, but contains less than one genome equivalent in the initial reaction volume. Second, RT-PCR does not provide any genotypic information about a patient’s infection beyond the causal organism. Such data can provide insight into the specific infecting strain and aid in tracing transmission within communities. Furthermore, the capacity to quickly and efficiently generate new viral genome data could expedite the generation of new diagnostics, vaccines and precise antivirals.
In principle, whole-genome DNA sequencing of SARS-CoV-2 has the potential to overcome these limitations. DNA sequencing can detect genome fragments even when a complete genome is not present in the sample. It can also extract extensive genotypic information about the viral genomes and genome fragments that are present. Notably, the SARS-CoV-2 genome is free of repeats, making it susceptible to complete characterization using short DNA reads.
To exploit this possibility, we have developed Pathogen-Oriented Low-cost Assembly & Re-sequencing (POLAR), which combines: (i) enrichment of SARS-CoV-2 sequence using a PCR primer library designed by the ARTIC Network3; (ii) tagmentation-mediated library preparation for multiplex sequencing on an Illumina platform; and (iii) SARS-CoV-2 genome assembly (Figure 1). We show that POLAR is a reliable, inexpensive, and high-throughput SARS-CoV-2 diagnostic. Specifically, POLAR makes it possible for a single person to process 192 patient samples in an 8-hour workday day at a cost of $31 per sample. Including time for sequencing and data analysis, POLAR still enables a 24-hour turnaround time. POLAR also achieves very high sensitivity, with a limit of detection of 84 genome equivalents per milliliter, outperforming nearly all diagnostic tests currently approved by the US Food and Drug Administration (FDA).
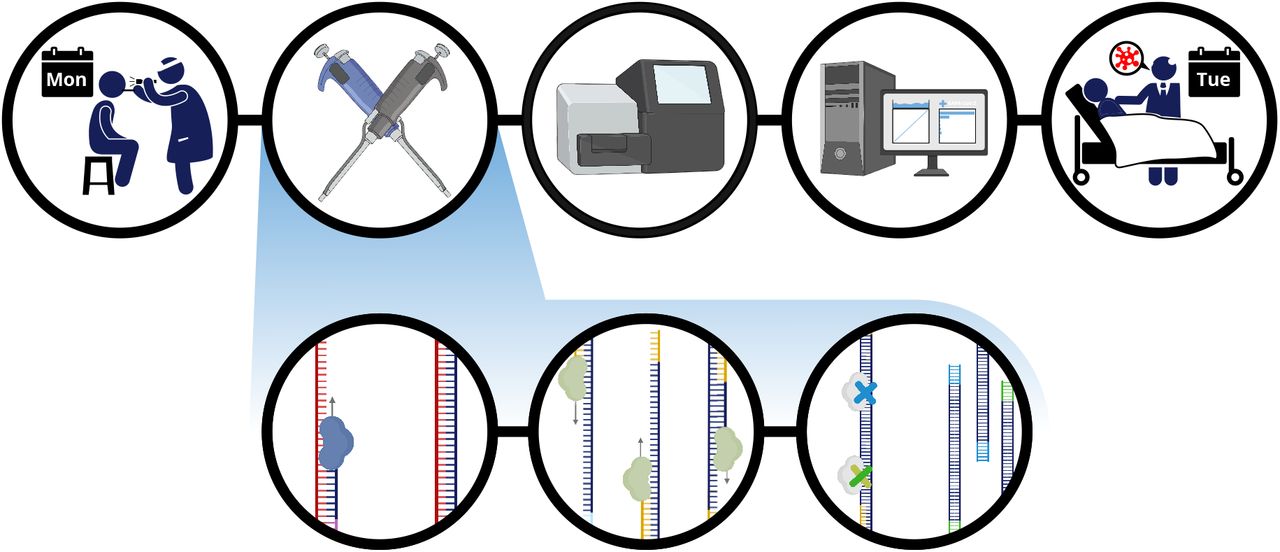
Patient is sampled in the clinic and total RNA from this sample is extracted and reverse transcribed into DNA. The sample is then enriched for SARS-CoV-2 sequence using a SARS-CoV-2 specific primer library. The amplicons then undergo a rapid tagmentation mediated library preparation. Data is then analyzed and used to report patient result the next day.
In POLAR, total RNA from a clinical sample is reverse transcribed into DNA. This is followed by a multiplex polymerase chain reaction using a SARS-CoV-2 specific primer library to generate 400bp amplicons that tile the viral genome with ~200bp overlap, enriching the library for SARS-CoV-2 sequence. The amplicons are then fragmented and ligated to adapters using a rapid tagmentation mediated library preparation, and barcoded to enable multiplex sequencing. Finally, the data is analyzed using a one-click analysis software package that we have created.
As part of this analysis, we determine whether a sample is infected by aligning the sequenced reads against a set of coronavirus reference genomes. Positive samples are identified as ones in which the sequenced reads cover more than 5% of the SARS-CoV-2 reference genome after primer sequences are filtered out. This diagnostic approach achieves a limit of detection of 84 genome equivalents per milliliter, making it more sensitive than nearly all methods currently approved by the FDA. When the viral concentration is higher (though still lower than, for instance, the limits of detection for the CDC SARS-CoV-2 tests), the data is also used to assemble an end-to-end, error-free SARS-CoV-2 genome from the sample, de novo.
Results
Whole-genome sequencing of SARS-CoV-2 yields a highly sensitive diagnostic
We began by evaluating the suitability of the POLAR protocol as a potential diagnostic methodology.
To do so, we created 5 successive 10-fold serial dilutions of a quantified SARS-CoV-2 genomic RNA sample obtained from the American Tissue Culture Society (ATCC), which is widely used as a reference standard for diagnostic development. Specifically, we prepared positive controls containing 840,000 genome equivalents/mL, 84,000 genome equivalents per milliliter, 8,400 genome equivalents per milliliter, 840 genome equivalents per milliliter and 84 genome equivalents per milliliter. We performed 20 replicates at each concentration.
We also prepared a series of negative controls: 2 replicates of nuclease-free water, processed separately from the positive samples; 2 replicates of HeLa RNA extract, and 2 replicates of K562 RNA extract. We additionally included 20 replicates of nuclease-free water, prepared side-by-side with the positive samples, to serve as cross-contamination controls. The side-by-side cross contamination controls were included in order to ensure that our method was not susceptible to false positives due to cross-contamination, a common error modality that is not well regulated in current FDA guidelines for diagnostic test development. In total, we performed the POLAR protocol on 26 different negative controls. Note that the above reflects the totality of experiments performed with our assay; we did not censor completed replicate experiments for any reason.
Each of the above 126 samples was processed using the POLAR protocol, and sequenced on a NextSeq550 Mid-Output Flow-cell. Note that, although a single technician can perform 192 experiments using the above workflow in an 8-hour shift, we did not perform all 192 experiments in the initial test. For these samples we generated 20 million paired-end 75bp reads of preliminary data.
To classify samples as positive or negative, we down sampled the data to 500 reads (2.5x coverage) per sample and checked to see if the breadth of coverage (the percentage of the target genome covered by at least 1 read, once primers are filtered out) was larger than 5% for each of the above samples (Figure 2A).

(A) Coverage tracks demonstrate sequencing depth across the SARS-CoV-2 genome produced by our protocol from samples with a range of starting SARS-CoV-2 genome concentrations. Red-highlighted regions represent virus sequence detected by qPCR-based COVID-19 diagnostics in use or development. (B) Scatter plot shows breath of coverage for all samples from all replicate dilution series. Dashed red line represents the empirically determined breadth of coverage threshold for positive samples.
Of the 100 true positives, we accurately classified 99 (99%), with a single false negative at the most dilute concentration, 84 genome equivalents per milliliter. All 80 higher-concentration samples (840 genome equivalents/mL or more) were accurately identified as positive with an average breadth of coverage of 71%; 95% of the samples at 84 genome equivalents/mL were accurately classified (19 of 20), with an average breadth of coverage of 19%. All but 1 of 26 true negatives were accurately classified as negative, with an average breadth of coverage of 1%; the single misclassification was one of the cross-contamination controls.
Taken together, these data highlight the accuracy of the diagnostic test even when the amount of sequence data generated is negligible. Furthermore, they establish that the limit of detection of our assay, using the FDA definition, is 84 genome equivalents per milliliter (see Figure 2A).
The POLAR protocol for whole-genome sequencing of SARS-CoV-2 is more sensitive than nearly all diagnostics currently approved by the US FDA
To compare POLAR to available diagnostic tests, we examined the 57 emergency use authorization summaries describing each of the 57 molecular SARS-CoV-2 diagnostic tests approved by the US FDA. For 44 tests, a limit of detection was clearly reported to the FDA in genome equivalents/milliliter (or, alternatively, genome copies/mL). For 41 of these 44 tests, the limit of detection was >= 100 genome equivalents/milliliter4–50. (Note that the LoD for the more sensitive of the two tests developed by the Center for Disease Control is 1000 genome equivalents per milliliter.) Thus, our test was significantly more sensitive than nearly all of the available tests.
We believe that this enhanced LoD is likely due to the fact that our method amplifies the entire viral genome, whereas RT-PCR only targets a handful of loci (Figure 2B). For instance, when examining the 21 different publicly available SARS-CoV-2 RT-PCR primer sets from the UCSC Genome Browser, we see that, even in aggregate, these primers amplify only 6.86% of the SARS-CoV-2 genome. At low starting concentrations of SARS-CoV-2, a sample can contain fragments of the viral genome that are detectable via whole genome sequencing, but which may not include the specific locus targeted by a particular RT-PCR assay.
The POLAR protocol for whole-genome sequencing of SARS-CoV-2 enables assembly of an end-to-end SARS-CoV-2 genome even from low-concentration samples
Next, we sought to determine if the sequencing data resulting from the POLAR protocol could be used to assemble de novo the SARS-CoV-2 viral genome.
To explore this question, we took 150,000 75–base pair paired-end Illumina reads (2 x 75bp) from each of 18 libraries, comprising 3 different dilution series and corresponding negative controls. For each library, we generated a de novo assembly using the memory efficient assembly algorithm MEGAHIT51 with default parameters. We first assessed the accuracy of these de novo assemblies by comparing them to the SARS-CoV-2 reference genome using a rescaled genome dot plot (Figure 3). The de novo assemblies showed very good correspondence with the SARS-CoV-2 reference, including the samples that contained only 84 genome equivalents per milliliter. We then quantified the accuracy and quality of these de novo SARS-CoV-2 assemblies. For the de novo assemblies that contained ≥8,400 equivalents per milliliter, 86.67% of assemblies consisted of a singular contig comprising 99.77% of the SARS-CoV-2 genome (Table 1). The remaining 0.23% of the SARS-CoV-2 genome corresponds to short regions at both ends of the genome, which are not amplified by the ARTIC primer set. While the de novo assemblies created from samples with 840 genome equivalents/mL and 84 genome equivalents/mL are less contiguous, we are able to recover on average 84.63% and 34.16% of the viral genome, respectively. Remarkably, 100% of the bases in 16 of these 20 de novo assemblies match the corresponding bases in the SARS-CoV-2 reference genome. Three of the remaining four de novo assemblies have only a single base pair difference as compared to the SARS-CoV-2 reference genome. Collectively, these data demonstrate that our method provides de novo SARS-CoV-2 genome assemblies at viral concentrations at or below the CDC RT-PCR limit of detection. Furthermore, at most of the concentrations examined, the SARS-CoV-2 genome assemblies produced by POLAR are gapless, and completely free of errors.
Assembly Statistics of Assembly SARS-Cov-2 Genome de novo Using Range of Starting SARS-Cov-2 Genome Concentrations.

Each rescaled genome dot plot (black boxes numbered 1 to 24) compares a de novo SARS-CoV-2 assembly (Y-axes) to the SARS-CoV-2 reference genome (X-axes). Columns contain replicate assemblies at a given SARS-CoV-2 concentration. The de novo assemblies displayed on the Y-axes have been ordered and oriented to match the reference viral genome in order to facilitate comparison. Each green line segment represents the position of an individual contig from the de novo assembly that aligned to the reference genome. Dotted red line represents the limit of detection for the Center for Disease Control qPCR tests currently used to detect SARS-CoV-2. For rescaled dot plots, contigs were sorted and unmapped contigs have been removed, leaving all remaining aligning contigs lying along the diagonal.
Protocol accurately assembles other coronaviruses, while distinguishing them from SARS-CoV-2
SARS-CoV-2 is one of many coronaviruses that commonly infect humans. We therefore sought to determine whether POLAR (which uses SARS-CoV-2 specific primers) could accurately distinguish between SARS-CoV-2 and other coronaviruses. To do so, we applied POLAR to samples containing genomic RNA from the following coronaviruses: Human Coronavirus NL63, Human Coronavirus strain 229E, Porcine Respiratory Coronavirus strain ISU-1 and Avian Coronavirus. (Genomic RNA was obtained from ATCC.)
Notably, for Porcine Respiratory Coronavirus strain ISU-1, Human Coronavirus strain 229E and Avian Coronavirus, our automated pipeline assembled the entire viral genome with no gaps (Figure 4). For Human Coronavirus NL63, there was a single gap. These assemblies covered at least 96.01% of their respective reference genome assembly, with a base accuracy of >99.9% (Table 2).
Assembly Statistics of Assembly of Other Coronavirus Genomes de novo.
Compilation of The Limit of Detection of FDA Approved SARS-Cov-2 Diagnostic Tests.

Genome dot plots comparing de novo assemblies and reference genomes for test samples spiked with non-SARS-CoV-2: Human Coronavirus strain 229E, Avian Coronavirus, Porcine Respiratory Coronavirus, and Human Coronavirus NL63. De novo assemblies on Y-axes, species matched reference genomes on X-axes. The de novo assemblies displayed on the Y-axes have been ordered and oriented to match the reference viral genomes in order to facilitate comparison.
At the same time, like our other SARS-CoV-2 negative controls, the data from these experiments had a breadth of coverage of <0.5% when the sequenced reads were aligned back to the SARS-CoV-2 reference genome. Thus, in all four cases, our pipeline accurately determined that these true negatives did not contain SARS-CoV-2. This highlights the potential of our approach for diagnosing other coronaviruses, including cases of co-infection by multiple coronaviruses including, but not limited to, SARS-CoV-2.
Automated analysis pipeline facilitates data analysis, providing one-click diagnostic report
To aid in the analysis of data produced by POLAR, we also developed a one-click open-source pipeline that takes the DNA reads produced from a sample and performs all the previously mentioned analyses, generating a document containing breadth of coverage statistics, a genome dot plot, and a test result (positive or negative) (Figure 5). The pipeline also reports the resulting SARS-CoV-2 reference genome, as well as any other genome assemblies that were generated. We confirmed that the pipeline can be run efficiently on a wide range of high-performance computing platforms and that the computational cost per test is negligible (<1¢). The pipeline, including documentation and test set, is publicly available at https://github.com/aidenlab/Polar.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The pipeline aligns the sequenced reads to a database of coronaviruses; if run on a cluster, this is done in parallel. Separately, the pipeline creates contigs from the sequenced reads. The resulting de novo assembly is then pairwise aligned to the SARS-CoV-2 reference genome. A custom python script then analyzes these data to determine the test result and compiles dot plots and alignment percentages into a single PDF. (B & C) Each report includes a genome dot plot of the de novo assembly against the SARS-CoV-2 reference genome, with a coverage track of sequenced reads aligned to the SARS-CoV-2 reference genome above the dot plot. The report also includes the breadth of coverage of sequenced reads aligned to 17 different coronaviruses. The diagnostic answer is given in the form of a “+” or “−” symbol and “Positive” or “Negative” for SARS-CoV-2 coronavirus in the top right corner of the report.
Discussion
Given the current need for SARS-CoV-2 testing, we developed a reliable, inexpensive, and high-throughput SARS-CoV-2 diagnostic based on whole genome sequencing. Our method builds off those developed by ARTIC Network for in-field viral sequencing in order to generate real-time epidemiological information during viral outbreaks52,53. We have demonstrated that this approach is sensitive, scalable, and reproducible, and consistent with US FDA guidelines for diagnostic testing for SARS-CoV-2.
The POLAR protocol has two key advantages over RT-PCR-based diagnostics.
First, it is highly sensitive, achieving a LoD of 84 genome equivalents per milliliter, which exceeds the reported LoD of all but three diagnostic tests54–56 approved by the US FDA. We believe that further refinements of the protocol will likely allow this to be further improved. By enhancing sensitivity, it may be possible to detect infection earlier in the course of infection – ideally, before a person is contagious – and to detect infection from a wider variety of sample types.
Second, it produces far more extensive genotype data than targeted, RT-PCR based diagnostics, including an end-to-end SARS-CoV-2 genome at concentrations that are beyond the limit of detection of many other assays. Having whole viral genomes from all diagnosed individuals enables the creation of viral phylogenies to better understand the spread of the virus in community and health care settings. It will further yield valuable understanding of the different strains and patterns of mutations of the disease. Finally, it will enable the discovery of additional testing, vaccine, and drug targets.
At the same time, the approach we describe also has several limitations as compared to other diagnostic tests. For example, our method does not provide any information regarding the SARS-CoV-2 viral load of a patient. This might be addressed by adding a synthetic RNA molecule with a known concentration into each patient sample in order to estimate viral load using relative coverage.
Another limitation is that our method is slower than other approaches, in the sense that it requires 24 hours from acquisition of a patient sample to a diagnostic result. By contrast, Abbott Labs (one example of many new diagnostic technologies developed in the past few months) has developed a diagnostic test capable of returning results in as little as 5 minutes for a positive result and 13 minutes for a negative result57. However, it is worth noting that the maximum number of diagnostic results an Abbot device could complete running 24 hours a day is roughly between 111 tests and 126 tests depending on the number of positive results58,59.
Beyond diagnosis of individual patients, POLAR can also be applied to SARS-CoV-2 surveillance, in settings such as municipal wastewater treatment plants60. In principle, such approaches could identify and characterize infection in a neighborhood or city very inexpensively, even for a large population, informing public policy decisions.
We note that multiple groups have been developing methods for sequencing whole SARS-CoV-2 genomes, and in some cases sharing the protocols ahead of publication on protocols.io (https://www.protocols.io/). Like POLAR, these methods often using the ARTIC primer set, with some of these approaches relying on long-read DNA sequencing61. Although long reads enable more contiguous genome assemblies when the underlying genome contains complex repeats, we find that such reads are not necessary for gapless assembly of SARS-CoV-2. As such, the use of long reads, which is costly, produces less accurate base calls, and hampers multiplexing, may be less applicable in a diagnostic context.
Other methods use short read DNA sequencing62–65. Most of these approaches partition individual samples into multiple wells, making them difficult to perform in a highly multiplex fashion63,64. One method enables extensive multiplexing, but does not include a random fragmentation step. As such, it requires 2 x 150bp paired-end reads in order to produce gapless assemblies, making a 24-hour turnaround impossible on extant Illumina devices65. Because POLAR includes a random fragmentation step requiring only 75bp reads, it can be performed on more rapid instruments such as NextSeq 550. However, at least one of these methods appears to enable both rapid turnaround and 9-fold multiplexing62. Although prior studies have not explored sensitivity and specificity of these protocols when deployed as a diagnostic, emerging work from many laboratories make it clear that whole-genome sequencing of SARS-CoV-2 is likely to be a promising modality that is well-suited for clinical use.
Methods & Materials
Collection of SARS-CoV-2
The quantified sample material used for the limit of detection was genomic RNA (gRNA) extracted from a cell line (Vero E6, ATCC® CRL-1586™) infected with SARS-related coronavirus 2 (SARS-CoV-2, isolate USA-WA1/2020, Lot: 70033700), using QIAamp® Viral RNA Mini Kit (Qiagen 52904) deposited by American Type Culture Collection (ATCC) and obtained from Biodefense and Emerging Infections Research Resources Repository (BEI Resources). The amount of viral genome RNA molecules per volume of total RNA including cellular nucleic acid and carrier RNA for the lot we received of SARS-CoV-2 gRNA was quantified as 5.5 × 104 genome equivalents/μL using a BioRad QX200 Droplet Digital PCR (ddPCR™) System.
Negative control RNA extraction
Approximately 1 million K562 cells and 1 million HeLa cells cultured in our lab were used as the starting material for RNA extraction using columns provided in the RNeasy Mini Kit (Cat no: 74104). The final elute was collected in 30μl of RNA-free waterThis elution was then split into 10μL aliquots and concentration was measured by using the GE Nanovue plus.
Performing the SARS-CoV LoD
The limit of detection (LoD) was determined by making five 10-fold serial dilutions using the SARS-CoV-2 gRNA from ATCC as a stock and nuclease-free water as a diluent. Each dilution was tested with two biological replicates each of which further had 10 technical replicates, so in total 20 replicates from each dilution of stock SARS-CoV-2 gRNA. For experiment,1μl of each dilution was spiked into a mix of 4.5μl of nuclease-free water, 0.5μl of 10mM dNTPs Mix (NEB, N0447L) and 0.5 μl of 50μM Random Hexamers (ThermoFisher, N8080127) to serve as the starting material, “RNA Extract”, for the protocol. The RNA, hexamers, and dNTPs mixture was incubated at 65°C for 5 minutes followed by a 1 minute incubation at 4°C in order to anneal hexamers to RNA. In order to reverse transcribe RNA into cDNA, we added 2μl of 5X SuperScript™ IV Reverse Buffer (ThermoFisher, 18090050), 0.5μl of SuperScript™ IV Reverse Transcriptase (200 U/μL) (ThermoFisher, 18090050), 0.5μl of 100mM DTT (ThermoFisher, 18090050), 0.5μl of RNaseOUT Recombinant Ribonuclease Inhibitor (ThermoFisher, 10777-019) to the hexamer annealed RNA. The reaction was then incubated at 42°C for 50 minutes followed by an incubation at 70°C for 10 minutes before holding at 4°C. For amplification of cDNA, we used SARS-CoV-2-specific version 3 primer set (total 218 primers) designed by Josh Quick from the ARTIC Network. Primers were purchased at LabReady concentration of 100μM in IDTE buffer (pH 8.0) from Integrated DNA Technologies (IDT). Multiplex-polymerase chain reaction (PCR) was performed in two separate reaction mixes prepared by combining 5μl of 5X Q5 Reaction Buffer (NEB, M0493S), 0.5μl of 10 mM dNTPs (NEB, N0447L), 0.25μl Q5 Hot Start DNA Polymerase (NEB, M0493S)with either 12.7μl Nuclease-free water (Qiagen, 129114) and 4.05μl of 10μM “Primer Pool #1” or, 12.77μl Nuclease-free water (Qiagen, 129114) and 3.98uL for 10μM “Primer Pool #2”. The final concentration of each primer in the reaction mix was 0.015μM in the PCR mix. 22.5μl of the corresponding mastermix (Pool #1 or Pool #2) was combined with 2.5μl of the reverse transcribed cDNA. The reaction was then incubated at 98°C for 30 seconds for 1 cycle followed by 25 cycles at 98°C for 15 seconds and 65°C for 5 minutes before holding at 4°C. Pool #1 or Pool #2 amplicons from each replicate were then mixed together and cleaned by adding 1:1 volume of sparQ PureMag beads (QuantaBio, 95196-060) and incubating at room temperature for 5 minutes. The beads were separated using a magnet and the supernatant was discarded, followed by two 200μl washes of freshly made 80% ethanol. Each sample was eluted in 11μl of 10mM Tris-HCl (pH 8.0) and incubated for 2 minutes at 37°C followed by separation on a magnet. The DNA was then quantified using a Qubit® High Sensitivity Kit (ThermoFisher, Q32851) as per manufacturer’s instructions and the concentrations were used to ensure 1ng of amplicon DNA in 4μl was carried per sample into library preparation.
Library preparation was performed using the Nextera XT DNA Library Preparation Kit (Illumina, FC-131-1096) and Nextera XT Index Kit v2 (Illumina, FC-131-2001/2002). 4μl of 1ng amplicon DNA was combined with a mix containing 1μl of Amplicon Tagment Mix (Illumina, FC-131-1096) and 5μl of Tagment DNA Buffer (Illumina, FC-131-1096) and incubated at 55°C for 5 minutes. Temperature was then lowered to 10°C followed by addition of 2.5μl of Neutralize Tagment Buffer immediately after the cooling started, mixed by pipetting, and incubated at room temperature for 5 minutes. After 5 minutes, the reaction was centrifuged at 280xG for 1 minute and the next reaction was set-up during centrifugation. 12.5μl of a mastermix containing 7.5μl of Nextera PCR Master Mix (Illumina, FC-131-1096) and 2.5μl of each Index primer i7 (Illumina, FC-131-2001/2002) and Index primer i5 (Illumina, FC-131-2001/2002) was combined with 12.5μl of the tagmented amplicon DNA. The reaction was then incubated on a thermal cycler with the following parameters: 1 cycle at 72°C for 3 minutes and 95°C for 30 seconds, 18 cycles at 55°C for 10 seconds, 72°C for 30 seconds, 72°C for 5 minutes followed by a 4°C hold. Post PCR clean up was done using 1:1.8 volume (45μL beads in 25uL reaction) of sparQ PureMag beads (QuantaBio, 95196-060), washed twice with 80% ethanol, eluted in 20μL of 10mM Tris-HCl (pH 8.0) followed by an incubation at 37°C for 2 minutes and separated on a magnetic plate. 10μl from each well of the plate was then transferred onto the corresponding well on a new midi plate. A Library Normalization (LN) (Illumina, FC-131-1096) master mix was created by combining two reagents in a 15μl conical tube. The reagents were multiplied by the number of samples being processed: 23μl of LNA1 and 4μl of LNB1. The mixture was then mixed by pipetting 10 times and then poured into a trough. Using a p200 multichannel pipette, 22.5μl of LN master mix was placed into each sample well. To mix, we sealed the plate, and vortexed using a plate shaker at 1800rpm for 30 minutes. The plate was then placed on a magnetic stand to separate the beads. Once the liquid on the plate was clear, without disturbing the beads, we discarded the supernatant. The beads were then washed twice by adding 22.5μl of LNW1 to each well, sealing the plate, using the plate shaker at 1800rpm for 5 minutes, then separating the beads on a magnetic plate and discarding the supernatant. After the washes, 15μl of 0.1N NaOH was added to each well. The plate was then sealed and vortexed at 1800rpm to mix the sample for 5 minutes. During the 5-minute mixing, 15μl of LNS1 was added to each well of a new 96-well PCR plate that was labeled as SGP. After the 5-minute elution step, the plate was placed on a magnetic stand, and 15μl of the supernatant was transferred to the corresponding well of the SGP plate. The plate was then sealed and spun at 1000xG for 1 minute.
Preparation of Illumina sequencing run
To prepare for the sequencing run, a “Mid-Output Kit” reagent cartridge (Illumina, 20024904) was removed from the −20°C freezer to thaw in a secondary container filled with room temperature deionized water about 1/3rd of its height. The cartridge was left in this water bath for 1 hour to completely thaw. 30 minutes into the thawing, the “Mid-Output Kit” flow cell (Illumina, 20024904) was removed from the 4°C refrigerator to warm up to room temperature for 30 minutes.
A tube of Hybridization Buffer (HT1) (Illumina, 20015892) was collected from the −20°C and thawed at room temperature. Once thawed, it was placed on ice. A thermomixer was pre-heated to 98°C. The libraries were pooled equally by using 2μl of all the normalized samples and the pool was then vortexed and centrifuged down to ensure proper mixing of the samples. 5μl volume from this pool was transferred into a new tube to which 995μl of ice-cold Hybridization Buffer (HT1) (Illumina, 20015892) was then added. The tube was quickly vortexed and centrifuged at 300xG for 1 minute. From this tube, 750μl was transferred to a new tube. To this new tube 750μl ice-cold Hybridization Buffer (HT1) (Illumina, 20015892) was added to further dilute the sample molarity. The tube was quickly vortexed, centrifuged and was placed at 98°C in the thermomixer for 2 minutes. Immediately after 2 minutes, the tube was placed on ice for 5 minutes. In a fresh 1.5mL tube, to bring the final concentration in the pool to 1.5pM, 97μl of the previous dilution and 1203μl of ice-Cold Hybridization buffer (HT1) (Illumina, 20015892) were mixed together. The final denatured library pool was then placed on ice until it was ready to load onto the reagent cartridge. Finally, the loaded reagent cartridge, along with the flow cell and buffer pack were inserted into the NextSeq500 for sequencing.
SARS-CoV-2 Coverage Analysis
To compare SARS-CoV-2 coverage across starting concentrations, FASTQs were aligned to the SARS-CoV-2 reference genome (NCBI Reference Sequence: NC_045512.2) using BWA66 with default parameters. Samtools67 was then used to sort, fixmates, dedup, and calculate depth per base. To filter out primer reads, we first discarded all depths per base below a threshold of >1, and then removed islands that had 50 or fewer consecutive positions covered. Remaining values are plotted by base position in the coverage track.
The rescaled dot plot below is generated by plotting contig alignment generate by Minimap268 to the reference genome. Contigs are sorted and non-mapped contigs have been removed, leaving all remaining aligning contigs lying along the diagonal.
RT-PCR primers regions were created by downloading RT-PCR primers from the UCSC genome browser69. Forward and reverse primers were paired to generate RT-PCR target regions for each pair. Bedtools70 was then used to merge these individual RT-PCR target regions into a single track in order to collapse the overlapping RT-PCR target regions.
Breadth of Coverage Scatter Plot
Breadth of coverage was determined after filtering out primer reads (described above). The number of positions with coverage after primer filtering was divided by the total length of the reference. This value is stored in the “stats.csv” file produced by the POLAR pipeline for all coronaviruses and used in the final report to create the bar charts and determine the result (positive or negative).
To create the scatter plot in Figure 2, data was plotted in R Studio using ggplot271, dplyr72 and forcats libraries. A position jitter was used to allow for better visualization of data points, which at high concentrations of SARS-CoV-2 often overlapped. The jitter parameters were calibrated to allow for optimal visualization of data points without changing the relative position of each data point.
Comparative Assembly Statistics
In order to determine the accuracy of our de novo assemblies, we compared our SARS-CoV-2 de novo assembly to the SARS-CoV-2 reference assembly (NCBI Reference Sequence: NC_045512.2), our Human coronavirus 229E de novo assembly to the Human coronavirus 229E reference assembly (NCBI Reference Sequence: NC_002645.1), our Avian Coronavirus to the Avian Coronavirus Massachusetts (formerly Avian Infectious Bronchitis Virus) (GenBank: GQ504724.1), our Human Coronavirus NL63 de novo assembly to the Human Coronavirus NL63 (GenBank: AY567487.2) reference assembly and our Porcine Respiratory Virus to the PRCV ISU-1 (GenBank: DQ811787.1) reference genome using MetaQuast73.
Per Sample Cost Breakdown of Reagents Needed to Perform the POLAR Method.
List of SARS-Cov-2 RT-PCR Primer Regions.
Benchmarking parameters for “Pipeline for POLAR: viral diagnostic for SARS-CoV2.”
Acknowledgments
This work was supported by the Thrasher Research Fund Early Career Award to a.p.a., a Howard Hughes Medical Institute Gilliam Fellowship to b.g.s., and an NSF Physics Frontier Center Grant. We thank Terry Leatherland, Grace Liu, Loic Fura and Victoria Nwobodo for access to a high RAM IBM E880 server. We thank Gary Schroth, Linda Ray, Erich Jaeger, Steph Craig and Mehdi Keddache of Illumina for flow cells, reagents, and constructive feedback. We also thank Dr. Joseph Petrosino of Baylor College of Medicine for fruitful discussions.
The benchmarking work was supported by resources provided by the University of Western Australia and the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia. We also gratefully acknowledge Microsoft and the WA technology company DUG for beta testing and benchmarking the pipeline on their systems.
We are also grateful to Dr. Clavia Ruth Wooton-Kee, Dr. David Cunningham, Ellen Busschers, and Dr. Dmitriy Khodakov and Dr. Christophe Herman for providing reagents.
References




