

Abstract
Primary cilia are sensory organelles that are built and maintained by intraflagellar transport (IFT) multi-protein complexes. Deletion of certain ciliary genes in Autosomal Dominant Polycystic Kidney Disease (ADPKD) mouse models markedly attenuates PKD severity, indicating that a component of cilia dysfunction may have potential therapeutic value. To broaden the role of ciliary dysfunction, here we investigate the role of global deletion of Ift-A gene, Thm1, in juvenile and adult ADPKD mouse models. In cyst-lining cells of both juvenile and adult ADPKD models, cortical collecting duct cilia lengths and cytoplasmic and nuclear levels of the nutrient sensor, O-linked β-Nacetylglucosamine (O-GlcNAc) were increased. Relative to juvenile Pkd2 conditional knock-out mice, deletion of Thm1 together with Pkd2 both increased and reduced cystogenesis in a tubule-specific manner without altering kidney function, inflammation, cilia lengths, and ERK, STAT3 and OGlcNAc signaling. In contrast, Thm1 deletion in adult ADPKD mouse models markedly attenuated almost all features of PKD, including renal cystogenesis, inflammation, cilia lengths, and ERK, STAT3 and O-GlcNAc signaling. These data suggest that differential factors in the microenvironments between renal tubules and between developing and mature kidneys influence cilia and ADPKD pathobiology. Further, since O-GlcNAcylation directly regulates ciliary homeostasis and the balance between glycolysis and oxidative phosphorylation, we propose that increased O-GlcNAcylation may promote certain key ADPKD pathological processes.
Introduction
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is among the most common, fatal monogenetic diseases, affecting approximately 1:1000 individuals worldwide1. ADPKD is characterized by the growth of large fluid-filled renal cysts, which cause injury and fibrosis and can lead to end-stage renal disease by the 6th decade of life. Tolvaptan is the only FDA-approved therapy, but has variable effectiveness and aquaresis side effects2, 3. Thus, the need to discover additional underlying disease mechanisms and design new therapeutic strategies continues.
Primary cilia are small, antenna-like sensory organelles that play an important role in ADPKD pathobiology via mechanisms that remain unclear. ADPKD is caused by mutations in PKD1 (≥80% of cases) or PKD2 (≥10% of cases), which encode polycystin 1 (PC1) and polycystin 2 (PC2), respectively4. PC1 and PC2 form an ion-channel receptor complex that functions at the primary cilium. While PC1 and PC2 also localize to other subcellular compartments, analyses of human ADPKD primary renal epithelial cells, of mouse models harboring human ADPKD mutations, and of an ethylnitrosourea (ENU)-induced Pkd2 mouse mutation that causes ciliary exclusion of PC2, indicate that deficiency of PC1 or PC2 from the cilium is sufficient to cause ADPKD5–7.
Primary cilia are synthesized and maintained via intraflagellar transport (IFT), which is the bidirectional transport of protein cargo along a microtubular axoneme. Two multiprotein complexes mediate IFT. The IFT-B complex interacts with the kinesin motor and mediates anterograde IFT, while the IFT-A complex together with cytoplasmic dynein mediates retrograde IFT. IFT-A proteins are also required for ciliary import of membrane and signaling molecules8–10. In mice, deletion of Ift-A or -B genes either perinatally or in the embryonic kidney results in renal cystic disease11–13. However, these mutants differ from ADPKD models in manifesting generally smaller renal cysts and greater fibrosis relative to cyst size14, 15. Additionally, Ift-A and –B mutants differ in cilia phenotype - shortened and absent cilia, respectively - and can also show opposing signaling phenotypes, reflecting the differing functions of IFT-A and -B12, 16–18. Intriguingly, deletion of Ift-B genes, Kif3a, Ift20 and Ift88, and of an IFT-A adaptor gene, Tulp3, in Pkd1 or Pkd2 conditional knock-out (cko) mice reduces severity of the PKD phenotype19–22, indicating that a component of cilia dysfunction has potential therapeutic value.
A commonly mutated IFT gene is THM1 (TPR-containing Hedgehog modulator 1; also termed TTTC21B). Causative and modifying mutations in THM1 have been identified in 5% of patients with ciliopathies, including nephronophthisis, Bardet Biedl syndrome, Meckel syndrome and Jeune syndrome14. THM1 encodes an IFT-A component, and its deletion impairs retrograde IFT, causing accumulation of proteins in bulb-like distal tips of shortened primary cilia16. Thm1 loss also impairs cilia entry of membrane-associated proteins, delays and reduces ciliogenesis, and promotes serum-induced cilia loss23. In mice, Thm1 deletion recapitulates many of the clinical manifestations of ciliopathies16, 24, 25. Perinatal global deletion of Thm1 results in renal cystic disease24, while deletion of Thm1 in adult mice does not result in a renal phenotype by 3 months of age, consistent with the developmental time-frame that determines whether loss of a cystogenic gene will cause rapid- or slow-progressing renal cystic disease26. To expand on the role of ciliary dysfunction in ADPKD, here we investigate the role of Thm1/ IFT-A deficiency in juvenile and adult ADPKD mouse models.
Methods
Generation of mice
Pkd1flox/flox, Pkd2flox/flox and ROSA26-Cre mice were obtained from the Jackson Laboratories (Stock numbers 010671, 017292 and 004847, respectively). Generation of Thm1 cko mice has been described previously24: Thm1aln/+; ROSA26CreERT+ male mice were mated to Thm1flox/flox females. Pkd1 floxed alleles were introduced into the colony to generate Thm1flox/flox;Pkd1flox/flox or Thm1flox/flox;Pkd1flox/+ females and Pkd1flox/flox; Thm1aln/+, ROSA26-CreERT/+ males, which were mated. Similarly, Pkd2 floxed alleles were introduced into the colony to generate Thm1flox/flox;Pkd2flox/flox or Thm1flox/flox;Pkd2flox/+ females and Pkd2flox/flox; Thm1aln/+, ROSA26-CreERT/+ males. To generate early-onset Pkd2 models, Thm1flox/flox;Pkd2flox/flox or Thm1flox/flox;Pkd2flox/+ nursing mothers mated to Pkd2flox/flox; Thm1aln/+, ROSA26-CreERT/+ males were injected intraperitoneally with tamoxifen (10mg/40g; Sigma) at postnatal day 0 (P0) to induce gene deletion. Offspring were analyzed at P21. To generate late-onset Pkd1 models, offspring from matings between Thm1flox/flox;Pkd1flox/flox or Thm1flox/flox;Pkd1flox/+ females and Pkd2flox/flox; Thm1aln/+, ROSA26-CreERT/+ males were injected intraperitoneally with tamoxifen (10mg/40g) at P35. To generate late-onset Pkd2 models, offspring from matings between Thm1flox/flox;Pkd2flox/flox or Thm1flox/flox;Pkd2flox/+ females and Pkd2flox/flox; Thm1aln/+, ROSA26-CreERT/+ males were injected intraperitoneally with tamoxifen (10mg/40g) at P28. Mice were analyzed at 6 months of age. All mouse lines were maintained on a pure C57BL6/J background (backcrossed 10 generations). All animal procedures were conducted in accordance with KUMC-IACUC and AAALAC rules and regulations.
Kidney and body weight measurements
Kidneys were dissected and weighed using a standard laboratory weighing scale. The kidney weight/body weight (KW/BW) ratio was calculated as the total kidney weights divided by body weight for each mouse.
Blood Urea Nitrogen Measurements
Mouse trunk blood was collected in Microvette CB 300 Blood Collection System tubes (Kent Scientific), and centrifuged at 1800g at room temperature for 10 minutes to collect serum. BUN was measured using the QuantiChrom Urea Assay Kit (BioAssay Systems) according to the manufacturer’s protocol.
Histology
Kidneys were bisected transversely, fixed in 10% formalin for several days, then processed in a tissue processor and embedded in paraffin. Tissue sections (7μm) were obtained with a microtome. Sections were deparaffinized, rehydrated through a series of ethanol washes, stained with hematoxylin and eosin (H&E), rehydrated, then mounted using Permount (ThermoFisher). Images were taken with a Nikon 80i microscope equipped with a Nikon DS-Fi1 camera. Cystic areas of H&E-stained sections were quantified using ImageJ.
Immunofluorescence
Following deparaffinization and rehydration, tissue sections were subjected to an antigen retrieval protocol. Tissue sections were steamed for 15□minutes in Sodium Citrate Buffer (10□mM Sodium Citrate, 0.05% Tween 20, pH 6.0), returned to room temperature, rinsed 10 times in distilled water, washed 5 minutes in PBS, incubated for 5 minutes in 1% SDS in PBS based on a method by Brown et al., 199627, then washed 3 times in PBS. Sections were blocked with 1% BSA in PBS for 1 hour at room temperature, and then incubated with primary antibodies against acetylated-α tubulin (1:4000; Sigma, T7451), IFT81 (1:200; Proteintech, 11744-1-AP), αSMA (1:500; Abcam, ab5694), F4/80 (1: 400, Cell Signaling, 30325) and PCNA (1:300; Cell Signaling Technology, 13110), P-ERK (1:200; Cell Signaling, 4370); THP (1:100; Santa Cruz Biotechnology, sc-271022) overnight at 4°C in the presence or absence of lectins, DBA (1:100; Vector Laboratories, FL-1031) or LTL (1:300, Vector Laboratories, FL-1321). Sections were washed three times in PBS, and then incubated with secondary antibodies conjugated to Alexa Fluor 488 (1:500; Invitrogen, A-11001 (anti-mouse) or A-11034 (anti-rabbit)) or Alexa Fluor 594 (1:500; Invitrogen, A-11005 (anti-mouse) or A-11012 (anti-rabbit)) for 1 hour at room temperature. After three washes of PBS, sections were mounted with DAPI Fluoromount-G (Electron Microscopy Sciences). Staining was visualized and imaged using a Nikon 80i microscope with a photometrics camera or a Nikon Eclipse TiE attached to an A1R-SHR confocal, with an A1-DU4 detector, and LU4 laser launch.
Immunohistochemistry
Tissue sections were deparaffinized, rehydrated, and subjected to antigen retrieval, which consisted of steaming sections for 25□minutes in Sodium Citrate Buffer (10□mM Sodium Citrate, 0.05% Tween 20, pH 6.0). Sections were treated with 3% hydrogen peroxide for 30 min to minimize background staining, washed in PBS, then blocked with 1% BSA for 1 hour. Tissue sections were incubated with primary antibodies against P-STAT3 (1:50; GeneTex, GTX118000), O-GlcNAc (1:200; ThermoFisher, RL2, MA1-072), OGT (1:200; a kind gift from Jerry Hart, UGA) and OGA (1:2000; a kind gift from Jerry Hart, UGA) overnight at 4°C. Sections were washed three times in PBS, then incubated with HRP-conjugated rabbit secondary antibody (Cell Signaling) for 30 minutes. Sections were washed three times in PBS, incubated with ABC reagent (Vector Laboratories), rinsed in PBS, then incubated with SigmaFAST DAB metal enhancer (Sigma) until desired signal/color was obtained. Sections were counterstained with haemotoxylin, dehydrated through an ethanol series, and mounted with Permount (ThermoFisher). Staining was visualized and imaged using a Zeiss A1 microscope with a Axiocam 105 color camera.
Scanning electron microscopy
Small pieces of kidney cortices were fixed in 4% paraformaldehyde/2% glutaraldehyde, then washed three times in 0.1 M Na-cacodylate, pH 7.4. Samples were post-fixed for 30 minutes with 1% OsO4 in 0.1 M Na-cacodylate buffer, then dehydrated using an ethanol series, followed by hexamethyldisilazane (HMDS; Electron Microscopy Sciences). Samples were mounted onto metal stubs and sputter coated with gold. Samples were viewed and imaged using a Hitachi S-2700 Scanning Electron Microscope equipped with a Quartz PCI digital capture.
qPCR
RNA was extracted using Trizol (Life Technologies), then reverse transcribed into cDNA using Quanta Biosciences qScript cDNA mix (VWR International). qPCR for Ccl2 and Oaz1, a housekeeping gene, was performed using Quanta Biosciences Perfecta qPCR Supermix (VWR International) in a BioRad CFX Connect Real-Time PCR Detection System. Primers used were mCcl2 (Forward: 5’-AAGCTCAACCCTGACTTCTTAC-3’; Reverse: 5’-CAACGTCTGAGAACTGGAGAAA-3’) and mOaz1 (Forward: 5’-GCCTGAGGGCAGTAAGGAC-3’; Reverse: 5’-GGAGTAGGGCGGCTCTGT-3’). qPCR was performed in duplicate using RNA lysates from five samples per genotype.
ADPKD and normal human kidney sections
Normal human kidneys (NHK) and ADPKD kidneys were obtained from the PKD Biomarkers, Biomaterials, and Cellular Models Core in the Kansas PKD Center. The protocol for the use of discarded human tissues complied with federal regulations and was approved by the Institutional Review Board at KUMC. Paraffin-embedded de-identified NHK, n=3 (K357, K402, K419), and ADPKD, n=3 (K386, K408, K423) tissues were sectioned. Sections were deparaffinized and rehydrated, steamed in Sodium Citrate Buffer (10□mM Sodium Citrate, 0.05% Tween 20, pH 6.0) for antigen retrieval, and immunostained for ARL13B (1:300; Proteintech, 17711-1-AP).
Statistics
GraphPad Prism 8 software was used to perform statistical analyses. Statistical significance was determined as p<0.05. For analysis of more than two groups, one-way ANOVA followed by Tukey’s test or a Kriskall-Wallis test were used to analyze data with or without normal distribution, respectively. For analysis of two groups, an unpaired t-test was used.
Results
Perinatal deletion of Thm1 in Pkd2 conditional knock-out mice reduces cortical cystogenesis, but does not improve kidney function
To examine the effect of IFT-A deficiency in an early-onset, rapidly progressing ADPKD mouse model, we deleted Thm1 together with Pkd2 at postnatal day (P) 0, and examined the renal phenotypes of control, Thm1 conditional knock-out (cko), Pkd2 cko and Pkd2;Thm1 double knock-out (dko) mice at P21. At this early stage, Thm1 cko kidneys on a pure C57BL6/J background appeared mostly intact morphologically, with some tubular dilations observed in the cortex and with kidney weight/body weight (KW/BW) ratios similar to control (Figures 1A and 1B). Yet BUN levels were elevated about 2-fold (Figure 1C). In Pkd2 cko mice, renal cysts are present in both cortex and medulla, and KW/BW ratios and BUN levels are increased 5- and 3-fold, respectively. In Pkd2;Thm1 dko mice, renal cysts are also present in the cortex and medulla, and KW/BW ratios and BUN levels are increased 4- and 3-fold, respectively. Thus relative to Pkd2 cko mice, Pkd2;Thm1 dko mice have reduced KW/BW ratios, but similar kidney dysfunction. Also relative to Pkd2 cko mice, Pkd2;Thm1 dko kidneys show decreased percent cystic index (Figure 1D), due to reduced cystogenesis in the cortex (Figure 1E), while cystogenesis in the medulla was similar (Figure 1F).
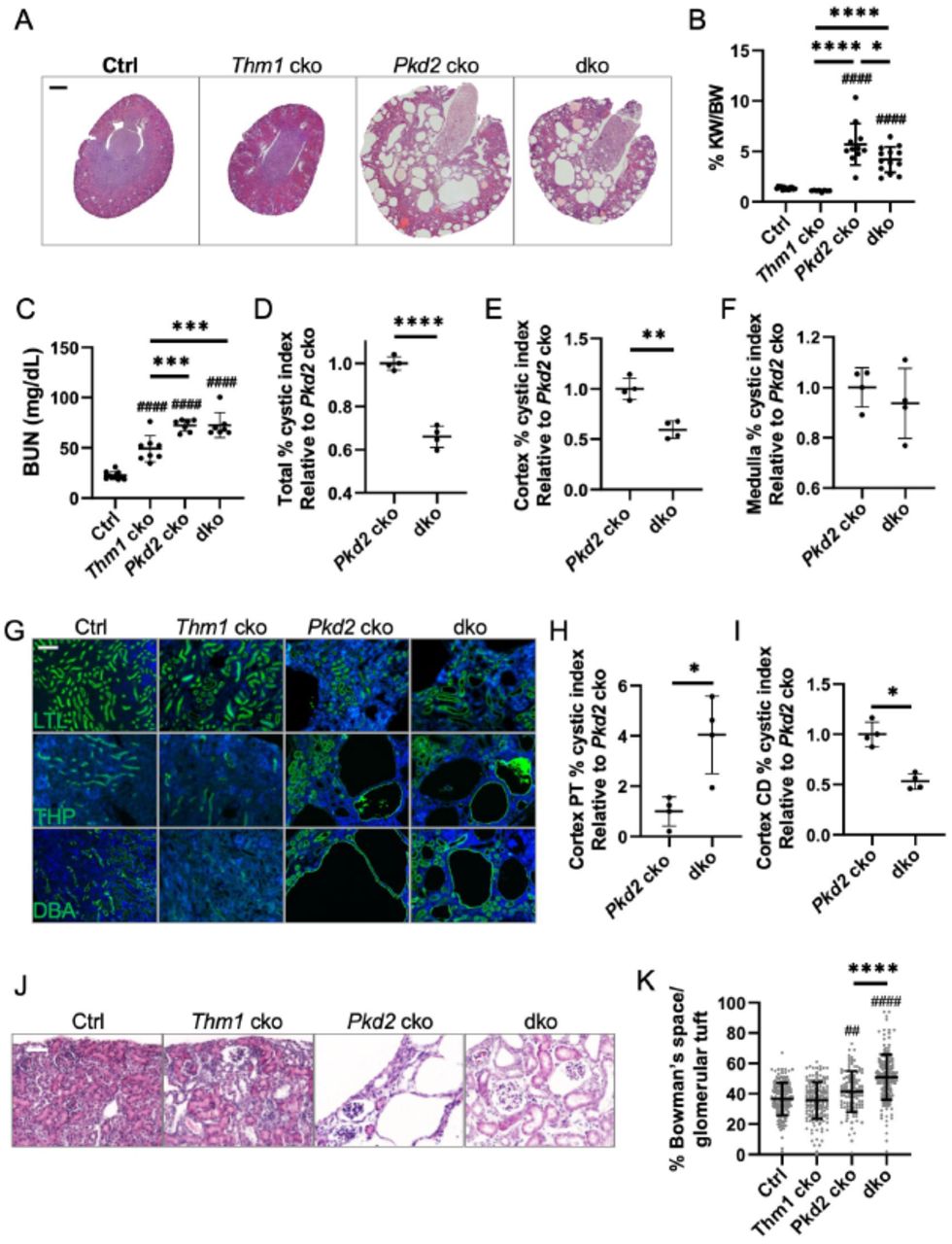
(A) Haemotoxylin and eosin staining of P21 kidney sections. Scale bar - 500μm. (B) Percent kidney weight/body weight (KW/BW) ratios, (C) Blood urea nitrogen (BUN) levels, (D) Percent cystic index, (E) Percent cortical cystic index, and (F) Percent medullary cystic index. Pkd2 cko percent cystic indices are set at 1. Bars represent mean ± SD. In (B) and (C), statistical significance was determined by one-way ANOVA followed by Tukey’s test. *p<0.05; ***p<0.001; ****p<0.0001; ####p<0.0001 compared to Ctrl. In (D), (E) and (F), statistical significance was determined by unpaired two-tailed t-test. **p<0.01; ****p<0.0001. Each data point represents a mouse. (G) Staining of kidney cortex with LTL, THP and DBA. Scale bar - 100μm (H) Percent LTL+ dilations (I) DBA+ percent cystic index in renal cortex. Pkd2 cko percent cystic indices are set at 1. Bars represent mean ± SD. Statistical significance was determined by unpaired two-tailed t-test. *p<0.05. (J) Haemotoxylin and eosin staining. Scale bar - 50μm (K) Area of Bowman’s space/area of Bowman’s capsule. Bars represent mean ± SD. Statistical significance was determined by one-way ANOVA followed by Tukey’s test. ****p<0.0001; ##p<0.01 compared to Ctrl; ####p<0.0001 compared to Ctrl
Perinatal deletion of Thm1 in Pkd2 conditional knock-out mice reduces cortical collecting duct cystogenesis, but increases proximal tubular and glomerular dilations
Since cortical cystogenesis was reduced in Pkd2;Thm1 dko kidneys relative to Pkd2 cko kidneys, we further analyzed the cortical cysts. At P21 in the Thm1 cko renal cortex, we observed some dilations, most of which were LTL+, marking proximal tubules, and fewer that were THP+ or DBA+, marking loop of Henle and collecting duct, respectively (Figure 1G). In Pkd2 cko renal cortex, LTL+ dilations, THP+ cysts, and multiple, large DBA+ cysts were observed. In Pkd2; Thm1 dko cortex, LTL+ dilations were increased relative to those of Pkd2 cko and Thm1 cko kidneys (Figures 1G and 1H); THP+ cysts were similar in size to those of Pkd2 cko kidneys (Figure 1G; relative percent THP+ cystic index of Pkd2 cko vs. dko mean ± sd is 1.0 ± 0.4 vs. 0.9 ± 0.1; Pkd2 cko mean value set at 1.0; n=4; not significant); and DBA+ cysts were decreased in size relative to those of Pkd2 cko kidneys (Figures 1G and 1I). We also observed glomerular dilations in the mutant kidneys (Figure 1J). To quantify glomerular dilations, percentage of area of Bowman’s space over the area of glomerular tuft was calculated. In Pkd2 cko kidneys, this percentage was increased, and in Pkd2;Thm1 dko kidneys, this percentage was further increased relative to Pkd2 cko kidneys, indicating that additional loss of Thm1 exacerbates Pkd2 cko glomerular dilations (Figure 1K). These differences reveal that the role of Thm1 deletion on a Pkd2 cko background is specific to the nephron segment.
Deletion of Thm1 alone increases inflammation
Proliferation is a cellular hallmark of ADPKD renal cystogenesis. We therefore immunostained for Proliferating cell nuclear antigen (PCNA) together with proximal tubule and collecting duct markers, LTL and DBA, respectively. In LTL+ tubules, PCNA levels were similar across control, Thm1 cko and Pkd2 cko kidneys, and slightly elevated in Pkd2;Thm1 dko kidneys (Figures 2A-2B). However, PCNA+ cells were increased in Pkd2 cko and Pkd2;Thm1 dko DBA+ tubules, and further increased in Pkd2 cko and Pkd2;Thm1 dko DBA+ cysts (Figure 2C). These data support that increased proliferation is an early driver of ADPKD renal cystogenesis.

(A) Immunostaining of kidney cortex for PCNA (red) together with LTL or DBA (green). Scale bar -10μm. (B) Percent PCNA+ cells per LTL+ and (C) DBA+ tubule. Each dot represents a tubule or a cyst from n=3 mice/genotype. Bars represent mean ± SD. Statistical significance was determined by Kruskal-Wallis test. ****p<0.0001; #p<0.05 compared to Ctrl; ###p<0.001 compared to Ctrl; ####p<0.0001 compared to Ctrl; &P<0.05 compared to Pkd2 cko Tubule; $P<0.05 compared to dko tubule. (D) Immunostaining of kidney cortex for αSMA and F4/80. Scale bar - 50μm. (E) Immunostaining of kidney cortex for acetylated α-tubulin (red) together with LTL (green). Scale bar -10μm. (F) Quantification of cilia length of LTL+ cells. (G) Immunostaining of kidney cortex for acetylated α-tubulin and IFT81 together with DBA. Scale bar - 10μm. n=3 mice/genotype. Quantification of cilia length of cortical DBA+ cells. Bars represent mean ± SD. Statistical significance was determined by one-way ANOVA followed by Tukey’s test. ****p<0.0001; #p<0.05 compared to Ctrl; ###p< 0.001 compared to Ctrl; ####p<0.0001 compared to Ctrl.
Cyst growth compresses surrounding parenchyma, leading to injury, inflammation, and fibrosis. We immunostained kidney sections for alpha smooth muscle actin (αSMA) and F4/80 to examine the presence of myofibroblasts and macrophages, respectively, which contribute to pro-inflammatory and pro-fibrotic processes. In control kidneys, αSMA was present around blood vessels and few F4/80+ cells were present (Figure 2D). In Thm1 cko kidneys, αSMA SMA localized around blood vessels, in addition to around glomeruli and tubular dilations, and number of F4/80+ cells was increased. In Pkd2 cko and Pkd2;Thm1 dko kidneys, even more αSMA+ and F4/80+ labelling was observed surrounding glomeruli, tubular dilations and cysts. Thus, deletion of Thm1 alone increases inflammatory processes, but its deletion on a Pkd2 cko background does not exacerbate ADPKD inflammation at P21.
Deletion of Pkd2 increases cilia length on renal epithelia
We next examined primary cilia on renal tubular epithelia by co-immunostaining for acetylated, α-tubulin together with LTL or DBA. In control kidneys, average cilia lengths were 3.0μm and 2.1μm for LTL+ and DBA+ cells, respectively (Figures 2E-2H). We also noted qualitative differences between LTL+ and DBA+ primary cilia, with the former cilia being thinner and longer, and the latter being thicker and more rod-like. Cilia lengths were increased in both Pkd2 cko LTL+ and DBA+ tubules. However, relative to Pkd2 cko tubules, cilia lengths were further increased in Pkd2;Thm1 dko LTL+ tubules, but similar in Pkd2;Thm1 dko DBA+ tubules. These differences reveal tubular-specific effects on cilia length.
Perinatal deletion of Thm1 increases ERK and STAT3 signaling
Primary cilia mediate signaling pathways. Thus, we next examined signaling pathways, ERK and STAT3, which promote disease progression in ADPKD28–31 and which we have observed can be regulated by Thm1 in vitro (data now shown). Low intracellular Ca2+ and high cAMP activates ERK signaling, which promotes cell proliferation30, 32 and also acts upstream of mTOR and AMPK pathways regulating cellular metabolism33. Full-length and cleaved PC1 can directly activate STAT3, which can have proliferative and inflammatory roles31, 34, 35. We immunostained for P-ERK together with either LTL or DBA. At P21, P-ERK did not localize with LTL, but localized with DBA (Figure 3A). While a similar number of P-ERK+ tubules was observed across control and the various mutant genotypes (Figure 3B), P-ERK intensity was increased in dilated tubules of Thm1 cko mice, and further increased in cyst-lining cells of Pkd2 cko and Pkd2;Thm1 dko mice (Figure 3C). These data support that ERK activation is a driver of renal cyst growth. Using immunohistochemistry, P-STAT3 was revealed to be increased in epithelial cells lining cortical dilations in Thm1 cko kidneys and to be further increased in cyst-lining cells of Pkd2 cko and Pkd2;Thm1 dko kidneys, both in the cortex and medulla (Figure 3E). Thus, dilations caused by Thm1 loss as well as Pkd2 cystic disease cause increased ERK and STAT3 signaling.

(A) Immunofluoresence for P-ERK together with LTL or DBA. Scale bar - 50μm. (B) Quantification of P-ERK+ tubules and (C) P+ ERK intensity. Each dot represents a tubule or a cyst from n=3 mice/genotype. Statistical significance was determined by oneway ANOVA followed by Tukey’s test. *p<0.05; #p<0.05 compared to Ctrl; ####p<0.0001 compared to Ctrl. (D) Immunohistochemistry for P-STAT3. Bottom and lower panels show cortex and medulla, respectively. n=3 mice/genotype. Scale bar - 25μm.
O-GlcNAcylation is increased in dilated tubules and cysts
Altered cellular metabolism has emerged as an important component of ADPKD pathobiology33, 36–38. One of these alterations includes the Warburg effect33, whereby cells preferentially convert the product of glycolysis, pyruvate, into lactate, even in the presence of oxygen. This pathway yields much less ATP than the entry of pyruvate in mitochondria, generating ATP via oxidative phosphorylation. The nutrient sensor, O-linked β-N-acetylglucosamine (GlcNAc) regulates the balance between glycolysis and oxidative phosphorylation39, as well as ciliary homeostasis40, 41, both of which are altered in ADPKD33, 42. We therefore examined O-GlcNAc signaling. In control kidneys, immunohistochemistry for O-GlcNAc revealed mostly nuclear staining in tubules of the cortex, and lesser staining in tubules of the medulla (Figure 4A). In Thm1 cko kidneys, O-GlcNAc expression was increased in nuclei of cells lining dilations in the cortex, but similar to control in the medulla. In Pkd2 cko kidneys, OGlcNAc staining was intense in cyst-lining cells in both the cortex and medulla. Relative to Pkd2 cko kidneys, Pkd2;Thm1 kidneys showed slightly reduced O-GlcNAc staining in cyst-lining cells of the cortex, but similar staining in the medulla. O-GlcNAC is regulated by two enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), which respectively transfers and removes the O-GlcNAc moiety on protein substrates. In control kidneys, low expression of OGT was present in the cytoplasm of tubular epithelia in the cortex and medulla (Figure 4B). In Thm1 cko kidneys, OGT was increased in the cytoplasm and nuclei of cells lining dilations in the cortex, while OGT levels were similar to control in the medulla. In Pkd2 cko and in Pkd2;Thm1 dko mice, intense OGT staining was present in cyst-lining cells in the cortex and medulla. In control kidneys, OGA staining was present in the cytoplasm of proximal tubule cells and even lighter cytoplasmic stain was present in the medullary tubules (Figure 4C). In Thm1 cko kidneys, OGA staining was similar to control. In Pkd2 cko and in Pkd2;Thm1 dko mice, OGA was increased in the nuclei and cytoplasm of cyst-lining cells in both the cortex and medulla. Together, these data suggest that misregulation of OGlcNAc signaling may contribute to renal cystogenesis.
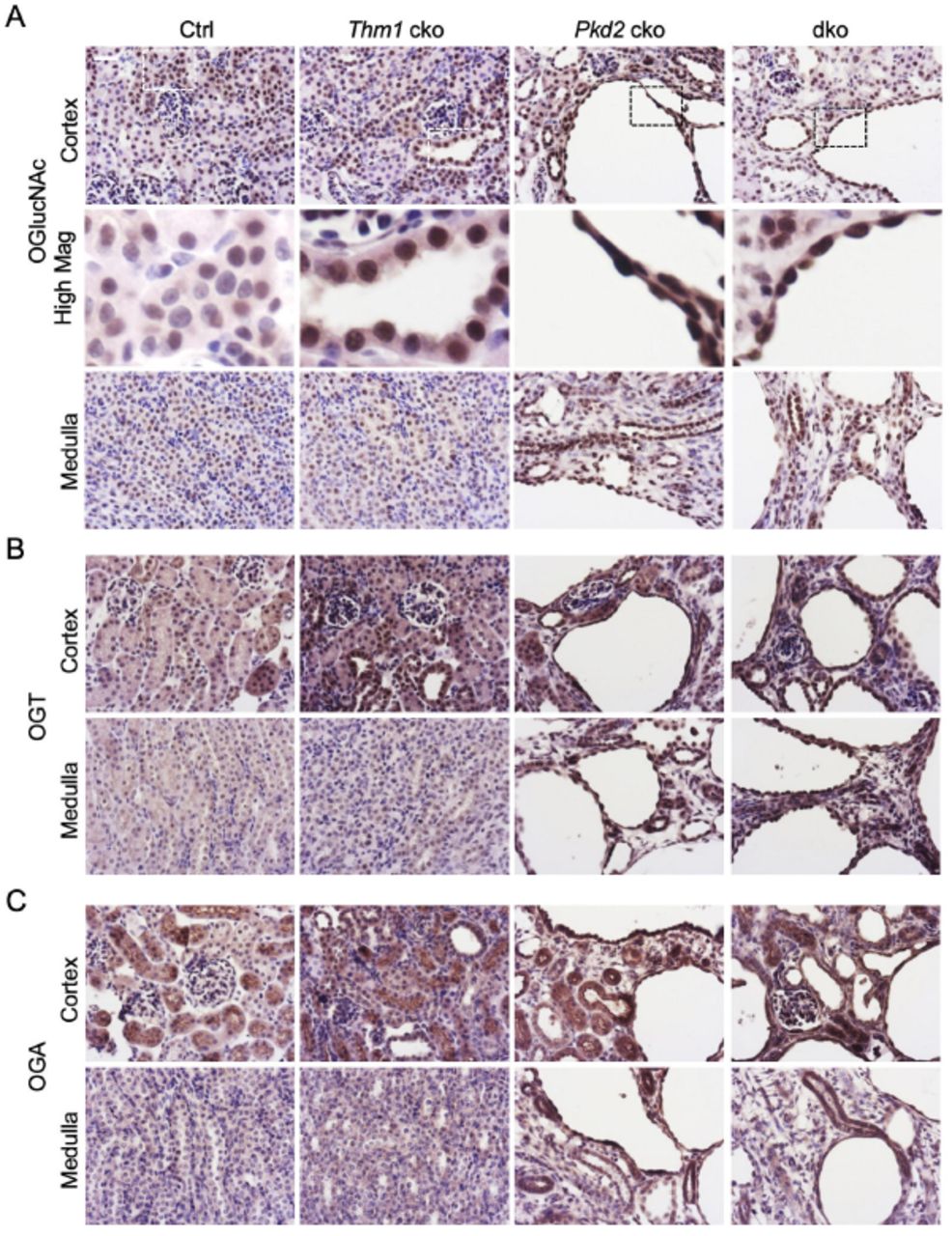
(A) Immunohistochemistry for O-GlcNAc; (B) OGT and (C) OGA. Bottom and lower panels show cortex and medulla, respectively. Scale bar - 25μm. n=3 mice/genotype.
Deletion of Thm1 in adult Pkd2 or Pkd1 conditional knock-out mice markedly attenuates ADPKD renal cystogenesis
We next examined the role of IFT-A deficiency in late-onset, slowly progressive adult ADPKD mouse models. We deleted Thm1 together with Pkd1 at P35 and examined the renal phenotypes at 6 months of age. Late-onset Thm1 cko kidneys have morphology and BUN levels similar to control kidneys (Figure S1A). In Pkd1 cko mice, renal cysts were mostly in the cortex, with the largest and most abundant cysts being DBA+ (Figure 5A). Fewer cysts were THP+, and only dilations, not cysts, were observed that were LTL+. Notably, all these features were reduced in Pkd1; Thm1 dko kidneys. KW/BW ratios were elevated in Pkd1 cko mice, and corrected in Pkd1;Thm1 dko mice (Figure 5B). Additionally, BUN levels were slightly elevated in Pkd1 cko mice, while BUN levels in Pkd1;Thm1 dko mice were similar to control (Figure 5C).
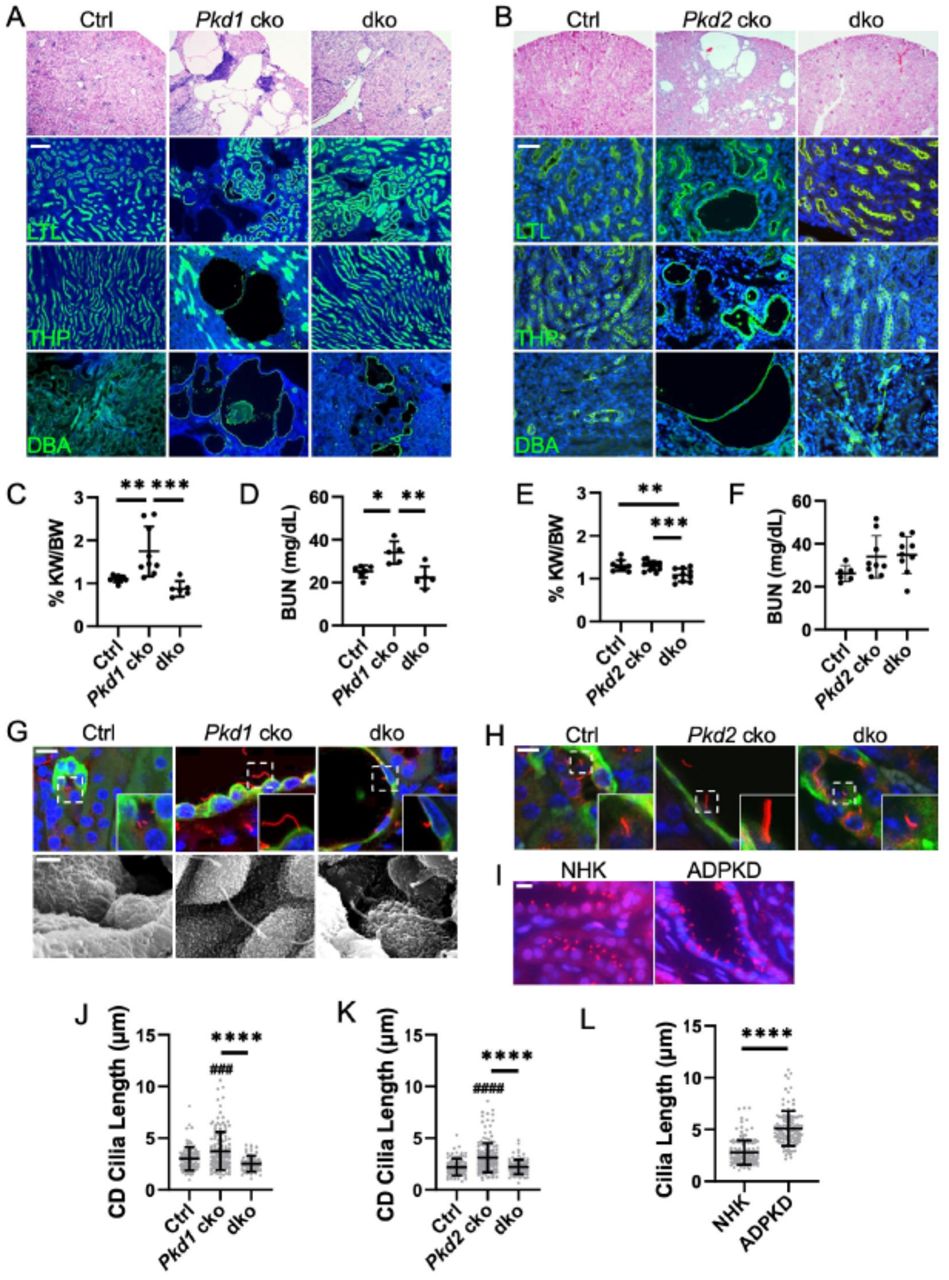
(A) Histology and lectin staining of Pkd1 cko kidney sections. Scale bar - 100μm. (B) Histology and lectin staining of Pkd2 cko kidney sections. Scale bar - 50μm. (C) KW/BW ratios and (D) BUN levels for Pkd1 cko mice and (E, F) for Pkd2 cko mice. Bars represent mean ± SD. Each dot represents an animal. Statistical significance was determined by one-way ANOVA followed by Tukey’s test. *p<0.05; **p<0.01; ***p<0.001. (G) Immunostaining of Pkd1 cko renal cortex for acetylated α-tubulin together with DBA. Scale bar - 10μm. Scanning electron micrographs of primary cilia. Scale bar – 1.5μm. n=3 mice/genotype. (H) Immunostaining of Pkd2 cko renal cortex for acetylated α-tubulin together with DBA. Scale bar - 10μm. n=3/genotype. (I) Immunostaining for ARL13B of normal human kidney (NHK) and ADPKD renal sections. Scale bar - 10μm. n=3 for each NHK and ADPKD. (J) Quantification of renal cilia lengths of Pkd1 cko mice, (K) Pkd2 cko mice, and (L) NHK and ADPKD tissue. Cilia lengths were quantified from immunofluorescence experiments of (G, H, I). Significance was determined using one-way ANOVA followed by Tukey’s test (J, K) or by an unpaired t-test (L). Bars represent mean ± SD. ###p< 0.001 compared to Ctrl; ####p<0.0001 compared to Ctrl; ****p<0.0001
Since PKD2 mutations result in less severe ADPKD, we deleted Thm1 together with Pkd2 at P28 and examined renal phenotypes at 6 months of age. Thm1 cko kidneys have morphology and BUN levels resembling those of control mice (Figure S1B). Pkd2 cko mice show renal cysts mostly in the cortex, with the largest cysts being DBA+, and smaller cysts being LTL+ or THP+ (Figure 5D). In contrast, in Pkd2;Thm1 dko mice, the Pkd2 cko cystic phenotype is largely corrected morphologically. In Pkd2 cko mice, KW/BW ratios were similar to control and BUN levels showed a trend toward a slight elevation, reflecting the mild disease induced in adulthood. In Pkd2;Thm1 dko mice, KW/BW ratios were slightly reduced relative to control and Pkd2 cko mice, due to increased body weight caused by global deletion of Thm125. In Pkd2;Thm1 dko mice, BUN levels showed a slight decreasing pattern relative to Pkd2 cko mice.
Deletion of Thm1 in adult ADPKD mouse models results in reduced cilia length of cortical collecting duct renal epithelia
Similar to juvenile ADPKD models, cilia lengths measured by immunostaining for acetylated α-tubulin, were increased in Pkd1 cko and Pkd2 cko DBA+ adult tubules (Figures 5G, 5H, 5J, and 5K). However, in contrast to juvenile models, cilia lengths of Pkd1;Thm1 and Pkd2;Thm1 dko DBA+ tubular epithelia were reduced relative to those of Pkd1 cko and Pkd2 cko epithelia and similar to control. Additionally, human ADPKD sections had longer epithelial cilia than NHK sections (Figures 5I and 5L), supporting that increased cilia length is also a feature of the human disease.
Deletion of Thm1 in adult Pkd1 conditional knock-out mice reduces proliferation, inflammation, P-ERK, P-STAT3, and O-GlcNAc
To further assess the extent of ADPKD attenuation by Thm1 deletion, we examined proliferation and inflammation in late-onset Pkd1 cko and Pkd1;Thm1 dko mice. In Pkd1 cko kidneys, PCNA was expressed in cyst-lining cells (Figure 6A), αSMA was expressed around blood vessels, cysts as well as glomeruli (Figure 6B), while F4/80 was expressed surrounding cysts and glomeruli. Notably, in Pkd1;Thm1 dko kidneys, PCNA was not expressed, αSMA was expressed around blood vessels, and F4/80+ cells were reduced and randomly dispersed throughout the kidney, resembling control. Similar to in juvenile mice, P-ERK was not expressed in LTL+ cells, but was expressed in DBA+ tubular and cyst-lining epithelia (Figure 6C). P-ERK staining was more intense in Pkd1 cko kidneys and reduced in Pkd1;Thm1 dko kidneys. P-STAT3 was also increased in cyst-lining cells of Pkd1 cko kidneys, and reduced in Pkd1;Thm1 dko kidneys (Figure 6D). Additionally, pro-inflammatory cytokine, Ccl243, was elevated in Pkd1 cko kidneys, but reduced in Pkd1;Thm1 dko kidneys (Figure 6E). Thus, proliferative and pro-inflammatory pathways are attenuated in late-onset ADPKD by deletion of Thm1.

(A) Immunofluorescence for PCNA (red) together with DBA (green). Scale bar - 100μm. (B) Immunostaining for αSMA (red) and F4/80 (red). Scale bar - 50μm. (C) Immunostaining for P-ERK (red) together with LTL (green) or DBA (green). Scale bar - 50μm. (D) Immunohistochemistry for P-STAT3. n=3 mice/genotype. (E) qPCR for Ccl2. Statistical significance was determined by one-way ANOVA followed by Tukey’s test. **p<0.01
Using immunohistochemistry, we examined O-GlcNAcylation. In control kidneys, light cytoplasmic staining of O-GlcNAc was present in tubules of both the cortex and medulla. In Pkd1 cko kidneys, increased O-GlcNAc occurred in cyst-lining epithelia of the cortex, in proteinaceous substances present in some of the cysts, and in tubules of the medulla (Figure 7A). In Pkd1;Thm1 dko kidneys, O-GlcNAc staining was reduced, with light cytoplasmic expression in cortical tubules, similar to control, and with increased expression still present in some medullary tubules. In control kidneys, OGT and OGA were lightly expressed in the cytoplasm of tubular epithelia of both cortex and medulla (Figures 7B and 7C). In Pkd1 cko kidneys, OGT and OGA were increased in the cytoplasm and nuclei of cyst-lining cells of the cortex and in some tubules of the medulla. In Pkd1;Thm1 dko kidneys, OGT and OGA expression were similar to control. These data suggest that increased O-GlcNAcylation is a feature of late-onset ADPKD, similar to early-onset disease. Further, deletion of Thm1 in late-onset ADPKD attenuates perturbation of this metabolic regulator.

(A) Immunohistochemistry for O-GlcNAc; (B) OGT and (C) OGA. Bottom and lower panels show cortex and medulla, respectively. Scale bar – 20μm. n=3 mice/genotype.
Discussion
Primary cilia are key organelles in ADPKD pathobiology, through mechanisms that are poorly understood. Deleting IFT-B genes attenuates PKD severity in both juvenile and adult ADPKD models19. In contrast, deleting Tulp3, an IFT-A adaptor, attenuated renal cystic disease in an adult ADPKD model only21,44. Since the attenuation of ADPKD with deletion of these ciliary genes is so striking, identifying the underlying mechanism could lead to a very effective therapeutic target.
Expanding the role of cilia dysfunction in ADPKD, we show that similar to Tulp3, deletion of IFT-A gene, Thm1, does not attenuate renal function in an early-onset ADPKD model, but markedly attenuates almost all aspects of ADPKD in a late-onset model (Figure 8). Yet distinct from Tulp344, global deletion of Thm1 in an early-onset model reduces KW/BW ratios and cortical collecting duct cystogenesis. Our data further reveal that the effects of Thm1 deletion on an early-onset ADPKD background are nephron-specific, with no effect on loop of Henle-derived cysts, and exacerbation of proximal tubular and glomerular dilations. This is the first demonstration that cilia dysfunction has differential effects on ADPKD tubules and may reflect varying microenvironments between nephron segments. The differential effects of Tulp3 or Thm1 deletion in early- versus late-onset ADPKD mouse models further suggest differences in developing versus mature renal microenvironments. Collectively, these data highlight the importance of renal context and examining molecules and pathways by cell type or at the single cell level in the kidney.

In an early-onset ADPKD mouse model, deletion of Thm1 causes nephron segment-specific effects, attenuating cortical collecting duct (CD) cystogenesis, but worsening proximal tubular and glomerular dilations, without affecting kidney function, cilia length, inflammation, and ERK, STAT3, and OGlcNAc signaling. In a late-onset ADPKD mouse model, deletion of Thm1 improves kidney function and reduces cystogenesis, inflammation, cilia length, and ERK, STAT3, and O-GlcNAc signaling.
Embryonic mutation or perinatal loss of either Kif3a, Ift88, Tulp3 or Thm1 causes renal cystic disease24, 21, 45, 46, suggesting that each of these genes is required for kidney development. Thus, their requirement for kidney development does not explain the differential effects in early-onset ADPKD. Instead, the differential effects may be due to differences in their specific cellular function, such as cilia length regulation, ciliary entry or transport of protein cargo, or regulation of signaling pathways. IFT-B and -A complexes regulate cilia length through different mechanisms. IFT-B genes mediate anterograde IFT, while IFT-A genes and Thm1 mediate retrograde IFT as well as ciliary entry of membrane and signaling proteins. In contrast, Tulp3 does not regulate cilia length, but is required for ciliary entry of membrane-associated and signaling molecules44. Additionally, IFT-B and -A mutants have shown opposing signaling phenotypes. Any of these functions can be explored to account for the differing early-onset dko phenotypes.
Increased cilia lengths on renal epithelia of several ADPKD mouse models, PKD1RC/RC, Pkd1 and Pkd2 cko mice47, 48, and recently, on human ADPKD tissue22, have been reported. Our data substantiate that loss of polycystin function causes increased cilia length in mouse models as well as in human ADPKD, suggesting that ciliary mechanisms in the disease are likely conserved between mouse and human. Further, our data show that in addition to genotype, cilia structure varies by renal tubule segment and maturation, suggesting that factors within a tubule’s microenvironment affect cilia length. Indeed, multiple factors including intracellular Ca2+ and cAMP, oxidative stress, cytokines, and fluid flow, affect cilia length of renal epithelial cells49–51, indicating that cilia length may be finely regulated in order to maintain renal tubular structure and function. In support of an ameliorative effect of reduced cilia length in ADPKD, inhibition of cilia disassembly in Pkd1 cko mice increased renal cilia length and exacerbated ADPKD52, while in the jck non-orthologous PKD mouse model, which also has increased renal cilia lengths, pharmacological shortening of primary cilia was associated with attenuated PKD53.
Reduced Ccl2 and Wnt signaling in Pkd; Ift-B dko mice20, 22, and reduced P53 enhancing cilia disassembly in Pkd; Ift-B dko cells42 are potential mechanisms by which ablation or shortening of primary cilia attenuate ADPKD. In addition to detecting ligands, primary cilia also detect mechanical cues. Although mechanosensing by primary cilia and the polycystins has been controversial, recent studies have renewed interest in a potential mechanosensory role for the polycystins, particularly regarding tissue microenvironment stiffness54, 55. If sensing of physical forces in the tissue microenvironment is essential to maintaining renal tubular function, then other mechanical cues that would change with cyst growth include shear stress and intraluminal pressure. Cilia length itself could then also be a possible contributing factor in PKD severity. Supporting a role for cilia length, a recent study has shown that primary cilia of proximal tubule epithelial cells transduce shear stress into metabolic pathways that culminate in oxidative phosphorylation56. Finally, by extrapolating findings of cilia studies from the cancer field57, cilia of not only renal tubular epithelial cells, but of interstitial cells might also affect signaling and disease severity.
Our data are the first to reveal increased O-GlcNAcylation in cyst-lining renal epithelial cells of both early- and late-onset ADPKD mouse models, suggesting increased O-GlcNAcylation may be a feature of ADPKD. Increased O-GlcNAcylation is a pathologic feature of diabetic nephropathy58, 59, and in rodent models, has shown to promote various aspects of chronic kidney disease60, 61 and also renal fibrosis62. In contrast, in a mouse model of contrast-induced acute kidney disease, an acute increase in O-GlcNAcylation was protective63, emphasizing important differences between chronic and acute increases in O-GlcNAcylation. While acute changes are adaptive and necessary to maintain cellular health and metabolism, chronic changes are likely to contribute to pathology64. Chronic elevation of O-GlcNAcylation in cancer cells promotes tumor growth, and cancer cells show the Warburg effect, suggesting similar cellular metabolic alterations between ADPKD and cancer. A recent study has demonstrated that O-GlcNAcylation of Phosphoglycerate kinase 1 (PGK1), which catalyzes the first ATP molecule in glycolysis, activates PGK1 to enhance lactate production and reduce mitochondrial oxidative phosphorylation, promoting the Warburg effect39. Perhaps a similar mechanism may occur in ADPKD cells.
OGT localizes to the pericentriolar region during the early phases of ciliogenesis56, and perturbation of O-GlcNAcylation affects cilia length40, 41. Further, the ciliary structural defects caused by OGT inhibition suggest impaired centriole formation and IFT56. However, studies have shown opposing effects of OGT deficiency or inhibition on cilia length40, 41, indicating further studies are required. Possible causes may include differences in cell type, degree of OGT deficiency, or feedback regulation between OGT and OGA64. Chronic hyper-O-GlcNAcylation in diabetic tissues results in ciliary defects65, demonstrating a causative link between misregulation of O-GlcNAcylation and defective ciliary homeostasis in a disease context. Since increased renal cilia lengths are associated with ADPKD renal cystogenesis47, 48, the regulation of O-GlcNAcylation on ciliogenesis could be another mechanism by which altered O-GlcNAcylation can affect ADPKD. Elucidating the mechanisms by which O-GlcNAc is upregulated in ADPKD and alters cellular metabolism and ciliogenesis could reveal novel mechanisms and therapeutic targets.
In summary, our data demonstrate for the first time the role of IFT-A in an ADPKD context, revealing differential effects between nephron segments and between developing and mature renal microenvironments. Our findings also reveal for the first time that O-GlcNAcylation is increased in ADPKD. We propose that as a regulator of ciliary homeostasis and of the balance between glycolysis and oxidative phosphorylation, increased O-GlcNAc may drive certain key pathological processes in ADPKD.
Disclosures
The authors declare no conflict of interest.
Contributions
WW, LMS, MAK, HHW, TSP, BAA, DTJ, RD, JTC, AC, MTP, MS, DPW, and PVT performed experiments. WW, LMS, MAK, HHW, TSP, BAA, DTJ, RD, JTC, AC, MTP, MS, CS, DPW, JPC and PVT analyzed and interpreted data. WW, LMS, BAA, and PVT designed research. WW, LMS, and PVT wrote the manuscript. All authors revised and approved the final manuscript.
Supplemental Material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Renal histology and (B) BUN levels of 6 month-old mice from the Pkd1;Thm1 colony and (C, D) from the Pkd2;Thm1 colony. Scale bar - 250μm
Acknowledgements
We thank members of the KUMC Department of Anatomy and Cell Biology and the Jared Grantham Kidney Institute for helpful discussions. We thank Jing Huang of the KUMC Histology Core, which is supported by the Intellectual and Developmental Disabilities Research Center NIH U54HD090216 and NIH P30GM122731. We also thank Pat St. John and Larysa Stroganova of the KUMC Electron Microscopy Research Laboratory, which is supported by NIH P20GM104936. This work was also supported by a K-INBRE Summer Student Award to JTC (K-INBRE P20GM103418), the PKD Biomaterials and Biomarkers Repository Core in the Kansas PKD Research and Translational Core Center (NIH P30DK106912 to JPC), R01DK108433 to MS, and R01DK103033 to PVT.
References




