

ABSTRACT
Coronavirus disease 2019 (COVID19) is a respiratory infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) originating in Wuhan China in 2019. The disease in notably severe in elderly and those with underlying chronic conditions. The molecular mechanism as to why the elderly are vulnerable and why children are resistant is largely unknown. Understanding these differences is critical for safeguarding the vulnerable and guiding effective policy and treatments. Here we show loading cells with cholesterol from blood serum using the cholesterol transport protein apolipoprotein E (apoE) enhances the endocytic entry of pseudotyped SARS-CoV-2. Super resolution imaging of the SARS-CoV-2 entry point with high cholesterol showed markedly increased apparent diameter (~10% to 100 nm) and almost twice the total number of viral entry points. The cholesterol concomitantly traffics angiotensinogen converting enzyme (ACE2) to the viral entry site where SARS-CoV-2 docks to properly exploit entry into the cell. Furthermore, we show cholesterol enhances binding of SARS-CoV-2 to the cell surface which increases association with the endocytic pathway. Decreasing cellular cholesterol has the opposite effect. Based on these findings and known loading of cholesterol into peripheral tissue during aging and inflammation, we build a cholesterol dependent model for COVID19 lethality in elderly and the chronically ill. As cholesterol increases with age and inflammation (e.g. smoking and diabetes), the cell surface is coated with viral entry points and optimally assembled viral entry proteins. Importantly our model suggests high levels of cholesterol is most alarming in the tissue, not the blood. In fact, rapidly dropping cholesterol in the blood may indicate severe loading of cholesterol in peripheral tissue and a dangerous situation for escalated SARS-CoV-2 infectivity. Polyunsaturated fatty acids (PUFAs) oppose the effects of cholesterol and provide a molecular basis for eating healthy diets to avoid severe cases of COVID19.
INTRODUCTION
In early 2020, COVID19 spread rapidly throughout the developed world leading to extensive death and an ongoing pandemic. The lethality of the virus is notably selective for elderly1,2 and those with chronic disease such as hypertension, diabetes, cardiovascular disease, and smoking3–5. Interestingly, almost all children are either asymptomatic or present with very minor symptoms, while elderly and those with underlying conditions experience very severe life-threatening symptoms. This disparity was also observed with SARS-CoV-1, a closely related virus which failed to kill any children under the age of 12, despite being much more lethal than SARS-CoV-2 in adults6. Understanding why young people are resistant in this class of viruses could help both healthy and chronically ill adults avoid severe symptoms of COVID19.
The ability of a virus to enter a cell is critical to its lethality. SARS-CoV-2 enters the cell by binding a receptor angiotensinogen converting enzyme 2 (ACE2)7,8. Initial theories suggested that the expression level of the ACE2 receptor determined the level of disease severity in children9. However, more recent studies in COVID-19 patients show conflicting results and even a paradoxical increase of ACE2 in young children4,10. This result raises the question, What controls SARS-CoV-2 entry?
The known viral entry mechanism for SARS-CoV-1, a closely related virus, helps answer this question. SARS-CoV-1 requires cholesterol and monosialotetrahexosylganglioside1 (GM1) lipid domains (also called GM-1 lipid rafts) for infectivity of cultured cells11. Cholesterol is needed to form GM1 lipid rafts, which are the entry point for the viruses via endocytosis11–14. GM1 lipid rafts partition away from polyunsaturated lipids or disordered lipids15 (see Supplemental Figure S1A).
Cholesterol is an insoluble eukaryotic lipid found in membranes throughout the human body, most notably in the plasma membrane. Its accumulation in tissue increases with age16–18. Furthermore, chronic inflammation causes cholesterol loading into macrophage rich tissue19. Pneumocytes in the lung load and unload cholesterol along with macrophages and this process is associated with lung diease20. For further discussion on cholesterol in peripheral tissue see Figure S1B.
Therefore, a key question is, Is SARS-CoV-2 infectivity also cholesterol dependent? Here we show that SARS-CoV-2 viral entry is cholesterol dependent and this dependence relates to proper localization and assembly of SARS-CoV-2 with ACE2 in GM1 lipid rafts.
RESULTS
Cholesterol dependent SARS-CoV-2 pseudovirus entry
To test the cholesterol dependence of SARS-COV-2 on viral entry we loaded cholesterol into HEK293T cells with apolipoprotein E (apoE) and blood serum and treated the cells with retroviruse pseudotyped with the SARS-CoV-2 S protein (CoV-2-PV)21,22. A segment of the S protein binds to the ACE2 and recapitulates viral entry23. ApoE is a cholesterol carrier protein that binds to low-density lipoprotein receptor and facilitates loading of cholesterol into cells when bound to lipids. When lipids are not present, apoE has the opposite effect; it facilitates efflux of cholesterol from the cell24 (see Figure 1A and Supplemental Figure S2).
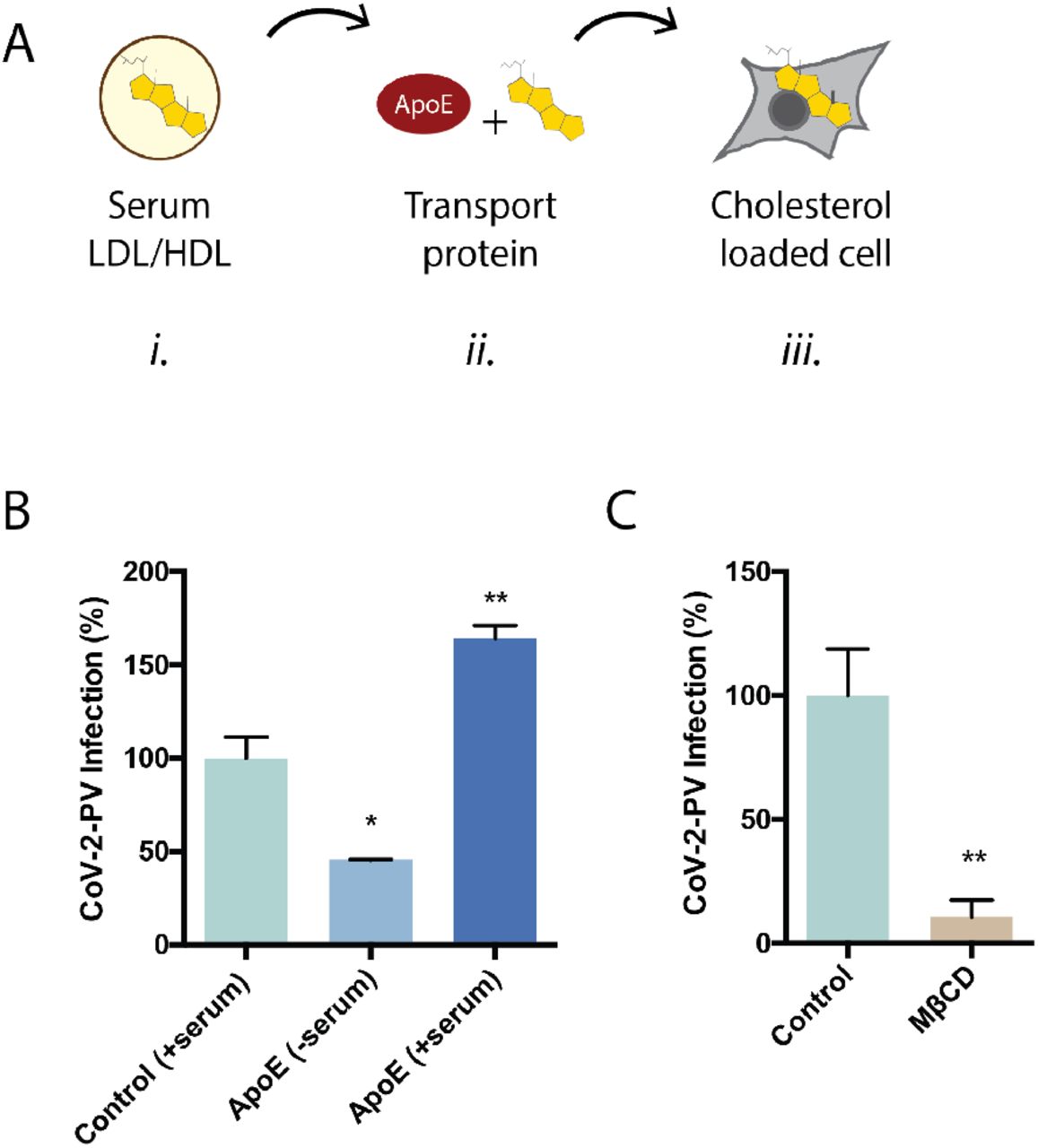
(A) Cartoon diagram showing the experimental setup for loading cultured cells with cholesterol. i., Cholesterol (yellow shading) is shown loaded into lipoprotein (e.g., low- and high-density lipoprotein (LDL and HDL respectively)) from blood serum. ii., Cholesterol free human apolipoprotein E (apoE, brown shading) a cholesterol transport protein is exposed to mixed with blood serum and iii. transports cholesterol in and out of cells (grey shading) (see also Supplemental Figure S1B). (B) SARS-CoV-2 pseudovirus (CoV-2-PV) entry assay. Cells were treated with a luciferase expressing retrovirus pseudotyped with the SARS-CoV-2 spike protein that recapitulates viral entry. Infectivity was monitored by a luciferase activity in cells treated with or without apoE. High cholesterol (apoE + serum) was more than 3-fold higher compared to low cholesterol. (C) Depletion of cellular cholesterol with methyl-beta-cyclodextrin (MβCD) blocked almost all viral entry measured by pseudotyped luciferase assay.
We found cholesterol loading significantly increased CoV-2-PV entry and unloading cholesterol had the opposite effect. CoV-2-PV entry increased 50% compared to control (serum only). Cholesterol was loaded with 4 ug/mL apoE and 10% serum. To confirm that the effect was from apoE transport of lipid from the serum, we added apoE without serum and found viral entry decreased more than 50% of control (Figure 1B). To further confirm the decreased viral entry is the result of cholesterol efflux, we treated cells with methyl-beta-cyclodextrin (MβCD), a chemical known to extract cholesterol from the membrane of cells. MβCD inhibited more than 90% of viral entry (Figure 1C). Same effect was observed with CoV-2-PV with mutated S protein (Figure S2). ACE2 labeling was robust in the presence of MβCD and serum free apoE, only GM1 labeling decreased (Figure 2A and S3A-B), suggesting the receptor expression does not account for the lack of entry.

(A) Representative images from dSTORM super resolution imaging. GM1 lipid raft (yellow) increase dramatically in the presence of blood serum when apoE is present (bottom panel), measured by CTxB labeling. ACE2 receptor (cyan) labeled with ACE2 antibody has very little overlap with GM1 lipid rafts when blood serum is removed (middle panel). Scale bar, 1 μm. (B) Cholesterol dependent trafficking of ACE2 in and out of lipid rafts. ApoE-mediated cholesterol influx translocates ACE2 from disordered regions into lipid rafts while cholesterol depletion by MβCD diminishes ACE2’s raft localization. (C) Model showing cholesterol-dependent raft localization of ACE2 enhances SARS-CoV-2 viral entry. (D) Raft size analysis suggests raft assembly is regulated by apoE mediated cholesterol transportation between serum and cell membrane. (E) ApoE transportation of cholesterol regulates raft density. Cholesterol influx from serum results in an increase in raft density. (F) Ripley’s K-Function (H(r)) showing raft separation after cholesterol depletion while distance between rafts gets shorter as apoE transport cholesterol from serum into the membrane. (G) Proposed model for infectivity in SARS-CoV-2 by age and chronic disease. When cholesterol is low there are very few entry points and their diameter is small (similar to children). With age, average cholesterol levels in the tissue increase, thereby increasing the number and size of viral entry points (similar to adults). In chronically ill patients, tissue cholesterol levels are the highest and all the ACE2 receptor is positioned for viral infectivity via the more potent endocytic mechanism (adults + chronic inflammation).
Cholesterol dependent trafficking of the ACE2 receptor to endocytic entry point
The cholesterol dependence of viral entry raises the question, What molecular mechanism causes the dependence? Previous studies showed ACE2 associates with detergent resistant membranes11, which are similar to GM1 lipid rafts, although not identical. We and many others have shown cholesterol is a major contributor to the apparent size, number, and trafficking of proteins into of GM1 lipid rafts in cellular membranes25–29. Logically, as the number size and number of infection sites increases, there is more opportunity for viral entry. Cholesterol can also enhance trafficking of the receptor ACE2 to the viral entry site. But experimental data showing a membrane protein translocation mechanism for ACE2 in a membrane is lacking.
To determine the amount of ACE2 positioned for viral entry and its dependence on cholesterol, we co-labeled ACE2 and GM1 lipid rafts in HEK293T cells with ACE2 antibody labelled with Cy3b and Alexa Fluor 647 conjugated fluorescent CTxB (a GM1 specific toxin), treated cells with apoE/serum, and imaged with super-resolution direct stochastical optical reconstruction microscopy (dSTORM)30–33. dSTORM is capable of visualizing nanoscale arrangements (i.e. sub-100 nm diameter lipid domain structures34) in intact cellular membranes.
Figure 2A shows representative dSTORM images of ACE2 and GM1 lipid rafts from apoE treated cells with and without serum exposure (i.e., cells loaded or not loaded with cholesterol, respectively). The association of ACE2 with GM1 lipid rafts was measured by pairwise correlation. Pairwise correlation measures the probability that two molecules are next to each other. For our experiments we compared the pair correlation between 0-5 nm.
We found pairwise correlation of ACE2 with GM1 lipid rafts increased more than 3-fold in cholesterol loaded cells. To confirm the effect depends on cholesterol and not another component of the serum or apoE regulation, we treated the cells with MβCD. In agreement with our viral entry assay, MβCD reduced ACE2 localization with GM1 lipid rafts by ~70%. These data suggest that cholesterol loading by apoE causes ACE2 to dock at the endocytic viral entry point (Figure 2B). We loaded cholesterol using 4 ug/mL apoE and 10% serum, the same as our viral entry assay.
We next investigated the effect cholesterol has on the apparent size and number of GM1 lipid rafts (i.e., the size and number of viral entry points for ACE2). Cells loaded with cholesterol (+apoE) increased in both apparent size (diameter) and the number of GM1 labeled rafts compared to not loaded (serum only) (Figure 2B-C). Cholesterol depletion by MβCD also robustly decrease the apparent raft size (Figure S3C). These decreases caused lipid rafts to separate from each other measured by Ripley’s H function. (See Figure 2F).
Cholesterol dependence in ACE2 localization and SARS-CoV-2 entry provides a possible explanation for age sensitivity in COVID19. As mentioned, cholesterol increases in cellular tissues with age16–18 and this accumulation directly links to disease24 including atherosclerosis35 and acute and chronic inflammation19. Remarkably, these same diseases are highly comorbid with COVID193,5. Based on these finding with cholesterol, we have proposed a model for age dependent infection of SARS-CoV-2 (Figure 2G). When cholesterol is low there are very few entry points and their diameter is small (similar to children). With age, average cholesterol levels in the tissue increase, thereby increasing the number and size of viral entry points (similar to adults). In chronically ill patients, tissue cholesterol levels are the highest and all the ACE2 receptor is positioned for viral infectivity (adults + chronic inflammation).
Cholesterol dependent binding
The reduced effectiveness of viral entry in the disordered region could be reduced binding to the receptor in the disordered region or reduced entry or both. To test SARS-CoV-2 binding, we applied the SARS-CoV-2 receptor binding domain (RBD) to HEK293T cells with and without MβCD. MβCD treatment reduced the binding of the RBD (Figure 3A). However, the effect was modest and suggests that entry is also less efficient in the disordered region.

(A) The receptor binding domains (RBD) of SARS-CoV-2 was added to cells treated with or without methyl-beta-cyclodextrin (MβCD). Binding was measured by fluorescence from an Alexa 647 labeled antibody to an FC tag on the RBD protein. (B) Overexpression of ACE2 decreases ACE2 pair correlation with GM1 lipid rafts suggesting additional ACE2 is added primarily to the disordered region, not GM1 lipid rafts. (C) Speculative model for decreases viral entry with increased ACE2 expression. In an environment where the viral titers are sub-saturating, high levels of ACE2 in the disordered region could cause the virus to attach away from the entry point and protect against viral entry. This scenario could occur in children with low cholesterol.
We also investigated whether elevated ACE2 expression and low cholesterol could contribute to a reduction in SARS-CoV-2 viral entry. We overexpressed ACE2 receptor and found that ACE2 colocalization with GM1 lipid rafts substantially decreased (Figure 3B). This suggest the excess ACE2 is localizing to the disordered region. Figure 3C depicts decreased viral entry based on surface entry in the disordered region rather than endocytosis.
Imaging of cholesterol dependent protease localization
In order for the virus to enter the cell it must be primed by proteases 36. In SARS-CoV-2 there is an efficient furin cleavage site between the S1 and S2 segments that is not found in SARS-CoV-137. This S1/S2 site primes the virus for entry. During the preparation of pseudovirus, this site appears to be mostly cut in the producing cells8. A second site (S2’) frees a fusion peptide and allows insertion of the virus into the host membrane. The S2’, is thought to be cleaved by TMPRSS2 or cathepsin proteases38–40. In SARS-CoV-1, TMPRSS2 is associated with a cell surface entry mechanism and cathepsin is associated with an endocytic mechanism36.
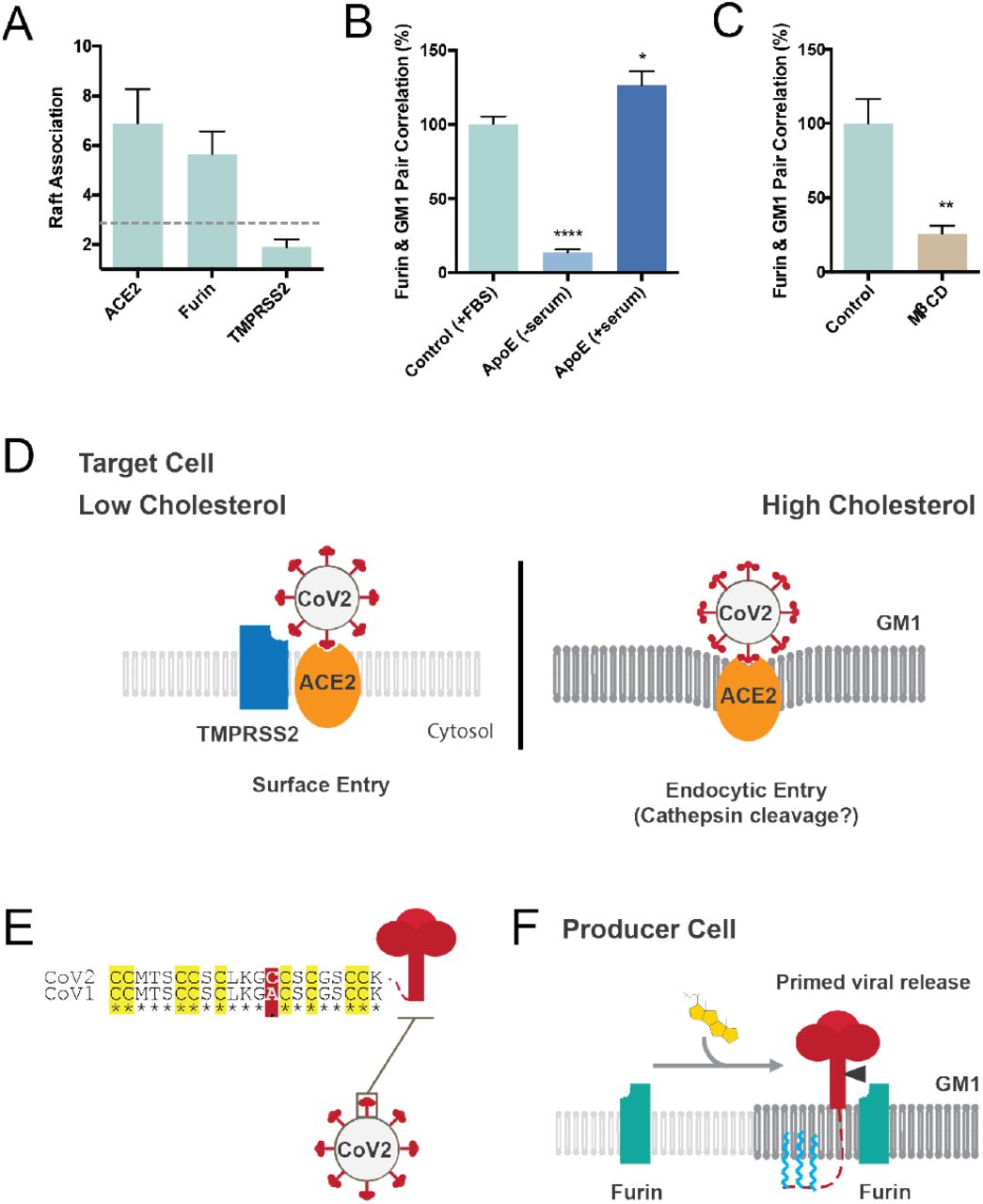
For the virus to interact with a protease it must be in proximity (i.e., the same lipid compartment). To test their potential proximity with the virus and define the nano-scale environment, we pairwise labeled furin and TMPRSS2 in combination with GM1 lipid rafts and imaged with dSTORM. Compared to ACE2, only furin was localized to GM1 lipid rafts (Figure 4A). Removing cholesterol with ether serum free apoE or MβCD caused furin to no longer associate with GM1 lipids (Figure 4B-C).

Furin and TMPRSS2 are proteases that facilitate viral entry37–40. (A) Super resolution imaging of furin and TMPRSS2 in GM1 lipid rafts. Raft association is shown as a measure of pair correlation, a unitless number see methods. A cutoff of approximately 3 times background is shown as a grey dotted line and interpreted as very little association with GM1 lipid rafts. ACE2 and furin are clearly associated with GM lipid rafts while TMPRSS2 sits in the disordered regions (<2 pair correlation with GM1 clusters). (B) Furin moves out of the raft under low cholesterol condition (apoE extracts cholesterol from membrane). Cholesterol loading by apoE causes an increased furin raft association. (C) Cholesterol depletion by MβCD traffics furin out of the GM1 lipid rafts into disordered regions. (D) Model showing the overall nanostructure of SARS-CoV-2 viral entry. In low cholesterol TMPRSS2 (blue shading) and ACE2 (orange oval) are in the disordered region (light grey lipids). When the cell is loaded with cholesterol the receptor and the protease translocate to GM1 lipid rafts (GM1, dark grey lipids). The translocation has a two-fold effect. First SARS-CoV-2 is positioned with more concentrate ACE2 which can directly facilitate viral entry and putative cathepsin cleavage, second furin would be available to hydrolyze any unprimed spike protein (not shown). (E) Sequence alignment of the palmitoylation site in SARS-CoV-1 (GenBank: ABD72995.1) and SARS-CoV-2 (GenBank: QII57161.1). Residues conserved are marked with an asterisk. All 9 putative palmitoylation sites identified in SARS-CoV-1 are conserved in SARS-CoV-2 (yellow highlight). A single mutation introduces a 10th putative palmitoylation site (red shading) (F) speculative role of palmitoylation in SARS-CoV-2 priming of the spike protein. In high cholesterol furin moves from the disordered lipids (light grey lipids) to ordered GM1 lipids (GM1, dark grey lipids). Cholesterol dependent membrane protein translocation is indicated by a grey arrow and the input of cholesterol (yellow shading).
Based on the positioning of ACE2 relative to TMPRSS2 and GM1 domains, we propose a model of infectivity regulated by high and low levels of cholesterol (Figure 4D). During low cholesterol ACE2 is in the disordered region and is cut by TMPRSS2, (surface entry). In high cholesterol, ACE2 translocates into the GM1 lipid raft and enters through the endocytic pathway, presumably to be cut by cathepsin similar to SARS-CoV-1. The virus surface is covered with spike protein and the binding of ACE2 may create enough affinity to pull GM1 lipids to envelope the virus and drive entry into an endosome.
Role of palmitoylation in viral priming
Lastly, we considered palmitoylation of SARS-CoV-2 spike protein. Palmitoylation is a post translation modification that attaches a 16 carbon saturated lipid to many proteins and traffics them to GM1 lipid rafts41. Palmitoylation of the cysteine-rich endodomain of the SARS-CoV-1 spike glycoprotein is important for spike-mediated cell fusion and viral entry42,43. Little is known about palmitoylation of SARS-CoV-2. We aligned SARS-CoV-1-2 and found that all 9 putative palmitoylation sites in SARS-CoV-1 are conserved in SARS-CoV-2 (Figure 4D). A single significant mutation (alanine to cysteine) introduces an additional putative palmitoylation site in SARS-CoV-2, which suggest SARS-CoV-2 remains under evolutionary pressure for palmitate driven viral entry.
In a viral producing cell palmitoylation is positioned on the intracellular leaflet suitable for targeting the nascent spike protein to GM1 lipid rafts. In high cholesterol, furin would translocate into GM1 lipid rafts, a position that would likely hydrolyze the spike protein at the S1/S2 site. Figure 4F shows a speculative model for S1/S2 priming in virus producing cells.
DISCUSSION
Taken together, we conclude that SARS-CoV-2 has at least three cholesterol-dependent mechanisms that likely contribute to differential infectivity in elderly with an underlying condition and with chronic inflammation. One, the size and number of entry sites are cholesterol dependent (Figure 2D-F). If there are few of the preferred entry points, the virus cannot enter the cell as efficiently(Figure 1B-C,). Two, the cholesteroldependent ability of the receptor, and presumably the virus, to dock into the more advantageous lipids is logically one of the most important steps to infectivity (Figure 1B). If the virus isn’t positioned in the membrane correctly it likely cannot enter the cell efficiently. Third the proteases that activate SARS-CoV-2 for membrane insertion are also cholesterol dependent and rely on a membrane protein translocation mechanism for activation (Figure 4) (i.e., lateral diffusion into lipid rafts in the membrane).
Based on our experiments the surface entry mechanism is less efficient and likely contributes to attenuated viral infection in young children. When cholesterol was depleted SARS-CoV-2 infectivity went down (Figure 1B-C). Further supporting this conclusion, the increased binding of the RBD domain in higher cholesterol suggests the virus has evolved to prefer the endocytic pathway. Over expression of TMPRSS2 does increase infectivity8. If TMPRSS2 is the preferred mechanism for children, they may not be as effective in elderly and chronically ill. However, our studies do not rule out that TMPRSS2 contributes to the endocytic pathway in pneumocytes or in higher cholesterol. Without a cholesterol source, most cell cultures likely represent the conditions seen in children, not the elderly and chronically ill. This may be important for development of treatments for COVID19.
Importantly, our model suggests high levels of cholesterol in the tissue are indicative of SARS-CoV-2 infectivity, not the levels of cholesterol in the blood. Most clinical diagnosis of cholesterol are done by analyzing blood samples, however these level do not necessary indicate the level of cholesterol in the tissue. This difference is particularly true of chronic inflammation where blood cholesterol is low due to inhibition of cholesterol efflux proteins in the peripheral tissue19 (see also supplementary material). Our model is consistent with a clinical study in China that showed LDL and HDL cholesterol were low in severe COVID19 patients, yet monocytes in the blood had high cholesterol44. The high cholesterol in monocytes is consistent with cholesterol loading into peripheral tissue during chronic inflammation.
In a speculative role, palmitoylation could aid SARS-CoV-2 priming by binding GM1 lipid rafts in the producing cells. This hypothesis is based purely on the known affinity of palmitate for GM-1 ordered lipids41 and the fact that the palmitoylation is on the cytosolic side of the membrane during viral production. This would increase the amount of virion cut at the S1/S2 position, especially in high cholesterol tissue. Alternatively, the high cholesterol could cause furin to be endocytosed and cycled back through the Golgi where it would interact with the spike protein as it traffics to the surface of the cell. More research is needed to understand the role of palmitoylation in SARS-CoV-2 life cycle.
In high cholesterol furin is likely located in the GM1 lipid rafts with ACE2 in the target membrane. For virions primed in the producing cells furin is likely of no consequence in the target membrane. However, since the trafficking of furin is cholesterol dependent (Figure 4B-C) priming could be different in young and old. In which case the availability of furin in high cholesterol could be another critical factor for age dependent infectivity.
The concept that molecules diffuse laterally in the plane of the membrane was first proposed by Roger Kornberg and Harden McConnell in 197145. Decades of research showed that both proteins and lipids contribute to restricted diffusion of lipid in the membrane46,47 and that the cells use the restricted movement for biological function25,26,28,48. The finding here that a virus would be highly dependent on such a process is not unexpected and our proposed membrane protein translocation mechanism could be significant for other viruses.
The use of super resolution imaging is an important advancement of this study. Previous studies used detergent resistant membranes (DRMs) to characterize localization of ACE2. In our experiments, ACE2 was very sensitive to raft disruption. Not surprisingly, conflicting reports showed ACE2 association with both disordered49,50 and ordered lipids11. DRMs are a good first approximation, but for proteins that move in and out, the detergent used to prepare the DRM’s likely biases proteins to the disordered regions.
Although Figure 1 shows only cholesterol controlling the trafficking of ACE2, it is highly likely that a specific lipid or peptide in the disordered region of the membrane influences the balance of ACE2 in or out of GM1 lipid rafts. The disordered region of the membrane is separate from GM1 lipid rafts and can have proteins or lipids that directly bind to ACE2 or palmitoylated SARS-CoV-2 and traffic ACE2 out of GM1 rafts. We have shown direct binding of phosphatidylinositol 4,5 bisphosphate (PIP2) opposes localization of a palmitoylated enzyme to GM1 lipid rafts26,51. PIP2 is polyunsaturated, cholesterol independent, and resides in the disordered region26,52,53. Binding of PIP2 to phospholipase D counteracted its cholesterol dependent location to GM1 lipid rafts26,54 and demonstrates that cholesterol can be in balance with other molecules in the membrane to regulate the trafficking of an enzyme in or out of a lipid raft. As cholesterol increases with age, the putative molecules that balance cholesterol also likely increase. Hence, simply removing cholesterol may have complications.
METHODS
Cell culture
Human embryonic kidney 293T (HEK293T) cells and HEK293T cells overexpressing human ACE2 (hACE2) were grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
HEK293T cells overexpressing hACE2 were generously provided by Dr. Michael Farzan (Department of Immunology and Microbiology, Scripps Research). Cells were generated by transduction with murine leukemia virus (MLV) pseudotyped with the vesicular stomatitis virus G protein and expressing myc-hACE2-c9, as previously described55. Briefly, HEK293T cells were co-transfected by PEI with three plasmids, pMLV-gag-pol, pCAGGS-VSV-G and pQCXIP-myc-hACE2-c9 at a ratio of 3:2:1, and medium was refreshed after overnight incubation of transfection mix. The supernatant with produced virus was harvested 72-hours post transfection and clarified by passing through 0.45μm filter. 293T-hACE2 cells were selected and maintained with medium containing puromycin (Sigma). hACE2 expression was confirmed by SARS2-PV entry assays and by immunofluorescence staining using mouse monoclonal antibody recognizing c-Myc.
Production of SARS-CoV-2 pseudoviruses
Retroviruses pseudotyped with the SARS-CoV-2 S proteins (SARS2-PV) was produced as previously described21,22 with modest modifications as described. HEK293T cells were transfected by X-tremeGENE™ 9 DNA Transfection Reagent (Millipore Sigma, #6365787001) at a ratio of 3:4:3 with a plasmid encoding murine leukemia virus (MLV) gag/pol proteins, a retroviral vector pQCXIX expressing firefly luciferase, and a plasmid expressing the spike protein of SARS-CoV-2 (GenBank YP_009724390). The Farzan lab (Scripps Research) generated a derivative where the two native arginines in the furin cleavage sites of SARS-CoV-2 S protein were mutated to serines (R682S R685S) to increase viral infection. All the viral plasmids were provided by Dr. Michael Farzan. Cell culture supernatant containing pseudoviruses was harvested at 72 hours post transfection.
Viral Entry Assay
HEK293T cells were cultured in 96-well flat culture plates with transparent-bottom (Corning™ Costar™, #3585) pre-coated with poly-D-lysine. Cells were incubated with media containing 4 μg/mL apoE with or without FBS supplementation overnight before viral exposure. For MβCD treatment, cells were incubated with 100μM MβCD for 30 min prior to virus infection. SARS-CoV-2 pseudoviruses were applied to the cells and allowed to infect at 37 °C for 24 hours. After viral infection, efficiency of viral entry was determined through a firefly luciferase assay. Specfically, cells were washed with PBS once and 16 μL Cell Culture Lysis Reagent (Promega, #E153A) were added into each well. The plate was incubated for 15 min with rocking at room temperature. 8 μL of cell lysate from each well was added into a 384-well plate (Corning™, #3574), followed by the addition of 16 μL of Luciferase Assay Substrate (Promega, #E151A). Luciferase activity measurement was performed on a Spark 20M multimode microplate reader (Tecan). All the infection experiments were performed in a biosafety level-2 (BSL-2) laboratory.
dSTORM Super-resolution Imaging
Fixed cell preparation
Cells were grown to 60% confluence. Cells were incubated with 4 μg/mL purified apoE protein with or without FBS supplementation (overnight) or 100μM MβCD (30 min) in media. Cells were rinsed with PBS and then fixed with 3% paraformaldehyde and 0.1% glutaraldehyde for 20 min to fix both proteins and lipids. Fixative chemicals were reduced by incubating with 0.1% NaBH4 for 7 min with shaking followed by three times 10 min washes with PBS. Cells were permeabilized with 0.2% Triton X-100 for 15 min and then blocked with a standard blocking buffer (10% bovine serum albumin (BSA) / 0.05% Triton in PBS) for 90 min at room temperature. For labelling, cells were incubated with primary antibody (anti-ACE2 antibody (Abcam, #ab189168), anti-Furin antibody (Abcam, #ab3467) or anti-TMPRSS2 antibody [EPR3861] (Abcam, #ab92323)) for 60 min in 5% BSA / 0.05% Triton / PBS at room temperature followed by 5 washes with 1% BSA / 0.05% Triton / PBS for 15 min each. Secondary antibody (donkey anti-rabbit cy3b and CTxB-647) was added in the same buffer as primary for 30 min at room temperature followed by 5 washes as stated above. Cells were then washed with PBS for 5 min. Cell labelling and washing steps were performed while shaking. Labeled cells were then post-fixed with fixing solution, as above, for 10 min without shaking followed by three 5 min washes with PBS and two 3 min washes with deionized distilled water.
dSTORM imaging
Images were recorded with a Zeiss ELYRA PS.1 microscope using TIRF mode equipped with a pilimmersion 63x objective. Andor iXon 897 EMCCD camera was used along with the Zen 10D software for image acquisition and processing. The TIRF mode in the dSTORM imaging provided low background high-resolution images of the membrane. A total of 10,000 frames with an exposure time of 18 ms were collected for each acquisition. Excitation of the Alexa Fluor 647 dye was achieved using 642 nm lasers and Cy3B was achieved using 561 nm lasers. Cells and brain tissues were imaged in a photo-switching buffer comprising of 1% β-mercaptoethanol (Sigma, #63689), and oxygen scavengers (glucose oxidase (Sigma, #G2133) and catalase (Sigma, #C40)) in 50mM Tris (Affymetrix, #22638100) + 10mM NaCl (Sigma, #S7653) + 10% glucose (Sigma, #G8270) at pH 8.0. Sample drift during acquisition was corrected by an autocorrelative algorithm.
Images were constructed using the default modules in the Zen software. Each detected event was fitted to a 2D Gaussian distribution to determine the center of each point spread function plus the localization precision. The Zen software also has many rendering options including removing localization errors and outliers based on brightness and size of fluorescent signals. Pair correlation and cluster analysis was performed using the Statistical Analysis package in the Vutara SRX software. Pair Correlation analysis is a statistical method used to determine the strength of correlation between two objects by counting the number of points of probe 2 within a certain donut-radius of each point of probe 1. This allows for localization to be determined without overlapping pixels as done in traditional diffraction-limited microscopy. Raft size estimation and raft density were calculated through cluster analysis by measuring the length and density of the clusters comprising of more than 10 particles with a maximum particle distance of 0.1 μm. Ripley’s H(r) analysis was performed to study the distribution of lipid raft clusters.
Receptor Binding Domain (RBD) Binding Assay
HEK293T cells were seeded into 96-well flat culture plates with transparent-bottom (Corning™ Costar™, #3585) one day before experiment. SARS-CoV-2 RBD with a human Fc fusion was a generous gift from Dr. Michael Farzan (Department of Immunology and Microbiology, Scripps Research). HEK293T cells were incubated with 10 μg/mL RBD peptide in cell culture medium overnight at 37°C in 5% CO2. Cells were washed with PBS once before fixation with 3% paraformaldehyde. After blocking with a standard blocking buffer (10% bovine serum albumin (BSA) / 0.05% Triton in PBS) for 90 min at room temperature, RBD binding to the cells were labelled with Alexa 647 goat anti-human IgG (Jackson ImmunoResearch, # 109-606-098) for 6 hours at 4°C. After labelling, cells were washed with PBS twice. Relative RBD binding was determined in quintuplicate for each condition by measuring fluorescent activity with a fluorescence microplate reader (Tecan Infinite 200 PRO) with excitation wavelength of 630 nm and an emission wavelength of 680 nm.
Statistical Analyses
All statistical calculations were performed in GraphPad Prism v6.0. For the Student’s t test, significance was calculated using a two-tailed unpaired parametric test with significance defined as p < 0.05, **P<0.01, ***P<0.001, ****P<0.0001. For the multiple comparison test, significance was calculated using an ordinary one-way ANOVA with Dunnett’s multiple comparisons test.
Supplemental Figures

Cholesterol transport and function in GM1 lipid rafts (A) The side view of a plasma membrane is shown (top extracellular). The membrane partitions into regions of ordered (saturated) and disordered (unsaturated) lipids. The ordered region is containing cholesterol and sphingolipids (Sph). The packing of cholesterol with the saturated lipids is thought to provide the order and makes ordered lipids thicker than disordered lipids. The disordered region contains unsaturated lipids including phosphatidylcholine (PC), phosphatidic acid (PA), and phosphatidylinositol 4,5-bisphosphate (PIP2). PIP2 and PA are signaling lipids and PIP2 forms its own lipid domains separate from GM1 lipid rafts. Palmitoylation of proteins typically occurs on the cytosolic portion of the membrane and inserts into the inner leaflet (red line with lipids attached). For a virus that exits the cell the palmitoylation may insert into the extracellular leaflet. (B) Cholesterol loading into lung pneumocyte’s and macrophages. In healthy individuals most cholesterol is produced in the liver. The effluxes cholesterol through ATB binding cassette (ABC) transporters (not shown) loaded into apolipoproteins (e.g., A and E; apoA and apoE). The cholesterol is then transported through the blood serum in the form of high- and low-density lipoproteins to the lung. In the lung receptors take up the cholesterol. In healthy individuals, excess cholesterol is effluxed from ABCA1 transporter in the lung loaded into apoA1 or apoE and transported back to the liver for elimination. During chronic inflammation reverse cholesterol transport is inhibited and cholesterol is loaded into macrophage rich tissues in the periphery.

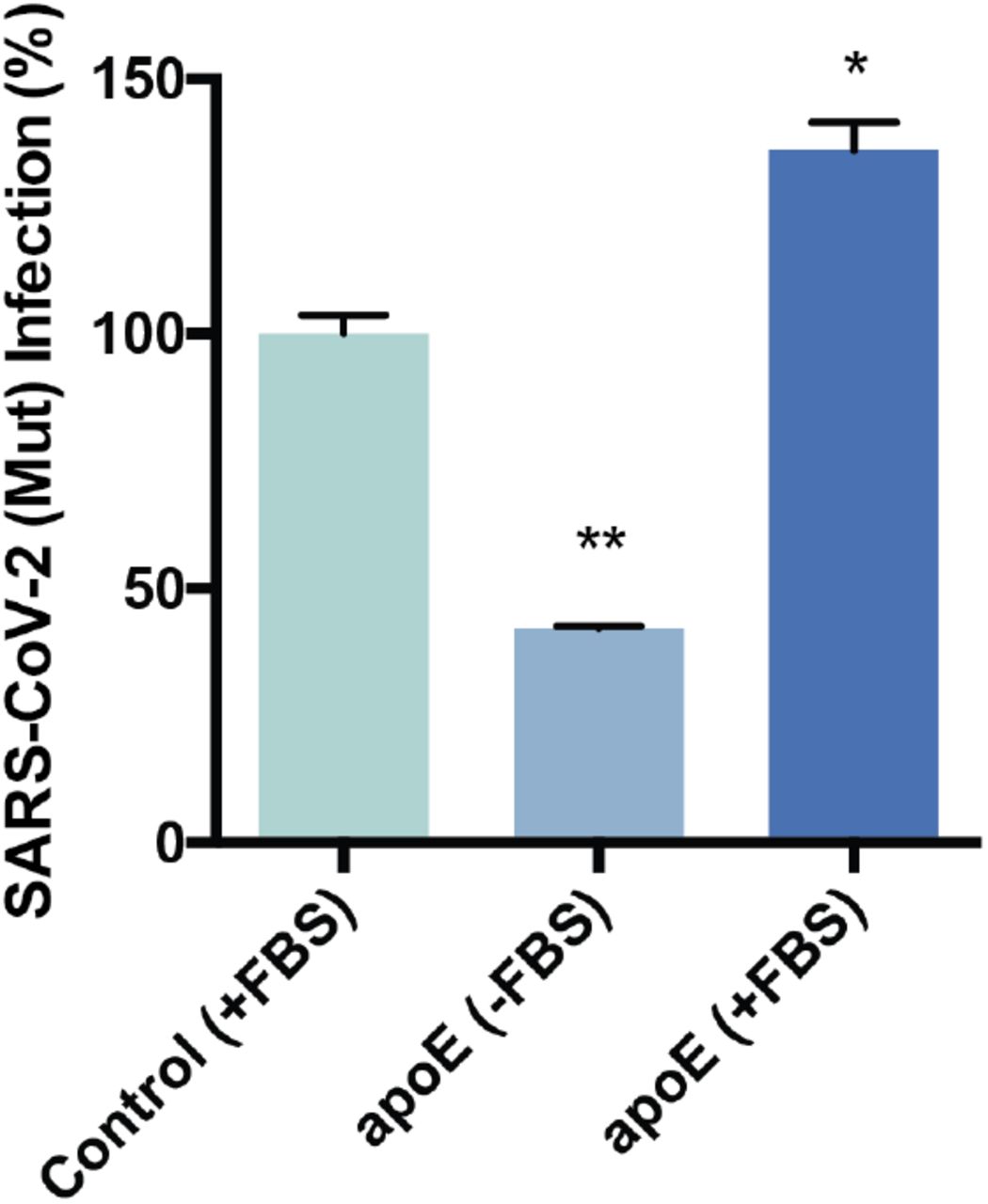
SARS-CoV-2 (Mut) pseudovirus (PV) entry assay. Cells were treated with a luciferase expressing retrovirus pseudotyped with the SARS-CoV-2 spike protein (mutant) that recapitulates viral entry. Infectivity was monitored by a luciferase activity in cells treated with or without apoE. High cholesterol (apoE + serum) was more than 3-fold higher compared to low cholesterol.

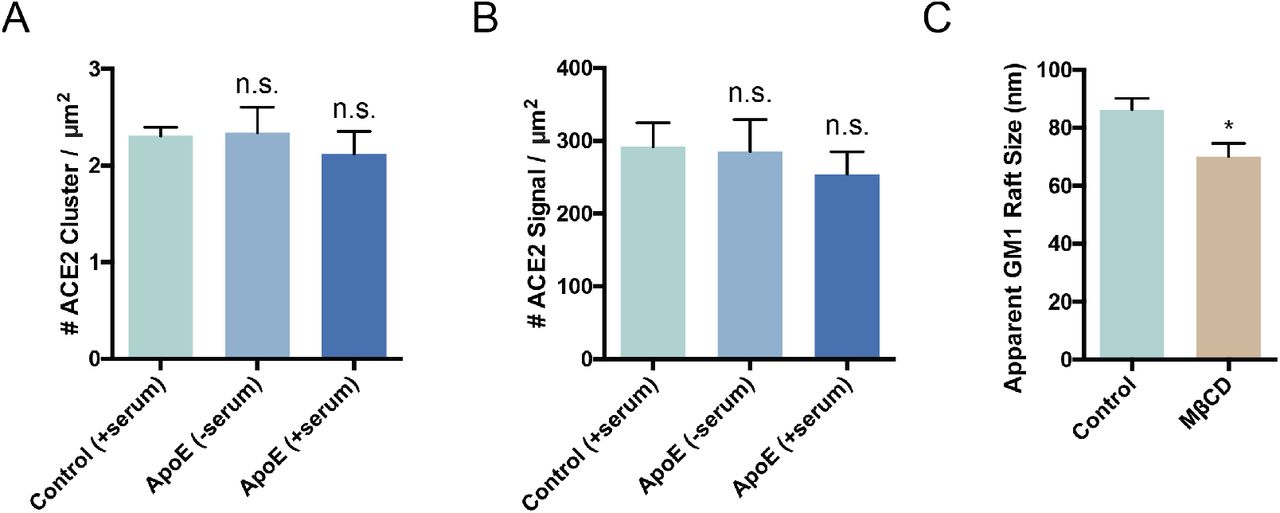
ACE2 surface expression and GM1 lipid raft density. Quantitation of ACE2 cluster (A) and signal (B) shows that the expression of ACE2 remains roughly constant when cells are loaded or unloaded with cholesterol by the cholesterol transport protein apoE. Although, while not statistically significant ACE2 expression appears to slightly decrease in high cholesterol (ApoE + serum) which agrees with the clinical findings of decreased ACE2 surface expression in older people. (C) Cholesterol depletion by methyl-beta-cyclodextrin (MβCD) robustly decreases the apparent size of GM1 lipid rafts.
Acknowledgements
We thank Michael Farzan for the SARS-CoV-2 spike plasmid, hACE2 overexpression HEK293T cell line, and RBD peptide; Andrew S. Hansen for helpful discussion and reading of the manuscript. This work was supported by the National Institutes of Health via a Director’s New Innovator Award to S.B.H. (DP2NS087943), R01 to S.B.H. (R01NS112534). We are grateful to the Iris and Junming Le Foundation for funds to purchase a super-resolution microscope, making this study possible.
Footnotes
Enlarged Figure 2-4.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}