

Abstract
Liposomes, aqueous vesicles enclosed by lipid bilayers, are widely used in research as simple, synthetic analogues of cell membranes. Membrane-binding DNA nanostructures have been developd whichh can modify the shape, porosity and reactivity of liposomes. Lipid-DNA binding is moderated using strands with hydrophobic or amphipathic chemical groups such as cholesterol. However, the factors that affect the binding interactions of cholesterol-modified DNA and membrane bilayers have not been systematically investigated. Here we characterise the effect of buffer and lipid composition and DNA structure near the cholesterol motif on the strength of DNA-lipid binding. We observed that DNA-membrane binding is inhibited at increasing ionic concentrations and that binding is severely reduced in strongly acidic conditions. Background membrane cholesterol content demonstrated a more varied effect, dependent on lipid composition. The composition of the DNA, whether simplex or duplex, showed little effect on binding, as did the presence or absence of a single-stranded ‘overhang’ to protect the cholesterol and prevent DNA strand aggregation. Our results inform the design and modelling of the membrane binding of cholesterolated DNA nanostructures.
Introduction
Liposomes are aqueous vesicles bound by one or more bilayers of lipids, a diverse group of amphipathic and hydrophobic small molecules. Due to their similarities to membrane bilayers, which are ubiquitous in nature and form the basis of biological compartmentalisation, liposomes have proven a powerful research tool for modelling cellular membranes in simplified synthetic systems. Liposomes can also be used for encapsulating therapeutic payloads in order to increase a drug’s circulation time and alter its distribution profile (Storm and Crommelin, 1998). Various strategies for engineering liposomes for therapeutic applications have been developed in order to increase circulation time, allow targeted payload release or deliver a payload to a cell’s cytosol (Dou et al., 2017; Veronese and Harris, 2002). A number of liposome-encapsulated ‘nanodrugs’ are FDA-approved (Bulbake et al., 2017).
DNA nanotechnology is a ‘bottom-up’ approach to designing and building nanometre-scale structures based on DNA based on Watson-Crick base pairing (Seeman and Sleiman, 2018). Since the development of DNA nanotechnology (Seeman, 1982), a variety of two and three-dimensional structures have been created and described (Wang et al., 2017) as well as environment-sensing mechanisms which allow DNA nanostructures to change state in response to an external trigger (Singh et al., 2018).
DNA and lipid nanotechnologies can be combined by modifying DNA with hydrophobic chemical groups such as cholesterol to enable membrane binding (Bell and Keyser, 2014). Using this approach, a variety of membrane-binding and membrane-spanning DNA nanostructures have been developed (Darley et al., 2019). Membrane-binding DNA nanostructures have been used to functionalise liposome surfaces (Akbari et al., 2017), control the shape of liposomes by inducing membrane curvature and tubulation (Franquelim et al., 2018; Grome et al., 2018) and form membrane-spanning nanopores (Burns et al., 2013; Langecker et al., 2012). Such nanpores can have dimensions which exceed those of natural protein pores (Diederichs et al., 2019) and feature gating mechanisms that can be triggered externally (Burns et al., 2016; Mendoza et al., 2017).
Despite the widespread use of cholesterol for DNA-lipid mediation, little is known about the kinetics and energetics of DNA nanostructure insertion in bilayers (Darley et al., 2019). Increasing the efficiency of membrane attachment is thus of great interest to the design and application of membrane-bound DNA nanostructures. In particular, currently large numbers of hydrophobic groups are necessary for spontaneous and stable membrane insertion to occur (Krishnan et al., 2016), and to overcome the substantial energy penalties associated with the insertion of membrane-spanning DNA nanopores (Göpfrich et al., 2016). It has been observed that both the quantity and position of TEG-cholesterol anchors on DNA nanostructures affects their affinity for lipid bilayers (Khmelinskaia et al., 2016; Langecker et al., 2012). Monovalent and divalent cations are necessary buffer components in order to assemble and maintain the stability of DNA duplexes and nanostructures (Kielar et al., 2018; Nakano et al., 1999), yet also are also known to affect the physical characteristics of membrane bilayers (Böckmann et al., 2003; Velikonja et al., 2013) and may affect the binding activity of cholesterol-modified DNA. Thus far, the optimal environmental conditions to promote binding interactions between cholesterol-modified DNA and have not been systematically investigated.
Here we have quantified the binding of cholesterol-modified DNA strands to synthetic liposomes using fluorescence microscopy. We examined the effects of pH, ion concentration and membrane cholesterol content on the binding of cholesterol-modified DNA strands to liposomes. We investigated three types of DNA motif: a single stranded form, a duplex, and a duplex with a short ssDNA ‘overhang’ proximal to the cholesterol group, recently proposed by Ohmann et al. to reduce aggregation during nanostructure assembly (Ohmann et al., 2019).
Materials and Methods
Preparation of Buffers and solutions
In order to investigate the effect of salts on DNA/Lipid interaction, Liposomes and DNA stocks were diluted in Liposome Buffer (210 mM D-Sorbitol [S1876, Sigma], 5 mM Tris-HCl [T3253, Sigma], pH 7.5) containing NaCl [AJA465, Ajax-Finechem] (12.5 mM to 400 mM) and MgCl2 [AJA296, Ajax-Finechem] (0 mM to 80 mM) as required.
To investigate the effect of pH on DNA/Lipid interaction we used a modified Liposome Buffer (210 mM D-Sorbitol, 100 mM NaCl) with pH adjusted to approximate pH values of 2, 4, 6, 7, 8 and 10. The pH was adjusted to +/- 0.2 of the target pH value by using 200 mM NaOH or HCl 3 hours prior to imaging.
Design and assembly of oligonucleotides and DNA duplexes
DNA strands used for colocalisation experiments were 23 nt-long, used alone (ssDNA), or hybridised to a complimentary oligo (dsDNA) or a complimentary sequence with a 5’ 6 nt single stranded ‘overhang’ (dsDNA-6 nt) (Supplementary Table 1). The oligonucleotide sequences were generated using NUPACK design software (Zadeh et al., 2011) and selected to prevent the formation of unwanted secondary structures. The 6 nt ‘overhang’ sequence introduced at the 5’ end of oligos was chosen from Ohmann, et al., 2019. Oligos were modified at the 3’ end with a tetraethylene glycol cholesterol moiety (TEG-cholesterol) and with a 5’ Alexa 647 fluorescent group respectively. All oligos were purchased from IDT (Integrated DNA Technologies, Inc., USA).
DNA stocks (100 μM, 1000x) were prepared using MilliQ water [Milli-Q, Millipore] and stored at 4°C. Alexa 647-labelled DNA was stored in foil at -20°C. DNA duplexes were annealed at 10 μM final concentration in duplex buffer. All oligos were heated to 90°C for five minutes then cooled in a termocycler at 5°C /minute for 15 minutes to a final temperature of 15°C, then stored at 4°C. For duplex assembly, unmodified complementary strands were added in a 3-fold excess. DNA was diluted in extrusion buffer to a final concentration of 100 nM after the melting and annealing steps.
Preparation of Liposomes
Liposomes were produced from two main lipid mixtures, DOPE/DOPC liposomes [49.9% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (DOPE 18:1, 850725 P, Avanti), 49.9% 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (DOPC 18:1, 850375 P, Avanti)] or DPhPC liposomes [99.8% 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC 850356P Avanti Polar Lipids)] (Supplementary Table 2). Both lipid mixtures were doped with 0.1% PE-rhodamine [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl, 810150P Avanti Polar Lipids] for fluorescence imaging and 0.1% PE-biotin [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-biotinyl, 870282P Avanti Polar Lipids] for surface tethering. All percentages indicate weight to weight ratios.
Liposomes with cholesterol were prepared by replacing either DPhPC or equal parts of DOPE and DOPC with cholesterol [700000P Avanti Polar Lipids]. All lipids stocks were dissolved in chloroform at 10 mg/mL and stored at -20 °C.
Large unilamellar vesicles (LUVs) were produced by extrusion using a Mini-Extruder kit (Avanti Polar Lipids Inc., USA). Briefly, lipid stocks were added to a round-bottom glass tube and dried into under gentle nitrogen flow into a thin film and resuspended in extrusion buffer to a final concentration of 1 mg/mL by vortex mixing and sonication. The resulting suspension was transferred to a 500 μL glass syringe (Hamilton Company, UK) and passed back and forth through a 100 nm polycarbonate filter (Whatman plc, USA) 41 times to produce a clear suspension of homogenous unilamellar liposomes. Liposomes were then diluted 100-fold in Liposome Buffer solution prior to loading onto tunnel slides for imaging.
Giant unilamellar vesicles (GUVs) were produced by electroformation using the Vesicle Prep Pro machine (Nanion Technologies GmbH, Germany). 30 μl of 3.5 mg/mL lipid dissolved in chloroform was added to a conductive indium tin oxide-coated glass slide and spread over a spot approximately 12 mm in diameter and allowed to air-dry for two minutes into a circular film. A 1.5 mm thick rubber gasket of 15 mm diameter was placed around the film, forming a well into which 250 μl of electroformation solution was added. A second indium tin oxide-coated glass slide was placed face-down on top of the gasket and clamped in place, creating a sealed chamber of liquid between the two slides. The machine was run using the default protocol of 3 V AC for 120 minutes to form GUVs. Giant unilamellar liposomes in electroformation solution were then diluted at a 1:1 ratio in buffer consisting of 210 mM sorbitol, 80 mM NaCl, 10 mM Tris-HCl, giving a final external solution of 210 mM sorbitol, 40 mM NaCl, 5 mM Tris-HCl. Liposome dissolution was tested by titration of increasing concentration of the detergent Polysorbate-20 (Supplementary Figure 4).
Construction of tunnel slides for microscopy
To form a tunnel slide for imaging DNA-liposome interactions, a 50 mm cover slip (#1 thickness) (Menzel Glaser GmbH) was fixed to a glass slide (Suzhou Upline Medical Products Co., China (PRC)) using two parallel strips of double-sided tape (Nichiban Co., Japan) approximately 150 μm thick placed 2 mm apart, forming a channel of approximately 15 μL volume. A thin layer of CoverGrip Coverslip Sealant (Biotium Inc., USA) was applied over remaining exposed tape to prevent the contamination of solutions by adhesive residue and left to cure for 24 hours. Solutions were added to one end of the channel with a pipette while simultaneously drawing solution from the opposite end with an absorbent paper wipe (Kimberly-Clark Professional, USA).
Tethering of liposomes for TIRF imaging
The imaging system was based on a protocol developed by Jungmann, et al. for DNA-PAINT super-resolution microscopy of cells (Jungmann et al., 2014), with modifications made to buffer compositions, volumes and solution concentrations.
First, 15 μL of a 9:1 mixture of bovine serum albumin (BSA) and biotinylated bovine serum albumin (BSA-biotin) in slide buffer at a combined concentration of 1 mg/mL was added to the channel and incubated for 10 minutes to block and coat the surface of the cover slip. Excess BSA and BSA-biotin in solution was then removed by flushing 60 μL of slide buffer through the channel.
Next, 15 μL of streptavidin at 0.1 mg/mL in buffer A was added to slide and incubated for 10 minutes. Unbound streptavidin remaining in solution was removed from the slide by flushing 60 μL of extrusion buffer through the slide. Afterwards, 15 μL of biotinylated liposome solution was introduced into the slide and incubated for 30 minutes to allow streptavidin-biotin conjugation. Finally, 15 μL of Alexa 647-labelled DNA solution (100 nM) in extrusion buffer was added to the slide and incubated for 30 minutes prior to imaging.
Fluorescence microscopy of extruded liposomes (Large Unilamellar Vesicles)
Surface-tethered liposomes were imaged using on a Zeiss Elyra PALM/SIM Microscope in Total Internal Reflection Fluorescence (TIRF) mode with a 63x/1.4 Oil Iris M27 oil immersion objective (Carl Zeiss AG, Germany) and Andor iXon 897 EMCCD camera (Oxford Instruments, United Kingdom).
Two-channel images were collected to visualize the fluorescence from liposomes (‘liposome channel’, 561 nm laser) and fluoreophore-tagged DNA (‘DNA channel’, 642 nm). Signal in the ‘liposome channel’ was imaged using an emission dichroic filter (570-650 nm band pass plus 750 nm long pass) with a camera integration time of 100 ms and line averaging of two. The ‘DNA channel’ was imaged using an emission dichroic filter (655 nm long pass) with a camera integration time of 33 ms and line averaging of two.
Fluorescence microscopy of electroformed liposomes (Giant Unilamellar Vesicles)
Binding interactions between DNA fluorophores and the surface of micron-scale GUVs were imaged using a Leica TCS SP8 DLS confocal microscope with a HC PL APO CS2 63 x oil immersion objective lens and Acousto-Optical Beam Splitter, a programmable crystal-based beam splitter (Leica Microsystems GmbH, Germany). Two-channel images were acquired showing Liposomes and DNA. In channel one, Rhodamine B-labelled liposomes were excited with a 561 nm laser and imaged between 569-611 nm. In channel two, DNA labelled with was excited with a 640 nm laser and imaged between 690-734 nm.
Quantifying DNA-liposome colocalisation
A custom macro script was developed using FIJI in ImageJ (Schindelin et al., 2012) to quantify the colocalisation of DNA and liposomes using a method inspired by Manders Overlap Coefficient (Dunn et al., 2011). Briefly, the pixel intensity data for the ‘liposome channel’ of all images within a dataset comprised of two-channel images (representative of a single experimental condition) was aggregated and analysed to determine the pixel intensity threshold used to define the boundary of liposomes against the slide background (Supplementary Methods, Supplementary Fig. 1). A unique binary mask of the liposome channel was then generated based on this threshold for each image in the dataset to separate liposome-covered section from the background. The method used showed no bias or correlation with liposome area (percentage coverage), in comparison with Pearson’s correlation, whichh did (Supplementary Fig. 2/3).
The mean pixel intensities of the DNA channel for each of these two sections was then compared to produce a ratiometric colocalisation score, C, indicating the relative fluorescent intesity of DNA attached to liposomes (within the binary mask) compared to the fluorescence of DNA in the background of the image (outside the binary mask). For example, a colocalisation score of C = 1 is indicative of an image where DNA fluorescence is evenly distributed across both sections and therefore displays an equal mean pixel intensity in both channels.
Results
Characterisation of DNA-liposome interactions through colocalisation analysis
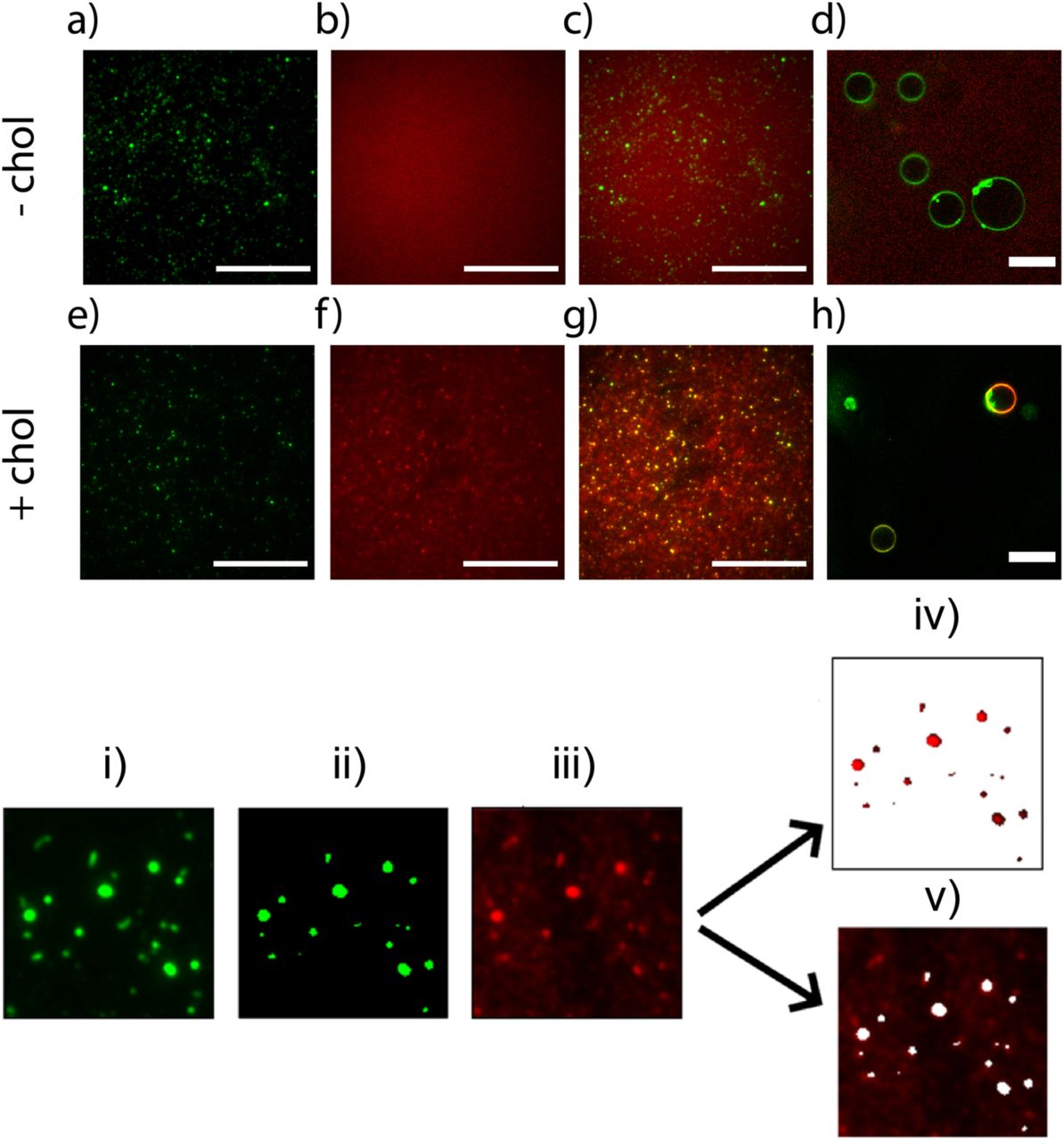
We immobilised a 100-fold dilution of extruded liposomes on the surface of a coverslip using biotin-avidin conjugation (Figure 1). Fluorescent DNA colocalised with fluorescent liposomes only when cholesterol tags were present (Figure 1e-h). DNA without a cholesterol tag did not colocalise with liposomes and was distributed evenly throughout the image independently of the position of liposomes (Figure 1a-d). The role of cholesterol tags in causing DNA-liposome colocalisation was further verified by confocal images of cholesterol-tagged DNA colocalising with giant unilamellar vesicles, while DNA with no cholesterol tag did not colocalise with liposomes (Figure 1d/1h)

Image acquisition and analysis of DNA-liposome interactions with fluorescence microscopy. Top row – without cholesterol: DNA (a) rhodamine-doped liposomes, (b) Alexa647-DNA (c) merge. (d) GUVs + Alexa647-DNA visualised using confocal fluorescence. Second row – with cholesterol (e) rhodamine-doped liposomes, (f) Alexa647-DNA, (g) merge, (h) GUVs + Alexa647 DNA. Bottom row: Image analysis process for quantifying DNA-liposome colocalization: the image of rhodamine-doped liposomes (i) is converted into a binary mask (ii). This mask is then used to partition the Alexa647-DNA image (iii) into two sections: liposomes (iv) and background (v). The mean pixel intensity of DNA in the liposome section (iv) is divided by the mean pixel intensity of DNA in the background section (v) to give a ratiometric colocalisation score. Scale bars: 20 µm.
We quantified the colocalisation of DNA to extruded liposomes by determining a colocalisation score. The liposome channel was first converted into a binary image according to a standardised pixel intensity threshold (SI). This binary image of the liposome channel was then used as a mask to divide the DNA channel into two sections: liposome and background, which were used to calculate a colocalisation score via: 
Where C is the reported colocalisation score, D is the mean pixel intensity of the fluorescent DNA in the liposome region of the DNA channel, and B is the mean pixel intensity of the fluorescent DNA in the background region of the DNA channel (Figure 1).
The effect of solution composition on DNA-liposome colocalisation
We investigated the effect of common DNA origami buffer components on the interactions of cholesterol-tagged DNA and lipid bilayers by quantifying the colocalisation of DNA and unilamellar liposomes in various conditions. For each condition, the co-localisation of ssDNA, dsDNA and dsDNA-6 nt was measured in order to compare the overall binding yield for each DNA configuration. Tests were repeated for 1:1 DOPE/DOPC liposomes and DPhPC liposomes to detect for lipid-dependant responses to variables.
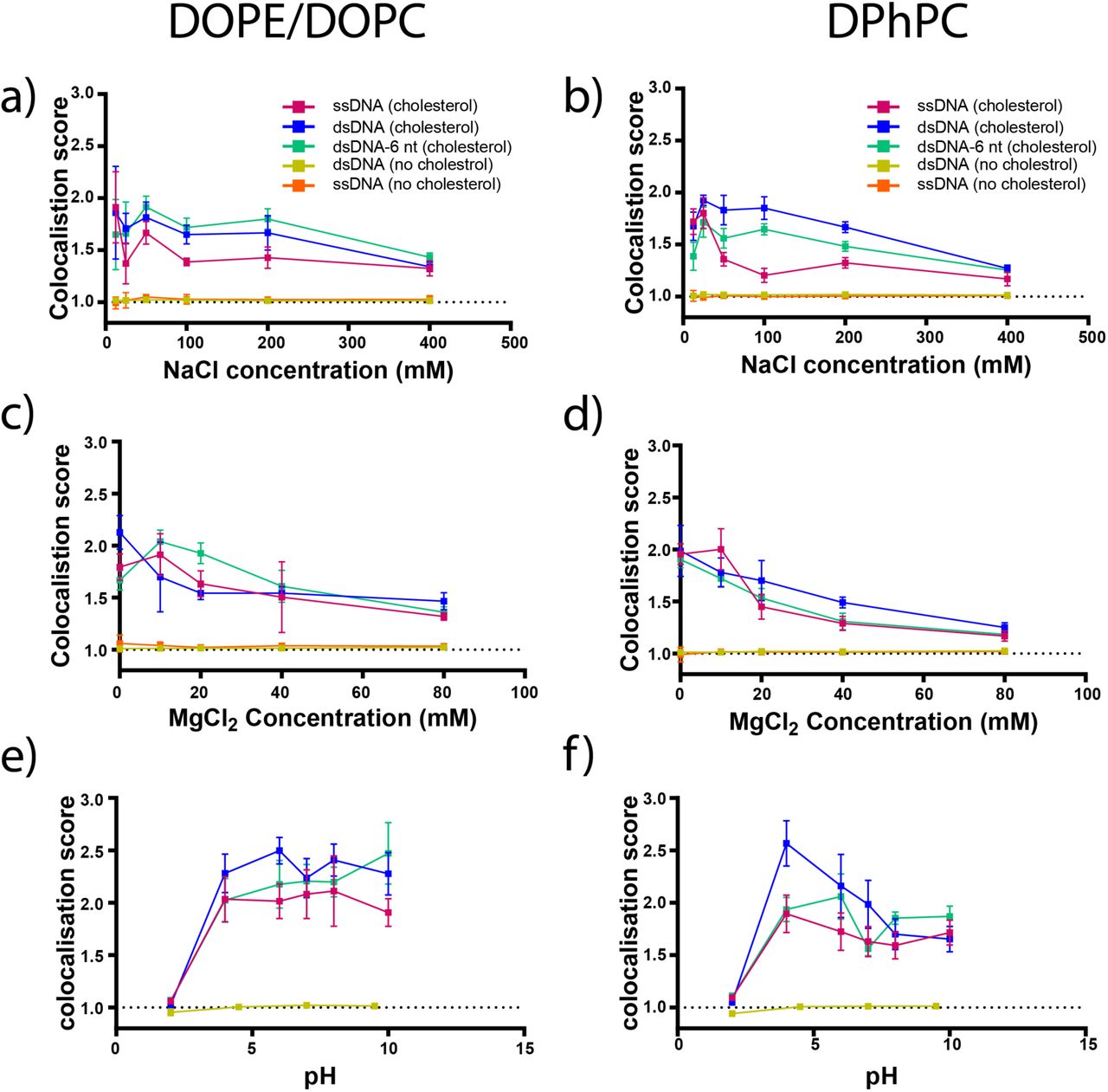
Imaging DNA-liposome interactions in extrusion buffer [NaCl] containing between 12.5 and 400 mM NaCl and extrusion buffer [MgCl2] containing between 0 and 80 mM MgCl2 revealed that both salts had a significant impact on DNA-liposome colocalisation. The colocalisation of cholesterol-tagged DNA and liposomes was inhibited at increasing concentrations of NaCl (Figure 2 A & B) and MgCl2 (Figure 2 C & D). For all three configurations of cholesterol-tagged DNA on both DOPE/DOPC liposomes and DPhPC liposomes, a significant decrease was observed in colocalisation scores between 12.5 mM and 400 mM NaCl (P < 0.05) and between 0 mM and 80 mM MgCl2 (P < 0.05). Linear regression analysis for all three configurations on both 1:1 DOPE/DOPC liposomes and DPhPC liposomes showed a significant (P < 0.05) trend of decreasing co-localisation scores with increasing NaCl and MgCl2 concentration.

The effect of NaCl, MgCl2 and pH on DNA-liposome colocalisation. Colocalisation scores and standard deviations are shown for Alexa647-labelled cholesterol-tagged single stranded DNA (ssDNA, pink), cholesterol-tagged double stranded DNA (dsDNA, blue) and cholesterol-tagged double stranded DNA with a 6 nt overhang (dsDNA-6nt, green) as well as dsDNA with no cholesterol tag (yellow) and ssDNA with no cholesterol tag (orange) and rhodamine-labelled DOPE/DOPC liposomes (left column, a/c/e) and DPhPC liposomes (right column, b/d/f). Conditions tested included extrusion buffer [NaCl] containing 12.5, 25, 50, 100, 200 and 400 mM NaCl (a/b), extrusion buffer [MgCl2] containing 0, 10, 20, 40 and 80 mM MgCl2 (c/d) and extrusion buffer [pH] adjusted to pH values of 2, 4, 6, 7, 8 and 10 (e/f).
Imaging DNA-liposome interactions in extrusion buffer adjusted to approximate pH values of 2, 4, 6, 7, 8 and 10 showed that the colocalisation of cholesterol-tagged DNA and liposome was inhibited in highly acidic conditions. At pH 2, the colocalisation of all three configurations of cholesterol-tagged DNA with both DOPE/DOPC liposomes (Figure 2e) and DPhPC liposomes (Figure 2f) was strongly inhibited and significantly less than at all other pH values (P < 0.05). For DOPE/DOPC liposomes, all configurations of cholesterol-tagged DNA produced similar colocalisation scores at pH values between 4 and 10. For DPhPC liposomes, cholesterol-tagged dsDNA showed increased binding at moderately acidic conditions which decreased with increasing pH, while ssDNA and dsDNA-6 nt showed similar binding at all pH values between 4 and 10.
DNA that was evenly distributed throughout a slide independently of liposome location would be expected to produce a colocalisation score of approximately 1.0, while membrane-binding DNA should produce a colocalisation score greater than 1.0. In the absence of cholesterol tags, we did not detect any significant colocalisation of DNA and liposomes at any NaCl concentration or pH value. For MgCl2, no significant colocalisation of DNA with no cholesterol tag was observed in 19 out of 20 samples. The remaining sample, ssDNA with no cholesterol tag on DOPE/DOPC liposomes at 40 mM, produced a colocalisation score of C = 1.01, significantly greater than one (P < 0.05) but far below those produced by cholesterol-tagged DNA (mean: 1.63 range: 1.18-2.12).
The effect of DNA configuration on DNA-liposome colocalisation
We compared the binding of cholesterol-tagged DNA in different configurations (ssDNA, dsDNA and dsDNA-6 nt) across all experiments to assess if one particular DNA configuration yielded higher colocalisation than other configurations. For each condition and configuration, 12 images and were recorded, giving a total of n = 204 colocalisation scores recorded for each DNA configuration on each liposome composition.
We tested for a difference in means of colocalisation scores for each pair of DNA configurations. This t-test was repeated for both DPhPC liposomes and DOPE/DOPC liposomes. Our results (Table 1) show there is are significant differences in mean DNA-liposome colocalisation dependant on DNA configuration. For DOPE/DOPC liposomes, we found DNA to colocalise in the order C(dsDNA) ≈ C(dsDNA-6 nt) > C(ssDNA). For DPhPC liposomes, we found DNA to bind in the order C(dsDNA) > C(dsDNA-6 nt) > C(ssDNA).
Mean differences in colocalisation scores for different configurations of DNA binding to DOPE/DOPC liposomes and DPhPC liposomes. Results are pooled from data shown in section 4.2. Paired t-tests of colocalisation scores were conducted comparing each DNA configuration pair-wise on both types of liposomes (n = 204) against the hypothesis ‘mean difference = 0’. For example, ‘ΔC(dsDNA) C(ssDNA)’ represents the mean difference in colocalisation scores of dsDNA and ssDNA.
The effect of membrane cholesterol content on DNA-liposome colocalistion
Colocalisation scores of DNA and liposomes in extrusion buffer showed different trends for the two lipid mixtures as cholesterol content was increased between 0% and 40% mass. For DOPE/DOPC liposomes, colocalisation scores of all three configurations of cholesterol-tagged DNA showed a significant increase between 0% and 40% cholesterol (P < 0.05). Linear regression analysis shows a gradient significantly greater than zero across the observed range of membrane cholesterol content (P < 0.05) (Figure 3A).
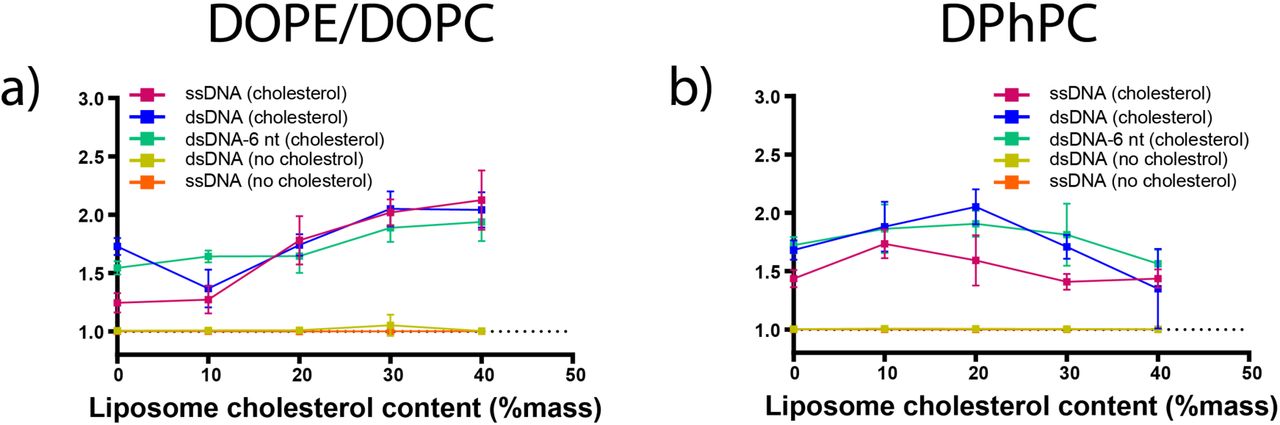
The effect of cholesterol content on DNA-liposome concentration. Colocalisation scores and standard deviations are shown for Alexa647-labelled cholesterol-tagged ssDNA (pink), cholesterol-tagged dsDNA (blue) and cholesterol-tagged dsDNA with a 6 nt overhang (green) as well as dsDNA with no cholesterol tag (yellow) and ssDNA with no cholesterol tag (orange) and rhodamine-labelled DOPE/DOPC liposomes (a) or DPhPC liposomes (b) prepared from lipid stocks containing 0, 10, 20, 30 or 40% cholesterol.
For DPhPC liposomes, colocalisation scores of cholesterol-tagged DNA increased to a maximum at 10%-20% membrane cholesterol, then decreased as membrane cholesterol content was increased above this point. All three configurations of cholesterol-tagged DNA (ssDNA, dsDNA and dsDNA-6 nt) showed both a significant increase in colocalisation between 0% and 20% (P < 0.05) and a significant decrease in colocalisation between 20% and 40% (P < 0.05).
Linear regression analysis for all three configurations showed a slight overall decreasing trend across the observed range (P < 0.05) (Figure 3 B).
Using the test C > 1.0, both ssDNA and dsDNA with no cholesterol tag did not show significant colocalisation with liposomes of any cholesterol content.
Discussion
The effect of solution composition on DNA-liposome colocalistion
Monovalent ions, divalent ions and pH known to modulate biophysical properties of membrane bilayers (Sachs et al., 2004) and the structural stability of DNA nanostructures (Douglas et al., 2009). Divalent cations such as Mg2+ are of particular interest to DNA nanotechnology applications to stabilise DNA duplexes and are required to facilitate the formation higher-order nucleic acids structures (Misra and Draper, 1998; Williams et al., 1989). Divalent cations also maintain the stability of large DNA nanostructures by inhibiting the electrostatic repulsion between DNA strands and are therefore considered essential in the assembly process (Douglas et al., 2009; Kielar et al., 2018). The sensitivity of DNA to salt and pH has been harnessed to design DNA nanostructures able to switch configuration in response to changes in ion concentration, allowing the development of DNA-based nanosensors (Singh et al., 2018). Despite these recent advances, the tolerance of DNA nanostructures to low-salt environments remains variable and design-dependent thus necessitating regular characterization and optimization (Hahn et al., 2014).
We sought to test whether changes to membrane density and diffusivity due to external buffer would affect cholesterol binding to the non-polar tail-group region of a membrane. Monovalent cations such as Na+ and divalent cations such as Mg2+ have been shown via modelling to promote lipid-lipid binding interactions within a bilayer (Böckmann et al., 2003) and affect the behaviour of water molecules at the membrane-water interface (Velikonja et al., 2013). The interaction between surface charges on membrane bilayers and the phosphate groups of DNA molecules is also affected by divalent cations (Antipina and Gurtovenko, 2016; Binder and Zschörnig, 2002). This interaction is dependent on the ratio of monovalent to divalent salts (Budker et al., 1980). In our tests we saw no membrane interactions between non-cholesterol tagged DNA at any MgCl2 concentration, suggesting that divalent cation-mediated binding was negligible under the conditions tested. Coarse-grained and atomistic simulations of DNA-lipid interactions could lead to a more detailed understanding of the effect of cations on the binding of DNA (Uusitalo et al., 2015; Yoo and Aksimentiev, 2015).
Binding was inhibited in acidic conditions (< pH 4). However, otherwise, there was no optimal pH for liposomes formed from either neutral DPhPC, or DOPE/DOPC, which contains zwitterionic phosphatidylethanolamine headgroups that are positively charged below pH 3.5 and negatively charged above pH 8 (Tsui et al., 1986). This suggests lipid ionisation does not play a significant role and variation in pH around physiological pH are not likely to affect membrane binding of cholesterol-tagged DNA. Hydronium ions (H3O+) have a similar effect on lipid bilayers as Na+ and Mg2+, and may explain the inhibition of binding in strongly acidic conditions (Deplazes et al., 2018).
For both liposome compositions, the colocalisation of ssDNA and liposomes was significantly reduced in comparison with double stranded configurations. This suggests the duplex dsDNA binds better to liposomes. Membrane-bound ssDNA has been observed through Förster Resonance Energy Transfer (FRET) (Roy et al., 2008) to lie close to the surface of lipid bilayer membranes, while dsDNA remains in a stable position protruding normal to the membrane surface (Ma et al., 2019). Thus, this improved binding may be due to the greater rigidity of dsDNA vs ssDNA.
The addition of a 6 nt overhang on cholesterol-tagged DNA strands has been postulated to assist during nanostructure assembly by inhibiting strand aggregation (Ohmann et al., 2019). We included a 6 nt overhang next to the cholesterol group on our dsDNA strand (dsDNA-6 nt) and observed a significant decrease in binding only on DPhPC liposomes. Lipid composition should, therefore, be considered when incorporating overhangs, but there would be no large penalty from routine incorporation on membrane-targetting nanostructures.
The effect of membrane cholesterol on DNA-liposome binding differed between phospholipid compositions. Cholesterol has been shown to affect the strength, fluidity and permeability of bilayers (Róg et al., 2009) as well as the organisation of ions and water molecules at the membrane-bilayer interface (Magarkar et al., 2014). The different response that we observed between different phospholipid types may be explained by differences in tail-group structure. Branched chain lipids such as DPhPC occupy a greater area per molecule within a bilayer compared to linear-chain lipids such as DOPE and DOPC (Tristram-Nagle et al., 2010).
Increasing the cholesterol content of lipid mixtures above 20% promoted the binding of DNA on DOPE/DOPC liposomes but inhibited binding on DPhPC liposomes. This was possibly due to the lower cholesterol saturation limit of DPhPC compared with DOPE (Huang et al., 1999). Here we controlled the cholesterol content during liposome preparation but did not quantify the exact cholesterol content in liposome membranes. Future work using fluorescent markers or high performance liquid chromatography analysis of liposome samples (Christie, 1985) could accurately quantify the membrane cholesterol content to better benchmark the role of cholesterol in DNA-lipid binding, and to account for any loss in cholesterol during liposome preparation.
Conclusion
In this work we have characterised the role of salt and pH during the assembly of DNA-liposome complexes. We tested different lipid species and DNA configurations to screen for optimal conditions to promote binding of DNA to liposomes. Our results suggest that lipid type, pH and DNA configuration are the most important parameters to consider when optimising for the binding of DNA nanostructures to liposomes, whereas mono- and divalent-salt concentration plays a minor role. These results will be helpful in experimental design and reagant choice for future experiments combining DNA and lipid nanotechnologies.
Footnotes
This revision has added Supplementary Methods, Figures and Tables.





{kind=link}
{kind=link}
{kind=link}