

Abstract
Despite its fundamental role as a model organism in plant sciences, the green alga Chlamydomonas reinhardtii entirely lacks genomic resources for any closely related species, obstructing its development as a study system in several fields. We present highly contiguous and well-annotated genome assemblies for the two closest known relatives of the species, Chlamydomonas incerta and Chlamydomonas schloesseri, and a third more distantly related species, Edaphochlamys debaryana. We find the three Chlamydomonas genomes to be highly syntenous with similar gene contents, although the 129.2 Mb C. incerta and 130.2 Mb C. schloesseri assemblies are more repeat-rich than the 111.1 Mb C. reinhardtii genome. We identify the major centromeric repeat in C. reinhardtii as an L1 LINE transposable element homologous to Zepp (the centromeric repeat in Coccomyxa subellipsoidea) and infer that centromere locations and structure are likely conserved in C. incerta and C. schloesseri. We report extensive rearrangements, but limited gene turnover, between the minus mating-type loci of the Chlamydomonas species, potentially representing the early stages of mating-type haplotype reformation. We produce an 8-species whole-genome alignment of unicellular and multicellular volvocine algae and identify evolutionarily conserved elements in the C. reinhardtii genome. We find that short introns (<~100 bp) are extensively overlapped by conserved elements, and likely represent an important functional class of regulatory sequence in C. reinhardtii. In summary, these novel resources enable comparative genomics analyses to be performed for C. reinhardtii, significantly developing the analytical toolkit for this important model system.
Introduction
With the rapid increase in genome sequencing over the past two decades, comparative genomics analyses have become a fundamental tool in biological research. As the first sets of genomes for closely related eukaryotic species became available, pioneering comparative studies led to refined estimates of gene content and orthology, provided novel insights into the evolution of genome architecture and the extent of genomic synteny between species, and enabled the proportions of genomes evolving under evolutionary constraint to be estimated for the first time (Mouse Genome Sequencing Consortium 2002; Cliften et al. 2003; Stein et al. 2003; Richards et al. 2005). As additional genomes were sequenced it became possible to produce multiple species whole-genome alignments (WGA) and to identify conserved elements (CEs) in noncoding regions for several of the most well-studied lineages (Siepel et al. 2005; Stark et al. 2007; Gerstein et al. 2010; Lindblad-Toh et al. 2011). Many of these conserved noncoding sequences overlap regulatory elements, and the identification of CEs has proved to be among the most accurate approaches for discovering functional genomic sequences (Alföldi and Lindblad-Toh 2013). As a result, CEs have frequently been used to enhance genome annotation projects and to study several aspects of regulatory sequence evolution (Mikkelsen et al. 2007; Lowe et al. 2011; Halligan et al. 2013; Williamson et al. 2014).
The ability to perform comparative analyses is contingent on the availability of genome assemblies for species that span a range of appropriate evolutionary distances. While this state has been achieved for the majority of model organisms, there remain several species of high biological significance that entirely lack genomic resources for any closely related species. Hiller et al. (2013) described such cases as ‘phylogenetically isolated genomes’, specifically referring to species for which the most closely related genomes belong to species divergent by one or more substitutions, on average, per neutrally evolving site. At this scale of divergence an increasingly negligible proportion of the genome can be aligned at the nucleotide-level (Margulies et al. 2006), limiting comparative analyses to the protein-level and impeding the development of such species as model systems in numerous research areas.
The unicellular green alga Chlamydomonas reinhardtii is a long-standing model organism across several fields, including cell biology, plant physiology and molecular biology, and algal biotechnology (Salomé and Merchant 2019). Because of its significance, the ~110 Mb haploid genome of C. reinhardtii was among the earliest eukaryotic genomes to be sequenced (Grossman et al. 2003; Merchant et al. 2007), and both the genome assembly and annotation are actively being developed and improved (Blaby et al. 2014). Despite its quality and extensive application, the C. reinhardtii genome currently meets the ‘phylogenetically isolated’ definition. The closest confirmed relatives of C. reinhardtii that have genome assemblies belong to the clade of multicellular algae that includes Volvox carteri, the Tetrabaenaceae-Goniaceae-Volvocaceae, or TGV clade. Collectively, C. reinhardtii and the TGV clade are part of the highly diverse order Volvocales, and the more taxonomically limited clades Reinhardtinia and core-Reinhardtinia (Nakada et al. 2008; Nakada et al. 2016). Although these species are regularly considered close relatives, multicellularity likely originated in the TGV clade over 200 million years ago (Herron et al. 2009), and C. reinhardtii and V. carteri are more divergent from one another than human is to chicken (Prochnik et al. 2010).
Without a comparative genomics framework, the wider application of C. reinhardtii as a model system is severely impeded. While this broadly applies to the general functional annotation of the genome as outlined above (e.g. refinement of gene models and annotation of CEs), it is particularly relevant to the field of molecular evolution. Although the evolutionary biology of C. reinhardtii has not been widely studied, the species has several features that have attracted recent attention to its application in this field. Its haploid state, high genetic diversity (~2% genome-wide (Craig et al. 2019)) and experimental tractability make it an excellent system to study the fundamental evolutionary processes of mutation (Ness et al. 2015; Ness et al. 2016), recombination (Liu et al. 2018; Hasan and Ness 2020), and selection (Böndel et al. 2019). However, without genomic resources for closely related species it is currently impossible to perform several key analyses, such as the comparison of substitution rates at synonymous and non-synonymous sites of protein-coding genes (i.e. calculating dN/dS), and the inference of ancestral states at polymorphic sites (a requirement of several population and quantitative genetics models (Keightley and Jackson 2018)).
Furthermore, V. carteri and its relatives in the TGV clade are extensively used to study the evolution of multicellularity and other major evolutionary transitions (e.g. isogamy to anisogamy), and five genomes of multicellular species spanning a range of organismal complexities have now been assembled (Prochnik et al. 2010; Hanschen et al. 2016; Featherston et al. 2018; Hamaji et al. 2018). These studies have often included analyses of gene family evolution, reporting expansions in families thought to be functionally related to multicellularity. While these analyses have undoubtedly made important contributions, they are nonetheless limited in their phylogenetic robustness, as C. reinhardtii is the only unicellular relative within hundreds of millions of years available for comparison. Thus, the availability of annotated genomes for unicellular relatives of C. reinhardtii will also serve as an important resource towards reconstructing the ancestral core-Reinhardtinia gene content, potentially providing novel insights into the major evolutionary transitions that have occurred in this lineage.
Here we present highly contiguous and well-annotated genome assemblies for the two closest known relatives of C. reinhardtii, namely Chlamydomonas incerta and Chlamydomonas schloesseri, and a more distantly related unicellular species, Edaphochlamys debaryana. Via comparison to the genomes of C. reinhardtii and the TGV clade species we present the first insights into the comparative genomics of Chlamydomonas, focussing specifically on the conservation of genome architecture between species and the landscape of sequence conservation in C. reinhardtii. While forming only one of the initial steps in this process, by providing the first comparative genomics framework for the species we anticipate that these novel genomic resources will greatly aid in the continued development of C. reinhardtii as a model organism.
Results & Discussion
The closest known relatives of Chlamydomonas reinhardtii
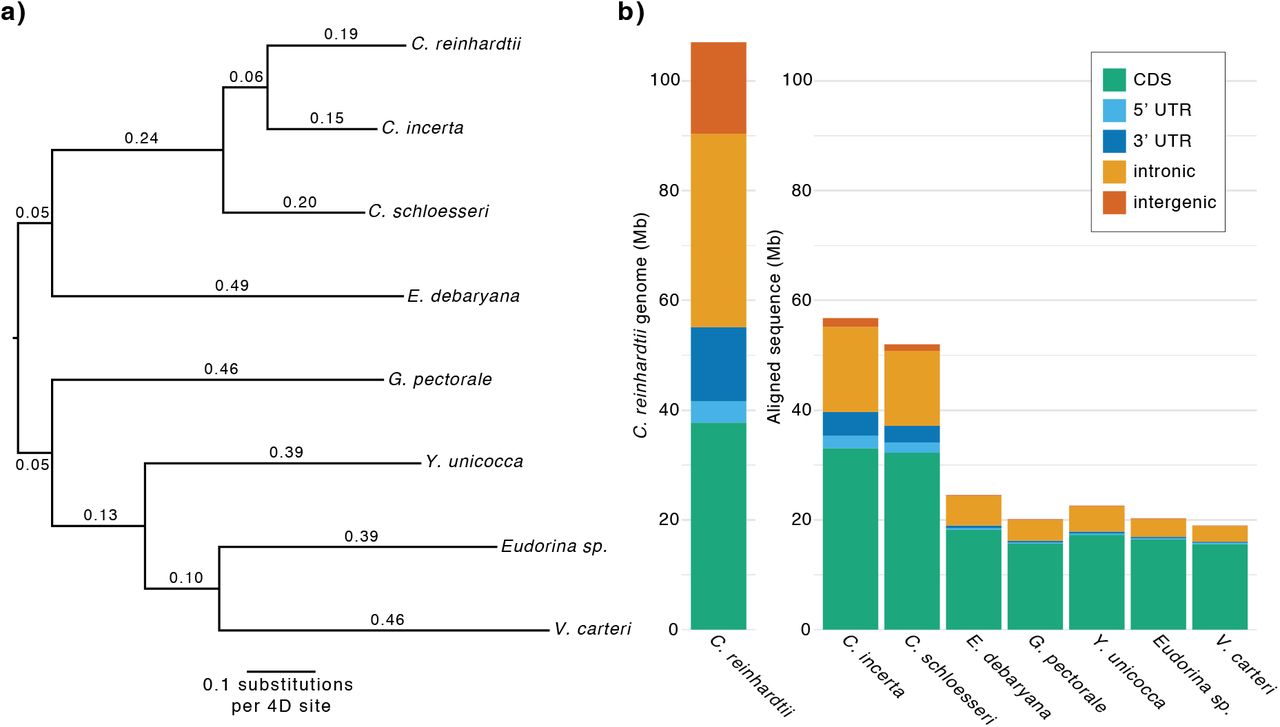
Although the genus Chlamydomonas consists of several hundred unicellular species it is highly polyphyletic (Pröschold et al. 2001), and C. reinhardtii is more closely related to the multicellular TGV clade than the majority of Chlamydomonas species. Given their more conspicuous morphology, the TGV clade contains ~50 described species (Herron et al. 2009), while the unicellular lineage leading to C. reinhardtii includes only two other confirmed species, C. incerta and C. schloesseri (Pröschold et al. 2005; Pröschold et al. 2018). As C. reinhardtii is the type species of Chlamydomonas, these three species collectively comprise the monophyletic genus (fig.1a, b, c), and Chlamydomonas will be used specifically to refer to this clade throughout.
C. incerta is the closest known relative of C. reinhardtii, and a small number of comparative genetics analyses have been performed between the two species (Ferris et al. 1997; Popescu et al. 2006; Smith and Lee 2008). C. incerta is known from only two isolates, and we selected the original isolate SAG 7.73 for sequencing. Unfortunately, although C. incerta SAG 7.73 is nominally from Cuba, the geographic origin of the isolate is uncertain due to a proposed historical culture replacement with C. globosa SAG 81.72 from the Netherlands (Harris et al. 1991). As the direction of replacement is unknown, the accepted taxonomic name of the species also remains undecided. C. schloesseri was recently described by Pröschold et al. (2018), and three isolates from a single site in Kenya exist in culture. We selected the isolate CCAP 11/173 for sequencing.
Beyond Chlamydomonas there are a substantial number of unicellular core-Reinhardtinia species with uncertain phylogenetic relationships (i.e. that may be part of the lineage including Chlamydomonas, the lineage including the TGV clade, or outgroups to both). Among these, the best studied species is E. debaryana, which was recently renamed from Chlamydomonas debaryana (Pröschold et al. 2018). Unlike the three described Chlamydomonas species, E. debaryana appears to be highly abundant in nature, with more than 20 isolates from across the Northern Hemisphere in culture, suggesting that it could be developed as a model for studying algal population structure and biogeography via the collection of further isolates. Draft genomes of the isolates NIES-2212 from Japan (Hirashima et al. 2016) and WS7 from the USA (Nelson et al. 2019) were recently assembled, while we selected the isolate CCAP 11/70 from Czechia for sequencing (fig. 1d).

Images of unicellular species. a) Chlamydomonas reinhardtii. b) Chlamydomonas incerta SAG 7.73. c) Chlamydomonas schloesseri SAG 2486 (=CCAP 11/173). d) Edaphochlamys debaryana SAG 11.73 (=CCAP 11/70). All images taken by Thomas Pröschold.
The genomes of Chlamydomonas incerta, Chlamydomonas schloesseri & Edaphochlamys debaryana
Using a combination of Pacific Biosciences (PacBio) long-read sequencing for de novo assembly (40-49x coverage, table S1) and Illumina short-read sequencing for error correction (43-86x coverage, table S2), we produced contig-level genome assemblies for C. incerta, C. schloesseri and E. debaryana. All three assemblies were highly contiguous, with N50s of 1.6 Mb (C. incerta), 1.2 Mb (C. schloesseri) and 0.73 Mb (E. debaryana), and L50s of 24, 30 and 56 contigs, respectively (table 1). Genome-mode BUSCO scores supported a high-level of assembly completeness, with the percentage of universal chlorophyte single-copy orthologs identified in each genome ranging from 95.9% to 98.1%. These metrics compare favourably to the best existing core-Reinhardtinia assemblies (table 1). Although the C. reinhardtii and V. carteri assemblies have greater scaffold-level N50 values than the three new assemblies, they are both considerably more fragmented at the contig level, with N50s of 215 kb and 85 kb, respectively. While this is not surprising given our application of long-read sequencing, it nonetheless demonstrates that these important model genomes could be substantially improved by additional sequencing effort. The contig-level N50s of the three new assemblies also exceeded those of the Gonium pectorale assembly (Hanschen et al. 2016), and the Pacbio-based assemblies of Yamagishiella unicocca and Eudorina sp. 2016-703-Eu-15 (hereafter Eudorina sp.) (Hamaji et al. 2018), and thus they currently represent the most contiguous assemblies in terms of uninterrupted sequence in the core-Reinhardtinia, and indeed the entire Volvocales (table S3).
Genome assembly metrics for eight high-quality core-Reinhardtinia genome assemblies.
Assembled genome size varied moderately across the eight species, ranging from 111.1 Mb (C. reinhardtii) to 184.0 Mb (Eudorina sp.) (table 1). Both C. incerta (129.2 Mb) and C. schloesseri (130.2 Mb) had consistently larger genomes than C. reinhardtii, and E. debaryana (142.1 Mb) had a larger genome than both Y. unicocca and V. carteri. Although additional genome assemblies will be required to fully explore genome size evolution in the core-Reinhardtinia, these results suggest that C. reinhardtii may have undergone a recent reduction in genome size. Furthermore, while earlier comparisons between multicellular species and C. reinhardtii led to the observation that certain metrics of genomic complexity (e.g. gene density and intron length, see below) correlate with organismal complexity, these results indicate that genome size, at least for these species, does not. Conversely, as proposed by Hanschen et al. (2016), GC content does appear to decrease with increasing cell number, with genome-wide values ranging from 64.1 to 67.1% for the unicellular species and from 64.5 to 56.1% in the TGV clade (table 1).
The larger genome sizes of the unicellular species, relative to C. reinhardtii, can largely be attributed to differences in the content of transposable elements (TEs) and large satellite sequences (defined as those with monomers >10 bp), with all three species containing greater total amounts (20.1-27.5 Mb) and higher genomic proportions (14.1-21.1%) of complex repetitive sequence than C. reinhardtii (15.3 Mb and 13.8%) (table 1). As discussed below, the larger genome size of E. debaryana can also be partly attributed to the substantially higher number of genes encoded by the species. For all three assemblies, repeat content was relatively consistent across contigs, with the exception of small contigs (<~100 kb), which exhibited highly variable repeat contents and likely represent fragments of complex genomic regions that have resisted assembly (fig. S1). The higher repeat contents of the three assemblies were broadly consistent across TE subclasses (fig. S2), although a direct comparison of the TEs present in each genome is complicated by phylogenetic bias in repeat masking and classification. The existence of a curated repeat library for C. reinhardtii directly contributes to masking and can improve homology-based classification of repeats in related species, however this effect will become increasingly negligible as divergence increases. This is likely to at least partly explain the lower repeat content and higher proportion of “unknown” classifications observed for E. debaryana compared to C. incerta and C. schloesseri (table 1, fig. S2).

Total repeat content per contig (TEs, satellites and simple/low-complexity repeats) plotted by contig length for a) Chlamydomonas incerta, b) Chlamydomonas schloesseri, and c) Edaphochlamys debaryana.

Repeat content per species by repeat subclass. Numbers within bars represent total sequence per subclass in Mb. LINE = long interspersed nuclear element, SINE = short interspersed nuclear element, LTR = long terminal repeat, DIRS = tyrosine recombinase encoding retrotransposons, PLE = Penelope-like elements, TIR = terminal inverted repeat (i.e. DNA transposons), RC = rolling-circle elements.
Nonetheless, based on manual curation of the most abundant TE families in each species, a qualitative comparison is possible. All curated TEs belonged to subclasses and superfamilies that are present in one or both of C. reinhardtii and V. carteri (the two species with existing repeat libraries), suggesting a largely common repertoire of TEs across the core-Reinhardtinia. Alongside the more widely recognised L1 LINE and Gypsy LTR elements, all species contained families of the comparatively obscure Dualen LINE elements (Kojima and Fujiwara 2005), PAT-like DIRS elements (Poulter and Butler 2015) and Helitron2 rolling-circle elements (Bao and Jurka 2013). We also identified families of Zisupton and Kyakuja DNA transposons, both of which were reported as potentially present in C. reinhardtii upon their recent discovery (Böhne et al. 2012; Iyer et al. 2014). These superfamilies are greatly understudied, and there are currently no Kyakuja elements deposited in either the Repbase (https://www.girinst.org/repbase/) or Dfam (https://www.dfam.org) repositories. Although not the main focus of this study, the annotation of elements from such understudied superfamilies highlights the importance of performing manual TE curation in phylogenetically diverse lineages. Alongside improving our understanding of TE biology, these elements are expected to contribute towards more effective repeat masking/classification and gene model annotation in related species, which will be of increasing importance given the large number of chlorophyte genome projects currently in progress (Blaby-Haas and Merchant 2019).
Phylogenomics of the core-Reinhardtinia and Volvocales
Due to the low number of available genomes and gene annotations, the phylogenetics of the Volvocales has almost exclusively been studied using ribosomal and plastid marker genes. These analyses have successfully delineated several broad clades (e.g. Reinhardtinia, Moewusinia, Dunaliellinia) (Nakada et al. 2008), but often yielded inconsistent topologies for more closely related taxa. Utilising both our own and several recently published genomic resources, we further explored the phylogenomic structure of the Reinhardtinia and Volvocales. As several genomes currently lack gene annotations, we first used an annotation-free approach based on the identification of chlorophyte single-copy orthologous genes with BUSCO (Waterhouse et al. 2018). This dataset consisted of 1,624 genes, present in at least 15 of the 18 included species (12 Reinhardtinia, three other Volvocales, and three outgroups from the order Sphaeropleales, table S3). For the 11 species with gene annotations (table S4), we produced a second dataset based on the orthology clustering of each species’ proteome, which yielded 1,681 single-copy orthologs shared by all species. For both datasets, we performed maximum-likelihood (ML) analyses using IQ-TREE (Nguyen et al. 2015). Analyses were performed on both concatenated protein alignments (producing a species-tree) and individual alignments of each ortholog (producing gene trees), which were then summarised as a species-tree using ASTRAL-III (Zhang et al. 2018).
All four of the resulting phylogenies exhibited entirely congruent topologies, with near maximal-support values at all nodes (fig. 2, S3). Rooting the tree on the Sphaeropleales species, the monophyly of the Volvocales, Reinhardtinia, and core-Reinhardtinia clades were recovered. Chlamydomonas was recovered with the expected branching order (Pröschold et al. 2018), as was the monophyly and expected topology of the TGV clade (Nakada et al. 2019). The most contentious phylogenetic relationships are those of the remaining unicellular core-Reinhardtinia, which include E. debaryana and also the recently published genomes of Chlamydomonas sphaeroides (Hirashima et al. 2016) and Chlamydomonas sp. 3112 (Nelson et al. 2019). In the most gene-rich analysis to date, E. debaryana was grouped in a weakly-supported clade with Chlamydomonas (termed metaclade C), while C. sphaeroides grouped with a small number of other unicellular species on the lineage including the TGV clade (Nakada et al. 2019). In our analysis, E. debaryana and C. sphaeroides were recovered as sister taxa on the lineage including Chlamydomonas, meeting the prior definition of metaclade C as the sister clade of the TGV clade and its unicellular relatives. Due to its recent discovery, C. sp. 3112 has not been included in previous phylogenetic analyses. This species is a member of the core-Reinhardtinia based on sequence similarity of ribosomal and plastid genes, and is likely a close relative of the described species Chlamydomonas zebra (table S5). Given its basal phylogenetic position relative to metaclade C and the TGV clade, species such as C. sp. 3112 could prove particularly useful in future efforts to reconstruct the ancestral gene content of the core-Reinhardtinia.


Phylogenomic analyses. a) ASTRAL-III species tree (15 Volvocales species and three outgroups) summarising 1,624 gene trees produced from individual protein alignments of chlorophyte BUSCO genes. b) ML phylogeny of nine Volvocales species and two outgroups inferred using LG+F+R5 model and a concatenated protein alignment of 1,681 putative single-copy orthologs identified by OrthoFinder. c) ASTRAL-III species tree summarising 1,681 gene trees produced from individual protein alignments of the OrthoFinder single-copy genes.

ML phylogeny of 15 Volvocales species and three outgroups inferred using LG+F+R6 model and concatenated protein alignment of 1,624 chlorophyte BUSCO genes. All bootstrap values >=99%. Species in bold have gene model annotations and were included in the OrthoFinder-based phylogenies (fig. S3b, c).
Synteny and conserved genome architecture in Chlamydomonas
Almost nothing is known about karyotype evolution and the rate of chromosomal rearrangements in Chlamydomonas and the core-Reinhardtinia. Prochnik et al. (2010) reported that the syntenic genomic segments identified between C. reinhardtii and V. carteri contained fewer genes than human and chicken syntenic segments, in part due to a greater number of small inversions disrupting synteny. As the longest contigs in our assemblies were equivalent in length to C. reinhardtii chromosome arms (6.4, 4.5 and 4.2 Mb for C. incerta, C. schloesseri and E. debaryana, respectively), and given the closer evolutionary relationships of the unicellular species, we explored patterns of synteny between the three species and C. reinhardtii. We used SynChro (Drillon et al. 2014) to identify syntenic segments, which first uses protein sequence reciprocal best-hits to anchor syntenic segments, before extending segments via the inclusion of homologs that are syntenic but not reciprocal best-hits. All three Chlamydomonas genomes were highly syntenous, with 99.5 Mb (89.5%) of the C. reinhardtii genome linked to 315 syntenic segments spanning 108.1 Mb (83.6%) of the C. incerta genome, and 98.5 Mb (88.6%) of the C. reinhardtii genome linked to 409 syntenic segments spanning 108.1 Mb (83.1%) of the C. schloesseri genome.
Given the high degree of synteny, it was possible to order and orientate the contigs of C. incerta and C. schloesseri relative to the assembled chromosomes of C. reinhardtii (fig. 3). A substantial proportion of the C. reinhardtii karyotype appeared to be conserved in C. incerta, with six of the 17 chromosomes (1, 3, 4, 7, 14 and 16) showing no evidence of inter-chromosomal rearrangements, and a further three (5, 13 and 15) showing evidence for only minor translocations <150 kb in length (fig. 3a). Consistent with its greater divergence from C. reinhardtii, C. schloesseri exhibited such one-to-one conservation between only four chromosomes (5, 7, 11 and 14) (fig. 3b). For both species, patterns of synteny indicated at least one inter-chromosomal rearrangement affecting the remaining chromosomes, although without additional scaffolding of contigs it is difficult to comment on the overall effect of such rearrangements on karyotype. Furthermore, by direct comparison to C. reinhardtii chromosomes, we may have overestimated karyotype conservation due to undetected chromosome fusion/fission events (i.e. if a C. reinhardtii chromosome is present as two chromosomes in one of the related species). Across both C. incerta and C. schloesseri, all chromosomes (except chromosome 15 in the C. incerta comparison) contained intra-chromosomal rearrangements relative to C. reinhardtii, with small inversions <100 kb in length comprising the vast majority (fig. S4a, b). Synteny was far weaker between C. reinhardtii and E. debaryana, with 58.6 Mb (52.8%) of the C. reinhardtii genome linked to 1,975 syntenic segments spanning 64.8 Mb (45.6%) of the E. debaryana genome (fig. S4c). Taken together with the previous assessment of synteny between C. reinhardtii and V. carteri, these results suggest that karyotype evolution in the core-Reinhardtinia is expected to be dynamic, with very high levels of synteny but a non-negligible rate of inter-chromosomal rearrangements present between closely related species, and likely far greater karyotypic diversity present between more distantly related species.



Dotplots representing syntenic genomic segments identified between C. reinhardtii and 50 largest contigs of a) Chlamydomonas incerta, b) Chlamydomonas schloesseri, and c) Edaphochlamys debaryana.

Circos plot (Krzywinski et al. 2009) representation of synteny blocks shared between a) C. reinhardtii and C. incerta, and b) C. reinhardtii and C. schloesseri. C. reinhardtii chromosomes are represented as coloured bands, and C. incerta / C. schloesseri contigs as grey bands. Contigs are arranged and orientated relative to C. reinhardtii chromosomes, and adjacent contigs with no signature of rearrangement relative to C. reinhardtii are plotted without gaps. Dark grey bands highlight putative C. reinhardtii centromeres, and asterisks represent rDNA. Note that colours representing specific chromosomes differ between a) and b).
Given the high-contiguity and synteny of the assemblies, it was possible to assess several complex features of genome architecture that regularly resist assembly in short-read assemblies. Telomeric repeats were observed in all three assemblies, with six C. incerta and 19 C. schloesseri contigs terminating in the satellite (TTTTAGGG)n, and 15 E. debaryana contigs terminating in (TTTAGGG)n (table S6). The Arabidopsis-type sequence (TTTAGGG)n is ancestral to green algae and was previously confirmed as the telomeric repeat present in E. debaryana, while the derived Chlamydomonas-type sequence (TTTTAGGG)n is found in both C. reinhardtii and V. carteri (Fulnečková et al. 2012). Given the phylogenetic relationships presented above (fig. 2), this implies either two independent transitions to the derived sequence or a reversion to the ancestral sequence in the lineage including E. debaryana, providing further evidence for the relatively frequent transitions that have produced extensive variation in telomere composition in green algae and land plants (Peska and Garcia 2020).
Ribosomal DNA repeats (rDNA) were assembled as part of three larger contigs in both C. incerta and C. schloesseri, but were found only as fragmented contigs entirely consisting of rDNA in E. debaryana. Although poorly assembled in C. reinhardtii, the rDNA arrays are located at subtelomeric locations on chromosomes 1, 8 and 14, where cumulatively they are estimated to be present in 250-400 tandem copies (Howell 1972; Marco and Rochaix 1980). The assembled C. incerta and C. schloesseri rDNA arrays (which are also not complete and are tandemly repeated at most five times) were entirely syntenous with those of C. reinhardtii, suggesting conservation of subtelomeric rDNA organisation in Chlamydomonas (fig. 3). rDNA arrays are commonly located in subtelomeric regions across several taxa, where among several other factors their location may be important for genomic stability (Dvořáčková et al. 2015).
Finally, we were able to assess the composition and potential synteny of centromeres in Chlamydomonas. The centromeric locations of 15 of the 17 C. reinhardtii chromosomes were recently mapped by Lin et al. (2018), who observed that these regions were characterised by multiple copies of genes encoding reverse-transcriptase. Upon inspection of these regions, we found that these genes are generally encoded by copies of the L1 LINE element L1-1_CR. Although these regions are currently not well-enough assembled to conclusively define the structure of centromeric repeats, L1-1_CR is present in multiple copies at all 15 putative centromeres and appears to be the major centromeric component (with chromosome-specific contributions from other TEs, especially Dualen LINE elements) (table S7, fig S5a). Remarkably, phylogenetic analysis of all curated L1 elements from green algae indicated that L1-1_CR is more closely related to the Zepp elements of Coccomyxa subellipsoidea than to any other L1 elements annotated in C. reinhardtii (fig. 4a). The divergence of the classes Trebouxiophyceae (to which C. subellipsoidea belongs) and Chlorophyceae (to which C. reinhardtii belongs) occurred in the early Neoproterozoic era (i.e. 700-1,000 million years ago) (Del Cortona et al. 2020), suggesting that L1-1_CR has been evolving independently from all other C. reinhardtii L1 elements for more than half a billion years. Zepp elements are of particular interest as they are thought to constitute the centromeres in C. subellipsoidea, where they are strictly present as one cluster per chromosome (Blanc et al. 2012). The clustering pattern of Zepp elements arises due to a nested insertion mechanism that targets existing copies, creating tandem arrays consisting mostly of the 3’ end of the elements (due to frequent 5’ truncations upon insertion) (Higashiyama et al. 1997). Chromosome-specific clustering of L1-1_CR was also evident in C. reinhardtii, with highly localised clusters observed at all 15 of the putative centromeres (fig. 4b). The double-peaks in L1-1_CR density present on chromosomes 2, 3 and 8, and the single sub-telomeric cluster present on chromosome 5, are all the result of the incorrect scaffolding of contigs in these highly repetitive regions (unpublished data). Thus, outside the putative centromeres, L1-1_CR appears to be entirely absent from the C. reinhardtii genome.

Mean densities of Zepp-like L1 LINE elements per 20 kb windows averaged over relevant chromosomes/contigs. Shaded areas represent 95% quantiles. a) Density of L1-1_CR elements relative to midpoint of 15 putative C. reinhardtii centromeres. b) Density of ZeppL-1_cInc elements relative to C. incerta contig ends syntenic to C. reinhardtii putative centromeres. c) Density of ZeppL-1_cSch and ZeppL-2_cSch elements relative to C. schloesseri contig ends syntenic to C. reinhardtii putative centromeres.

a) ML phylogeny of chlorophyte L1 LINE elements inferred using the LG+F+R6 model and alignment of endonuclease and reverse transcriptase protein domains. Bootsrap values <=70% are shown. Species are given by the element name suffix as follows: _CR = C. reinhardtii, _VC = V. carteri, _cInc = C. incerta, _cSch = C. schloesseri, _eDeb = E. debaryana, _eud = Eudorina sp. 2016-703-Eu-15. b) Density (0-100%) of L1-1_CR in 50 kb windows across C. reinhardtii chromosomes. Dark bands represent putative centromeres, x-axis ticks represent 100 kb increments and y-axis ticks represent 20% increments. Plot produced using karyoploteR (Gel and Serra 2017).
Every centromeric location in C. reinhardtii coincided with breaks in syntenic segments and the termination of contigs in C. incerta and C. schloesseri (fig. 3), suggesting that these regions are also likely to be repetitive in both species. The phylogenetic analysis revealed the presence of one and two L1-1_CR homologs in C. incerta and C. schloesseri, respectively, which we term Zepp-like (ZeppL) elements (fig. 4a). Of the 30 contig ends associated with the 15 C. reinhardtii centromeres, 28 contigs in both species contained a ZeppL element within their final 20 kb (fig. S5b, c), and genome-wide the ZeppL elements exhibited similarly localised clustering to that observed in C. reinhardtii (fig. S6a, b). Thus, it appears that both the location and composition of the C. reinhardtii centromeres are likely to be conserved in both C. incerta and C. schloesseri. We further identified two families of ZeppL elements in the E. debaryana genome and one family of ZeppL elements in the Eudorina sp. genome, although we did not find any evidence for ZeppL elements in either Y. unicocca or V. carteri. Given the lack of synteny between C. reinhardtii and E. debaryana it was not possible to assign putatively centromeric contigs. Nonetheless, highly localised genomic clustering of ZeppL elements was observed for both E. debaryana and Eudorina sp. (fig. S6c, d), suggesting that these elements may play a similar role to that in Chlamydomonas.




Genome-wide density of Zepp-like elements. Contigs are represented by grey bands and ordered by size. Dark grey bars above/below contigs represent contig ends inferred as syntenic with C. reinhardtii centromeres. Axis ranges from 0-100%. a) Chlamydomonas incerta. b) Chlamydomonas schloesseri. c) Edaphochlamys debaryana. d) Eudorina sp. 2016-703-Eu-15.
As sequencing technologies advance it is becoming increasingly clear that TEs, alongside satellite DNA, contribute substantially to centromeric sequence in many species (Chang et al. 2019; Fang et al. 2020). Given the evolutionary distance between C. subellipsoidea and Chlamydomonas, it is tempting to predict that ZeppL elements may be present at the centromeres of many other species of green algae. However, it is unlikely that centromeres are conserved between species from the Trebouxiophyceae and Chlorophyceae. Firstly, it has recently been shown that the centromeric repeats in the Chlorophyceae species Chromochloris zofingiensis consist of LTR elements from the Copia superfamily (Roth et al. 2017). Secondly, the apparent absence of ZeppL elements from Y. unicocca and V. carteri suggest that these elements are not required for centromere formation in these species. Instead, it is possible that the propensity for Zepp and ZeppL elements to form clusters may play a role in their recruitment as centromeric sequences, which is likely to have happened independently in C. subellipsoidea and Chlamydomonas. As more highly contiguous chlorophyte assemblies become available, it will be important to search these genomes for ZeppL clusters to assess whether these elements can be used more generally as centromeric markers.
Gene and gene family evolution in the core-Reinhardtinia
Gene model annotation was performed for each species using 7.4-8.2 Gb of stranded RNA-seq (table S8). Protein mode BUSCO scores supported a high level of annotation completeness across all three species (97.0-98.1% chlorophyte genes present), although relative to genome mode scores there was an increase in the proportion of genes identified as fragmented (4.0-5.9%) (table 2). C. incerta and C. schloesseri had comparable gene counts to C. reinhardtii, although lower gene densities as a result of their larger genomes. With 19,228 genes, the E. debaryana genome contained substantially more genes than any other currently annotated core-Reinhardtinia species. As reported by Hanschen et al. (2016), several metrics appeared to correlate with organismal complexity. Relative to the unicellular species, gene density was lower, and median intergenic and intronic lengths were longer, in G. pectorale and V. carteri. Presumably this is at least partly due to an increase in the amount of regulatory sequence in these genomes, although this has not yet been explored.
Gene annotation metrics for core-Reinhardtinia species.
Across all species, both mean intron lengths (discussed below) and intron numbers per gene were very high for such compact genomes. For the unicellular species, the mean number of introns per gene coding sequence ranged from 7.7-9.3, with slightly lower counts in G. pectorale (6.2) and V. carteri (6.7). These numbers are more comparable to vertebrates such as human (8.5) than to other model organisms with similar genomes sizes, such as Caenorhabditis elegans (5.1), Drosophila melanogaster (3.0), and Arabidopsis thaliana (4.1). Modelling of intron evolution across the breadth of eukaryota has predicted that a major expansion of introns occurred early in chlorophyte evolution (prior to the divergence of Chlorophyceae and Trebouxiophyceae), and that high intron densities have since been maintained in certain lineages by a balance between intron loss and gain (Csuros et al. 2011). It has been hypothesised that the relative roles of DNA double-strand break repair pathways play a major role in the dynamics of intron gain and loss, as the homologous recombination (HR) pathway is thought to cause intron deletion, while non-homologous end-joining (NHEJ) may result in both intron gain and loss (Farlow et al. 2011). In is interesting to note that HR occurs at an extremely low rate in C. reinhardtii (Plecenikova et al. 2014), and if this is shared across Chlamydomonas and the core-Reinhardtinia, it may contribute to the maintenance of such high intron numbers. Alternatively, introns could be maintained by other forces, such as selection. Intron gains and losses caused by NHEJ are expected to possess specific genomic signatures (Farlow et al. 2011; Sun et al. 2015), and thus it should now be possible to test this hypothesis by exploring patterns of intron gain and loss across the three Chlamydomonas species.
To explore gene family evolution in the core-Reinhardtinia, we performed orthology clustering using the six available high-quality gene annotations (98,342 total protein-coding genes), which resulted in the delineation of 13,728 orthogroups containing 86,446 genes (fig. 5). The majority of orthogroups (8,532) were shared by all species, with the second most abundant category (excluding genes unique to a single species) being those present in all species except G. pectorale (868 orthogroups). Given the lower BUSCO score observed for G. pectorale (table 2) it is likely that a proportion of these orthogroups are also universal to core-Reinhardtinia species. The next most abundant category was the 859 orthogroups present only in Chlamydomonas. Unfortunately, essentially nothing is known about the biology and ecology of C. incerta and C. schloesseri, and even for C. reinhardtii we have a minimal understanding of its biology in natural environments (Sasso et al. 2018; Craig et al. 2019). Although many of the Chlamydomonas-specific orthogroups are associated with functional domains, this scenario currently precludes the formation of any clear hypotheses to test. In contrast to Chlamydomonas, only 51 orthogroups were unique to the two multicellular species. This may be an underestimate due to the relative incompleteness of the G. pectorale annotation, and it will be important to re-visit this analysis as more annotations become available (e.g. for Y. unicocca and Eudorina sp.). Nonetheless, the availability of our three new high-quality annotations for unicellular species will provide a strong comparative framework to explore the relative roles of gene family birth versus expansions in existing gene families in the transition to multicellularity.

Upset plot (Lex et al. 2014) representing the intersection of orthogroups between six core-Reinhardtinia species. Numbers above bars represent the number of orthogroups shared by a given intersection of species.
Finally, we explored the contribution of gene family expansions to the high gene number observed in E. debaryana. The E. debaryana genome contained more species-specific genes (3,556) than any other species, however this figure was not substantially higher than the unassigned gene counts for G. pectorale and V. carteri (fig. 5). We quantified E. debaryana gene family expansion and contraction by calculating per orthogroup log2-transformed ratios of the E. debaryana gene count and the mean gene count for the other species. Arbitrarily defining an expansion as a log2-transformed ratio >1 (i.e. a given orthogroup containing more than twice as many E. debaryana genes than the mean of the other species) and a contraction as a ratio <−1, we identified E. debaryana-specific expansions in 294 orthogroups and contractions in 112. Although 192 of the expanded orthogroups were associated with functional domains (table S9), it is once again very difficult to interpret these results without any knowledge of the biological differences between unicellular species. Given that E. debaryana has been found across the Northern Hemisphere it is possible that the species experiences a greater range of environments than the Chlamydomonas species, however this is currently entirely speculative.
Evolution of the mating-type locus in Chlamydomonas
Across core-Reinhardtinia species, sex is determined by a haploid mating-type locus (MT) with two alleles, termed MT+ or female, and MT- or male, in isogamous and anisogamous species, respectively. The C. reinhardtii MT is located on chromosome 6, spanning >400 kb and consisting of three domains, the T (telomere-proximal), R (rearranged) and C (centromere-proximal) domains. While both the T and C domains exhibit high synteny between the MT alleles, the R domain contains the only MT limited genes (Ferris and Goodenough 1997) and harbours substantial structural variation, featuring several inversions and rearrangements (Ferris et al. 2002; De Hoff et al. 2013). Crossover events are suppressed across the entire MT, although genetic differentiation between gametologs is reduced as a result of widespread gene conversion (De Hoff et al. 2013; Hasan et al. 2019). Comparative analyses of MT+/female and MT-/male haplotypes between core-Reinhardtinia species, and particularly between TGV clade species, have revealed highly dynamic MT evolution, with extensive gene turnover and structural variation resulting in a complex and discontinuous evolutionary history of haplotype reformation (Ferris et al. 2010; Hamaji et al. 2016b; Hamaji et al. 2018). This is most strikingly illustrated by the MT male R domains of V. carteri and Eudorina sp., the former being ~1.1 Mb in length and relatively repeat-rich, while the latter is just 7 kb and contains only three genes (Hamaji et al. 2018). Only one MT limited gene is common to all species, the minus dominance gene MID, which determines MT-/male gametic differentiation (Ferris and Goodenough 1997; Yamamoto et al. 2017).
To explore whether MT evolution is similarly dynamic between the more closely related Chlamydomonas species, we used a reciprocal best-hit approach to identify C. reinhardtii MT orthologs in C. incerta and C. schloesseri. The sequenced isolates of both species were determined to be MT-based on the presence of MID, as was previously reported for C. incerta (Ferris et al. 1997). Although we were able to map the entire C. reinhardtii MT-haplotype to single contigs in both the C. incerta and C. schloesseri assemblies, it is important to state that it is currently impossible to define the R domain boundaries for either species without sequencing their MT+ alleles. Unfortunately, it is currently unknown if any of the one (C. incerta) or two (C. schloesseri) other isolates are MT+, and as no isolate from either species has been successfully crossed it is not even known if they are sexually viable (Pröschold et al. 2005). We also determined the sequenced isolate of E. debaryana to be MT-via the identification of MID, although we did not explore MT evolution further given the evolutionary distance to C. reinhardtii. At least one heterothallic mating pair of E. debaryana are in culture, and a future comprehensive study of MT in the species is therefore possible.
In C. incerta, gene order was entirely syntenic across the C domain, with the exception of MT0828, which did not yield a hit anywhere in the genome. Conversely, both T and R domain genes have undergone several rearrangements and inversions relative to C. reinhardtii MT-(fig. 6a). Furthermore, the T domain genes SPP3 and HDH1 were present on separate contigs in C. incerta and do not appear to be MT-linked (table S10). Synteny otherwise continued well into the adjacent autosomal sequence, in line with the genome-wide patterns of synteny described above. We observed even less synteny between C. reinhardtii and C. schloesseri MT-genes, with both the T and C domains showing two large inversions each (fig. 6b). However, gene order in the surrounding autosomal sequence was also largely collinear. As in C. incerta, SPP3 was located elsewhere in the C. schloesseri assembly, suggesting a relatively recent translocation to the T domain in C. reinhardtii. Finally, the C domain gene 97782 was also located on a different contig, while the genes MT0796, MT0828 and 182389 did not yield hits anywhere in the C. schloesseri genome. While the C. reinhardtii MT-limited gene MTD1 was found in both C. incerta and C. schloesseri, we found no hits for the MT+ limited genes FUS1 and MTA1, suggesting that these genes (assuming they exist) are also expected to be MT+ limited in both species.

Synteny representation between genes of the C. reinhardtii MT-haplotype and flanking autosomal sequence, and a) inferred C. incerta MT-haplotype and flanking sequence genes (contig C0033), or b) inferred C. schloesseri MT-haplotype and flanking sequence genes (contig C0045). The T, R and C domains of the C. reinhardtii MT-are highlighted. Note that C0045 does not contain the initial ~260 kb of chromosome 6.
The lack of collinearity relative to the C. reinhardtii T domain may be indicative of an extended R domain in these species, especially in C. schloesseri, where we observe multiple rearrangements in all three domains. We do not, however, observe dramatic variation in MT size; whereas C. reinhardtii MT-is ~422 kb, if NIC7 and MAT3 are taken as the boundaries of the locus (De Hoff et al. 2013), C. incerta MT-is ~329 kb and C. schloesseri MT-is ~438 kb. In all, while we do find evidence of MT-haplotype reformation within Chlamydomonas, this is mostly limited to rearrangements, with far less gene turnover and MT size variation than has been observed between more distantly related core-Reinhardtinia species. While MT evolution has previously been explored in the context of transitions from unicellularity to multicellularity and isogamy to anisogamy, our data suggest that MT haplotype reformation is still expected to occur between closely related isogamous species, albeit at a reduced scale.
Alignability and estimates of neutral divergence
In order to facilitate the identification of conserved elements (CEs), we produced an 8-species core-Reinhardtinia whole-genome alignment (WGA) using Cactus (Armstrong et al. 2019). Based on the alignment of C. reinhardtii four-fold degenerate (4D) sites extracted from the WGA, we estimated putatively neutral branch lengths across the topology connecting the eight species using the GTR substitution model (fig. 7a). Divergence between C. reinhardtii and C. incerta, and C. reinhardtii and C. schloesseri, was estimated as 34% and 45%, respectively. Divergence between C. reinhardtii and E. debaryana was estimated as 98%, while all four TGV clade species were saturated relative to C. reinhardtii (i.e. on average, each 4D site is expected to have experienced more than one substitution). To put these estimates within a more recognisable framework, divergence across Chlamydomonas is approximately on the scale of human-rodent divergence (Lindblad-Toh et al. 2011), while divergence between Chlamydomonas and the TGV clade is approximately equivalent to that of mammals and sauropsids (birds and reptiles), which diverged ~320 million years ago (Alföldi et al. 2011). Our estimates corroborate a previous estimate of synonymous divergence between C. reinhardtii and C. incerta of 37% (averaged over 67 orthologous genes) (Popescu et al. 2006), and are broadly in line with the divergence time estimate of ~230 million years ago between the TGV clade and their unicellular ancestors (Herron et al. 2009). Finally, it is important to note that we have likely underestimated neutral divergence, as 4D sites are unlikely to be evolving neutrally due to selection acting on codon usage, which has been shown to decrease divergence between C. reinhardtii and C. incerta (Popescu et al. 2006).
a) Estimates of putatively neutral divergence under the GTR model, based on the topology of figure 2 and 1,552,562 C. reinhardtii 4D sites extracted from the Cactus WGA. b) A representation of the C. reinhardtii genome by site class, and the number of aligned sites per C. reinhardtii site class for each other species in the Cactus WGA.
As expected, genome-wide alignability (i.e. the proportion of bases aligned between C. reinhardtii and a given species in the WGA) decreased substantially with increasing divergence, with 53.0% of the C. reinhardtii genome aligned to C. incerta, 48.6% to C. schloesseri, and on average only 19.9% to the remaining five species (fig. 7b). The majority of C. reinhardtii CDS was alignable within Chlamydomonas (87.7% and 85.5% to C. incerta and C. schloesseri, respectively), indicating that it will be possible to perform molecular evolutionary analyses on CDS between the three species. CDS also constituted the majority of the aligned sequence to the other five species, comprising on average 78.3% of the aligned bases despite CDS forming only 35.2% of the C. reinhardtii genome. In contrast, far less non-exonic sequence was alignable, especially at evolutionary distances beyond Chlamydomonas. Substantial proportions of intronic bases were aligned to C. incerta (44.1%) and C. schloesseri (38.8%), with on average 11.3% aligned to the other five species. Less than 10% of intergenic sequence was aligned to any one species, and on average less than 1% was aligned to non-Chlamydomonas species. Distributions of intergenic tract lengths across the core-Reinhardtinia are highly skewed (fig. S7), so that in C. reinhardtii tracts shorter than 250 bp constitute 63.5% of tracts but just 5.5% of total intergenic sequence. The sequence content of tracts >250 bp is highly repetitive (total repeat content 63.4%), while tracts <250 bp are relatively free of repeats (4.3% repeat content) and as a result are far more alignable to C. incerta and C. schloesseri (40.8% and 32.0% of bases aligned, respectively). This suggests that at least for introns and short intergenic tracts it is feasible to explore the landscape of non-exonic evolutionary constraint, primarily utilising alignment data from Chlamydomonas, supplemented by what is likely the alignment of only the most conserved sites at greater evolutionary distances.

Distribution of intergenic tract lengths across six core-Reinhardtinia species. G. pectorale distribution differs due to the lack of UTR annotation for this species.
Missing genes in Chlamydomonas reinhardtii
One of the major successes of comparative genomics has been the refinement of gene annotations and identification of novel gene models and exons (e.g. Lin et al. (2008); Mudge et al. (2019)). Prior to identifying conserved sequences and classifying them as coding or noncoding, we attempted to identify novel C. reinhardtii genes absent from the latest annotation (v5.6) using patterns of Chlamydomonas synteny and the core-Reinhardtinia WGA. A de novo C. reinhardtii gene annotation yielded 433 novel gene models, 142 of which were retained based on the presence of a syntenic homolog in one or both of the Chlamydomonas species, and/or a phyloCSF (Lin et al. 2011) score >100. PhyloCSF assesses the protein-coding potential of a multi-species alignment using patterns of substitution at putative synonymous and non-synonymous sites, and so is not reliant on gene annotations in other species. Of the 142 supported genes, 90 had significant BLASTp hits (e-value <1×10−5, >=80% protein length) to C. reinhardtii proteins from annotation version 4.3 and likely represent models that were lost during the transition from v4 to v5 of the genome.
The genomic landscape of sequence conservation in Chlamydomonas reinhardtii
Based on the core-Reinhardtinia WGA, we identified 265,006 CEs spanning 33.8 Mb or 31.5% of the C. reinhardtii genome. The majority of CE bases overlapped CDS (70.6%), with the remaining bases overlapping 5’ UTRs (2.9%), 3’ UTRs (4.4%), introns (20.0%) and intergenic sites (2.0%) (table 3). Relative to the site class categories themselves, 63.1% of CDS bases, 24.8% of 5’ UTR bases, 11.0% of 3’ UTR bases, and 19.2% of intronic bases were overlapped by CEs. Only 4.1% of intergenic bases were covered by CEs, however when splitting intergenic tracts into those <250 bp (short tracts) and >250 bp (long tracts), a more appreciable proportion of short tract bases (14.1%) were covered by CEs. As would be predicted given the expectation that CEs contain functional sequences, C. reinhardtii genetic diversity (π) was 39.5% lower for CEs (0.0134) than non-CE bases (0.0220), a result that was relatively consistent across site classes with the exception of long intergenic tracts (table 3). It is, however, important to state that the CEs we have identified are likely to contain a proportion of non-constrained sites. While this is always to be expected to some extent (e.g. CDS is generally included in CEs despite the presence of synonymous sites), given a mean length of 128 bp our CE dataset should be cautiously interpreted as regions containing elevated proportions of constrained sites.
Overlap between conserved elements and C. reinhardtii genomic site classes.
Given the compactness of the C. reinhardtii genome (82.1% genic, median intergenic tract length 134 bp), it is expected that a high proportion of regulatory sequence will be concentrated in UTRs and intergenic sequences immediately upstream of genes (i.e. promoter regions). Relatively little is known about the genome-wide distribution of regulatory elements in C. reinhardtii, although analyses based on motif modelling have identified putative cis-regulatory elements enriched in these regions (Ding et al. 2012; Hamaji et al. 2016a). Presumably many CEs overlapping UTRs and promoter regions harbour regulatory elements, and the CEs we have identified could be used in future studies to validate potential functional motifs. Due to our inability to align longer intergenic tracts, it remains an open question whether functional elements are present at non-negligible abundances in these regions. Although the lack of alignment could in itself be taken for a lack of constraint, the highly repetitive nature of these regions may disrupt the alignment of functional sequences present among repeats. It is noticeable that C. reinhardtii genetic diversity is lower in long intergenic tracts than all site classes except CDS (table 3), which could be due the presence of functional sequences or alternatively an as of yet unknown evolutionary mechanism.
All six annotated core-Reinhardtinia species contained conspicuously long introns (median lengths 198-343 bp, table 2). As reported previously for C. reinhardtii (Merchant et al. 2007), the distribution of intron lengths for core-Reinhardtinia species lacked the typical peak in intron lengths at 60-110 bp that is present in several model organisms with similarly compact genomes (fig. 8a, b). In D. melanogaster, short introns (<80 bp) appear to largely consist of neutrally evolving sequence, while longer introns that form the tail of the length distribution contain sequences evolving under evolutionary constraint (Halligan and Keightley 2006). To explore the relationship between intron length and sequence conservation in C. reinhardtii, we ordered introns by length and divided them into 50 bins, so that each bin contained an approximately equal number (~2,667) of introns. Mean intron length per bin was significantly negatively correlated with the proportion of bases overlapped by CEs (Pearson’s r = −0.626, p <0.01) (fig. 8c). This was particularly pronounced for introns <100 bp (~5% of introns), for which 48.1% of bases were overlapped by CEs, compared to 18.5% for longer introns. Therefore, it appears that in a reverse of the situation found in D. melanogaster, the minority of introns in C. reinhardtii are short and potentially functionally important, while the majority of introns are longer and contain far fewer conserved bases. The tight peak in the distribution of intron lengths combined with the lack of sequence constraint in D. melanogaster short introns led Halligan and Keightley (2006) to hypothesise that intron length was under selection, but not the intronic sequence itself, and that introns had essentially evolved to be as short as possible. It is possible that C. reinhardtii introns >100 bp are similarly evolving under selection to be bounded within certain length constraints, although the selective advantage of maintaining intron lengths substantially longer than the minimum remains unknown. Given that atypical intron length distributions are common to all core-Reinhardtinia species, whatever mechanism is driving intron length is likely to be evolutionarily ancient.

a) Intron length distributions for five model organisms (Cele = C. elegans, Dmel = D. melanogaster, Ncra = Neurospora crassa, Athal = A. thaliana, Esil = Ectocarpus siliculosus). The brown alga E. siliculosus is included as an example of an atypical distribution. b) Intron length distributions for six core-Reinhardtinia species, note different y-axis scale (Crei = C. reinhardtii, Cinc = C. incerta, Csch = C. schloesseri, Edeb = Edaphochlamys debaryana, Gpec = G. pectorale, Vcar = V. carteri). c). Correlation between mean intron length per bin and the proportion of bases overlapped by CEs. Introns were ordered by length and separated into 50 bins, so that each bin contained the same number of introns.
There are at least two leading explanations for why shorter C. reinhardtii introns may be functionally important. Firstly, intron retention (IR) has been shown to occur significantly more frequently in shorter genes (median = 181 bp) (Raj-Kumar et al. 2017). IR is the most common form of alternative splicing (AS) detected in C. reinhardtii (~30% of AS events), although AS in the species has not yet been extensively characterised and only ~1% of introns are currently annotated as alternatively retained. Furthermore, as only ~20% of IR events produce a functional protein (Raj-Kumar et al. 2017), not all retained introns are expected to be evolving under coding constraint. It is therefore difficult to assess the overall contribution of IR to intronic sequence conservation. Secondly, short introns may be enriched for regulatory sequences. Short introns <100 bp represent the first intron in a gene approximately four-fold more frequently (44.6%) than longer introns (10.3%) (fig. S8a). Introns <100 bp were also significantly more likely to occur closer to the transcription start site (mean intron positions relative to transcript length for introns <100 bp = 24.2% and introns >100 bp = 39.5%; independent-samples t-test t=-54.0, p<0.01) (fig. S8b). For many genes, introns within the first 1 kb have strong regulatory effects on gene expression (Rose 2018), and in C. reinhardtii it has been shown that the addition of a specific first intron to transgenes substantially increases their expression (Baier et al. 2018). It is also important to emphasise that a non-negligible proportion of sites in introns >100 bp are overlapped by CEs (18.5%), and thus longer introns are also likely to harbour some functional sites. Alongside regulatory sequences there are several possible explanations for this, including the presence of intronic RNA genes (Chen et al. 2008; Valli et al. 2016) and other categories of AS (e.g. alternative acceptor or donor splice sites) (Raj-Kumar et al. 2017).

a) Relationship between the proportion of introns that are the first intron of a gene and the mean intron length per bin (see main text). b) The relationship between the mean intron position relative to transcript length (e.g. an intron at position 500 of a 2000 bp transcript equals 25%) and mean intron length per bin.
Finally, we further identified 5,611 ultraconserved elements (UCEs) spanning 356.0 kb of the C. reinhardtii genome, defined as sequences >=50 bp exhibiting 100% sequence conservation across the three Chlamydomonas species. A subset of just 55 UCEs exhibited >=95% sequence conservation across all eight species, indicating that hardly any sequence is expected to be conserved to this level across the core-Reinhardtinia. The vast majority of UCE bases (96.0%) overlapped CDS, indicating constraint at both nonsynonymous and synonymous sites. There are several reasons why synonymous sites may be subject to such strong constraint, including interactions with RNA processing or formation of RNA secondary structures, the presence of exonic regulatory elements, or selection for optimal codon usage. Noticeably, 15 of the 55 core-Reinhardtinia UCEs overlapped ribosomal protein genes, which are often used as a standard for identifying optimal codons given their extremely high gene expression (Sharp and Li 1987), and several of the other genes overlapped by UCEs are also expected to be very highly expressed (e.g. elongation factors) (table S11). Although considered to be a very weak evolutionary force, this indicates that coordinated selection for optimal codons across the core-Reinhardtinia may be driving extreme sequence conservation. UCEs have proved to be excellent phylogenetic markers across several taxa (Faircloth et al. 2012; Faircloth et al. 2015). Given the lack of nuclear markers and the current difficulty in determining phylogenetic relationships in the core-Reinhardtinia, the 55 deeply conserved elements could potentially be used to provide additional phylogenetic resolution.
Conclusions
Via the assembly of highly contiguous and well-annotated genomes for three of C. reinhardtii’s unicellular relatives, we have presented the first nucleotide-level comparative genomics framework for this important model organism. These resources are expected to enable the continued development of C. reinhardtii as a model system for molecular evolution. Furthermore, by providing insights into the gene content and genomic architecture of unicellular core-Reinhardtinia species, they are also expected to advance our understanding of the genomic changes that have occurred during the transition to multicellularity in the TGV clade.
Despite such advances, these genome assemblies have only now raised C. reinhardtii to a standard that had been achieved for many other model organisms ten or more years ago. Many of the analyses we have performed could be greatly enhanced by the inclusion of additional Chlamydomonas species, but addressing this is a question of taxonomy rather than sequencing effort. This is somewhat analogous to the past situation for Caenorhabditis, where only very recent advances in ecological knowledge have led to a rapid increase in the number of sampled species and sequenced genomes (Stevens et al. 2019). We hope that this study will encourage Chlamydomonas researchers to increase sampling efforts for new species, fully enabling the power of comparative genomics analyses to be realised for the species.
Methods
Nucleic acid extraction and sequencing
Isolates were obtained from the SAG or CCAP culture centres, cultured in Bold’s Basal Medium, and where necessary made axenic via serial dilution, plating on agar, and isolation of single algal colonies. High molecular weight DNA was extracted using a customised extension of an existing CTAB/phenol-chloroform protocol (file S1). One SMRTbell library (sheared to ~20 kb, with 15-50 kb size selection) was prepared per species, and each library was sequenced on a single SMRTcell on the PacBio Sequel platform. PacBio library preparation and sequencing were performed by Edinburgh Genomics.
DNA for Illumina sequencing was extracted using a phenol-chloroform protocol (Ness et al. 2012). Across all species a variety of library preparations, read lengths, insert sizes and sequencing platforms were used (table S2). RNA was extracted from four-day liquid cultures using Zymo Research TRI Reagent (product ID: R2050) and the Direct-zol RNA Miniprep Plus kit (product ID: R2070) following user instructions. One stranded RNA-seq library was prepared for each species using TruSeq reagents, and sequencing was performed on the Illumina HiSeq X platform (Chlamydomonas incerta 150 bp paired-end, Chlamydomonas schloesseri and Edaphochlamys debaryana 100 bp paired-end). All Illumina sequencing and library preparations were performed by BGI Hong Kong.
De novo genome assembly
Detailed per-species methods and command line options are detailed in file S2. We first identified and removed reads derived from any contaminants by producing taxon-annotated GC-coverage plots with BlobTools v1.0 (Laetsch and Blaxter 2017a). Assemblies were produced using Canu v1.7.1 (Koren et al. 2017), with three iterative round of error-correction performed with the PacBio reads and the GenomicConsensus module Arrow v2.3.2 (https://github.com/PacificBiosciences/GenomicConsensus). All available Illumina data for each species was subsequently used to perform three iterative rounds of polishing using Pilon v1.22 (Walker et al. 2014). Assemblies of the plastid and mitochondrial genomes were produced independently and will be described elsewhere.
Annotation of genes and repetitive elements
A preliminary repeat library was produced for each species with RepeatModeler v1.0.11 (Smit and Hubley 2008-2015). Repeat models with homology to Chlamydomonas reinhardtii v5.6 and/or Volvox carteri v2.1 transcripts (e-values <10−3, megablast (Camacho et al. 2009)) were filtered. The genomic abundance of each repeat model was estimated by providing RepeatMasker v4.0.9 (Smit et al. 2013-2015) with the filtered RepeatModeler output as a custom library, and any TEs with a cumulative total >100 kb were selected for manual curation, following Suh et al. (2014). Briefly, multiple copies of a given TE were retrieved by querying the appropriate reference genome using megablast, before each copy was extended at both flanks and aligned using MAFFT v7.245 (Katoh and Standley 2013). Alignments were then manually inspected, consensus sequences were created, and TE families were classified following Wicker et al. (2007) and Kapitonov and Jurka (2008). This procedure was also performed exhaustively for C. reinhardtii (i.e. curating all repeat models regardless of genomic abundance), which will be described in detail elsewhere. Final repeat libraries were made by combining the RepeatModeler output for a given species with all novel curated TEs and V. carteri repeats from Repbase (Bao et al. 2015) (files S3 and S4). TEs and satellites were soft-masked by providing RepeatMasker with the above libraries. In line with the most recent C. reinhardtii annotation (Blaby et al. 2014), low-complexity and simple repeats were not masked as the high GC-content of genuine coding sequence can result in excessive masking.
Adapters and low-quality bases were trimmed from each RNA-seq dataset using Trimmomatic v0.38 (Bolger et al. 2014) with the parameters optimised by Macmanes (2014). Trimmed reads were mapped to repeat-masked assemblies with the 2-pass mode of STAR v2.6.1a (Dobin et al. 2013). Gene annotation was performed with BRAKER v2.1.2 (Hoff et al. 2016; Hoff et al. 2019), an automated pipeline that combines the gene prediction tools Genemark-ET (Lomsadze et al. 2014) and AUGUSTUS (Stanke et al. 2006; Stanke et al. 2008). Read pairs mapping to the forward and reverse strands were extracted using samtools v1.9 (Li et al. 2009) and passed as individual BAM files to BRAKER, which was run with the “--UTR=on” and “--stranded=+,-” flags to perform UTR annotation. Resulting gene models were filtered for genes with internal stop codons, protein sequences <30 amino acids, or CDS overlapped by >=30% TEs/satellites or >=70% low-complexity/simple repeats.
Proteins were functionally annotated via upload to the Phycocosm algal genomics portal (https://phycocosm.jgi.doe.gov). Phycocosm uses an array of tools to add detailed annotation (gene ontology terms, Pfam domains, etc.), and additionally provides a genome browser interface to enable visualisation.
Phylogenomics analyses
Genome and gene annotations for all available Reinhardtinia species and selected outgroups (tables S3, S4) were accessed from either Phytozome (if available) or NCBI. For annotation based analyses, protein clustering analysis was performed with OrthoFinder v2.2.7 (Emms and Kelly 2015), using the longest isoform for each gene, the modified BLASTp options “-seq yes, −soft_masking true, −use_sw_tback” (following Moreno-Hagelsieb and Latimer (2008)) and the default inflation value of 1.5. Protein sequences from orthogroups containing a single gene in all 11 included species (i.e. putative single copy-orthologs) were aligned with MAFFT and trimmed for regions of low-quality alignment using trimAl v1.4.rev15 (“-automated1”) (Capella-Gutiérrez et al. 2009). A ML species-tree was produced using concatenated gene alignments with IQ-TREE v1.6.9 (Nguyen et al. 2015), run with ModelFinder (“-m MFP”) (Kalyaanamoorthy et al. 2017) and ultrafast bootstrapping (“-bb 1000”) (Hoang et al. 2018). ASTRAL-III v5.6.3 (Zhang et al. 2018) was used to produce an alternative species-tree from individual gene-trees, which were themselves produced for each aligned single copy-ortholog using IQ-TREE as described above, with any branches with bootstrap support <10% contracted as recommended.
Annotation-free phylogenies were produced from a dataset of single-copy orthologous genes identified by BUSCO v3.0.2 (Waterhouse et al. 2018) run in genome mode with the pre-release Chlorophyta odb10 dataset (allowing missing data in up to three species). For each BUSCO gene, proteins were aligned and trimmed, and two species-trees were produced as described above.
General comparative genomics and synteny analyses
Basic genome assembly metrics were generated using QUAST v5.0.0 (Gurevich et al. 2013). Repeat content was estimated by performing repeat masking on all genomes as described above (i.e. supplying RepeatMasker with the RepeatModeler output for a given species + manually curated repeats from all species). Assembly completeness was assessed by running BUSCO in genome mode with the Eukaryota odb9 and Chlorophyta odb10 datasets. Each species was run with C. reinhardtii (-sp chlamy2011) and V. carteri (-sp volvox) AUGUSTUS parameters, and the run with the most complete BUSCO genes was retained.
Synteny segments were identified between C. reinhardtii and the three novel genomes using SynChro (Drillon et al. 2014) with a block stringency value (delta) of 2. To create the input file for C. reinhardtii, we combined the repeat-filtered v5.6 gene annotation (see below) with the centromere locations for 15 of the 17 chromosomes, as defined by Lin et al. (2018). The resulting synteny blocks were used to check the C. incerta and C. schloesseri genomes for misassemblies, by manually inspecting breakpoints between synteny blocks on a given contig that resulted in a transition between C. reinhardtii chromosomes (see file S2). This resulted in four C. incerta and two C. schloesseri contigs being split due to likely misassembly.
A ML phylogeny of L1 LINE elements was produced from the endonuclease and reverse transcriptase domains (i.e. ORF2) of all known chlorophyte L1 elements. Protein sequences were aligned, trimmed and analysed with IQ-TREE as described above. All C. incerta, C. schloesseri and E. debaryana elements were manually curated as part of the annotation of repeats (see above). The Yamagishiella unicocca, Eudorina sp., and V. carteri genomes were searched using tBLASTn with the L1-1_CR protein sequence as query, and the best hits were manually curated to assess the presence or absence of ZeppL elements in these species.
Gene annotation metrics and gene family evolution
The C. reinhardtii v5.6 gene models were manually filtered based on overlap with the novel repeat library (files S3 and S4), which resulted in the removal of 1,085 putative TE genes. For all species, annotation completeness was assessed by protein mode BUSCO analyses using the Eukaryota odb9 and Chlorophyta odb10 datasets. Gene families were identified using OrthoFinder as described above with the six core-Reinhardtinia species with gene annotations (C. reinhardtii, C. incerta, C. schloesseri, E. debaryana, G. pectorale and V. carteri). Protein sequences for all species were annotated with InterPro domain IDs using InterProScan v5.39-77.0 (Jones et al. 2014). Domain IDs were assigned to orthogroups by KinFin v1.0 (Laetsch and Blaxter 2017b) if a particular ID was assigned to at least 20% of the genes and present in at least 50% of the species included in the orthogroup.
Mating-type locus evolution
As the three novel genomes are all MT- and the C. reinhardtii reference genome is MT+, we first obtained the C. reinhardtii MT-locus and proteins from NCBI (accession GU814015.1) and created a composite chromosome 6 with an MT-haplotype. A reciprocal best hit approach with BLASTp was used to identify orthologs, supplemented with tBLASTn queries to search for genes not present in the annotations. To visualise synteny, we used the MCscan pipeline from the JCVI utility libraries v0.9.14 (Tang et al. 2008), which performs nucleotide alignment with LAST (Kiełbasa et al. 2011) to identify orthologs. We applied a C-score of 0.99, which filters LAST hits to only reciprocal best hits, while otherwise retaining default parameters. We manually confirmed that the LAST reciprocal hits were concordant with our BLASTp results. Scripts and data for this analysis are available at: https://github.com/aays/MT_analysis
Core-Reinhardtinia whole-genome alignment and estimation of putatively neutral divergence
An 8-species core-Reinhardtinia WGA was produced using Cactus (Armstrong et al. 2019) with all available high-quality genomes (C. reinhardtii v5, C. incerta, C. schloesseri, E. debaryana, Gonium pectorale, Y. unicocca, Eudorina sp. and V. carteri v2). The required guide phylogeny was produced by extracting alignments of 4D sites from single-copy orthologs identified by BUSCO (genome mode, Chlorophyta odb10 dataset). Protein sequences of 1,543 BUSCO genes present in all eight species were aligned with MAFFT and subsequently back-translated to nucleotide sequences. Sites where the aligned codon in all eight species contained a 4D site were then extracted (250,361 sites), and a guide-phylogeny was produced by supplying the 4D site alignment and topology (extracted from the Volvocales species-tree, see above) to phyloFit (PHAST v1.4) (Siepel et al. 2005), which was run with default parameters (i.e. GTR substitution model).
Where available the R domain of the MT locus not included in a given assembly was appended as an additional contig (extracted from the following NCBI accessions: C. reinhardtii MT-GU814015.1, G. pectorale MT+ LC062719.1, Y. unicocca MT-LC314413.1, Eudorina sp. MT male LC314415.1, V. carteri MT male GU784916.1). All genomes were softmasked for repeats as described above, and Cactus was run using the guide-phylogeny and all genomes set as reference quality. Post-processing was performed by extracting a multiple alignment format (MAF) alignment with C. reinhardtii as the reference genome from the resulting hierarchical alignment (HAL) file, using the HAL tools command hal2maf (v2.1) (Hickey et al. 2013), with the options –onlyOrthologs and –noAncestors. Paralogous alignments were reduced to one sequence per species by retaining the sequence with the highest similarity to the consensus of the alignment block, using mafDuplicateFilter (mafTools suite v0.1) (Earl et al. 2014).
Final estimates of putatively neutral divergence were obtained using a method adopted from Green et al. (2014). For each C. reinhardtii protein-coding gene, the alignment of each exon was extracted and concatenated. For the subsequent CDS alignments, a site was considered to be 4D if the codon in C. reinhardtii included a 4D site, and all seven other species had a triplet of aligned bases that also included a 4D site at the same position (i.e. the aligned triplet was assumed to be a valid codon, based on its alignment to a C. reinhardtii codon). The resulting alignment of 1,552,562 sites were then passed to phyloFit with the species tree, as described above.
Identification of novel Chlamydomonas reinhardtii genes
De novo gene annotation was performed on the C. reinhardtii v5 genome using BRAKER (without UTR annotation) and all RNA-seq datasets produced by Strenkert et al. (2019). Potential novel genes were defined as those without any overlap with CDS of v5.6 genes. To determine if any novel predictions had syntenic homologs within Chlamydomonas, SynChro was re-run against C. incerta and C. schloesseri using updated C. reinhardtii input files containing the potential novel genes. Coding potential was assessed by passing CDS alignments extracted from the WGA to phyloCSF (Lin et al. 2011), which was run in “omega” mode using the neutral branch length tree from phyloFit.
Identification and analyses of conserved elements
CEs were identified from the 8-species WGA using phastCons (Siepel et al. 2005) with the phyloFit neutral model (described above) and the standard UCSC parameters “--expected-length=45, --target-coverage=0.3, --rho=0.31”. Parameter tuning was attempted, but it proved difficult to achieve a balance between overly long CEs containing too many non-constrained bases at one extreme, and overly fragmented CEs at the other, and the standard parameters were found to perform as adequately as others.
C. reinhardtii site classes were delineated using the repeat-filtered v5.6 annotation, augmented with the 142 novel genes identified (file S5). To assess the genomic distribution of conserved bases, site classes were called uniquely in a hierarchical manner, so that if a site was annotated as more than one site class it was called based on the following hierarchy: CDS, 5’ UTR, 3’ UTR, intronic, intergenic. Overlaps between site classes and CEs were calculated using BEDtools v2.26.0 (Quinlan and Hall 2010). Genetic diversity was calculated from re-sequencing data of 17 C. reinhardtii field isolates from Quebec, as described by Craig et al. (2019). For analyses of intron length and conservation, all introns were called based on longest isoforms as they appear in the annotation (i.e. no hierarchical calling was performed as described above).
Supplementary files
supplementary_tables.xlsx
file_S1.pdf: high molecular weight DNA extraction protocol for Chlamydomonas.
file_S2.pdf: detailed genome assembly methods.
file_S3.fa: Volvocales curated TE library.
file_S4.xlsx: Volvocales curated TE annotation notes.
file_S5.gff3: C. reinhardtii v5.6 gene annotation, filtered for TE/repeat genes and with newly identified genes added.
file_S6.txt: OrthoFinder gene clustering used for phylogenomics analyses.
file_S7.fa: aligned and trimmed OrthoFinder single-copy orthologs used for phylogenomics analyses.
file_S8.nwk: IQ-TREE phylogeny produced from OrthoFinder single-copy orthologs.
file_S9.nwk: ASTRAL-III phylogeny produced from OrthoFinder single-copy orthologs.
file_S10.fa: aligned and trimmed chlorophyte BUSCO genes used for phylogenomics analyses.
file_S11.nwk: IQ-TREE phylogeny produced from chlorophyte BUSCO genes.
file_S12.nwk: ASTRAL-III phylogeny produced from chlorophyte BUSCO genes.
file_S13.tsv: C. reinhardtii – C. incerta synteny blocks.
file_S14.tsv: C. reinhardtii – C. incerta syntenic orthologs.
file_S15.tsv: C. reinhardtii – C. schloesseri synteny blocks.
file_S16.tsv: C. reinhardtii – C. schloesseri syntenic orthologs.
file_S17.tsv: C. reinhardtii – E. debaryana synteny blocks.
file_S18.tsv: C. reinhardtii – E. debaryana syntenic orthologs.
file_S19.fa: chlorophyte L1 LINE proteins.
file_S20.fa: aligned and trimmed chlorophyte L1 LINE proteins.
file_S21.nwk: IQ-TREE phylogeny of chlorophyte L1 LINE proteins.
file_S22.txt: OrthoFinder gene clustering of six core-Reinhardtinia species.
file_S23.txt: InterProScan raw output for genes of six core-Reinhardtinia species.
file_S24.tsv: InterPro domains associated with core-Reinhardtinia orthogroups.
file_S25.bed: phastCons conserved elements in C. reinhardtii v5 coordinates.
file_S26.bed: ultraconserved elements in C. reinhardtii v5 coordinates.
Data availability
The 8-species core-Reinhardtinia Cactus WGA and all genome assemblies and annotations are available from the Edinburgh Datashare repository (doi: https://doi.org/10.7488/ds/2847). All sequencing reads, genome assemblies and gene annotations will shortly be available from NCBI under the BioProject PRJNA633871. Code and bioinformatic pipelines are available at: https://github.com/rorycraig337/Chlamydomonas_comparative_genomics
Author contributions
RJC performed analyses and wrote the first draft of the manuscript, with the exception of the analyses and manuscript section on mating-type evolution, which were performed and written by ARH. RJC and RWN performed laboratory work. RJC, RWN and PDK conceived the study. All authors read and commented on the final draft version of the manuscript.
Acknowledgements
We are indebted to Lewis Stevens, Dominik Laetsch and Mark Blaxter for their guidance and advice on all genomics matters. We thank Alexander Suh and Valentina Peona for their invaluable guidance on TE curation, Thomas Pröschold for providing isolate images and for useful discussions on taxonomy, and Olivier Vallon for useful discussions on chlorophyte genomics. We thank Susanne Kraemer and Jack Hearn for their work on an earlier version of the C. incerta genome. Rory Craig is supported by a BBSRC EASTBIO Doctoral Training Partnership grant. PacBio sequencing was funded by a NERC Biomolecular Analysis Facility Pilot Project Grant (NBAF1123).





{kind=link}
{kind=link}
{kind=link}
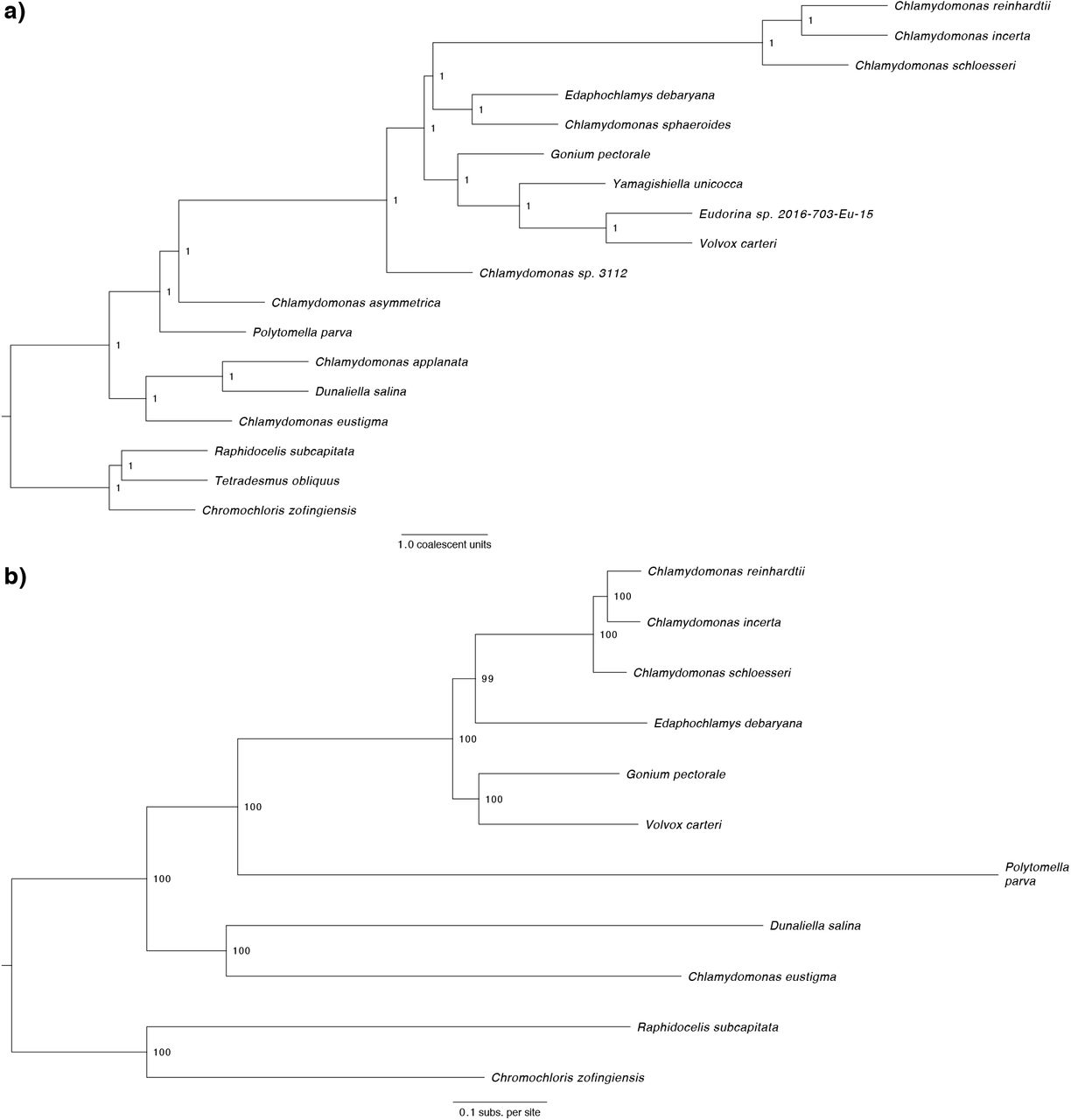{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
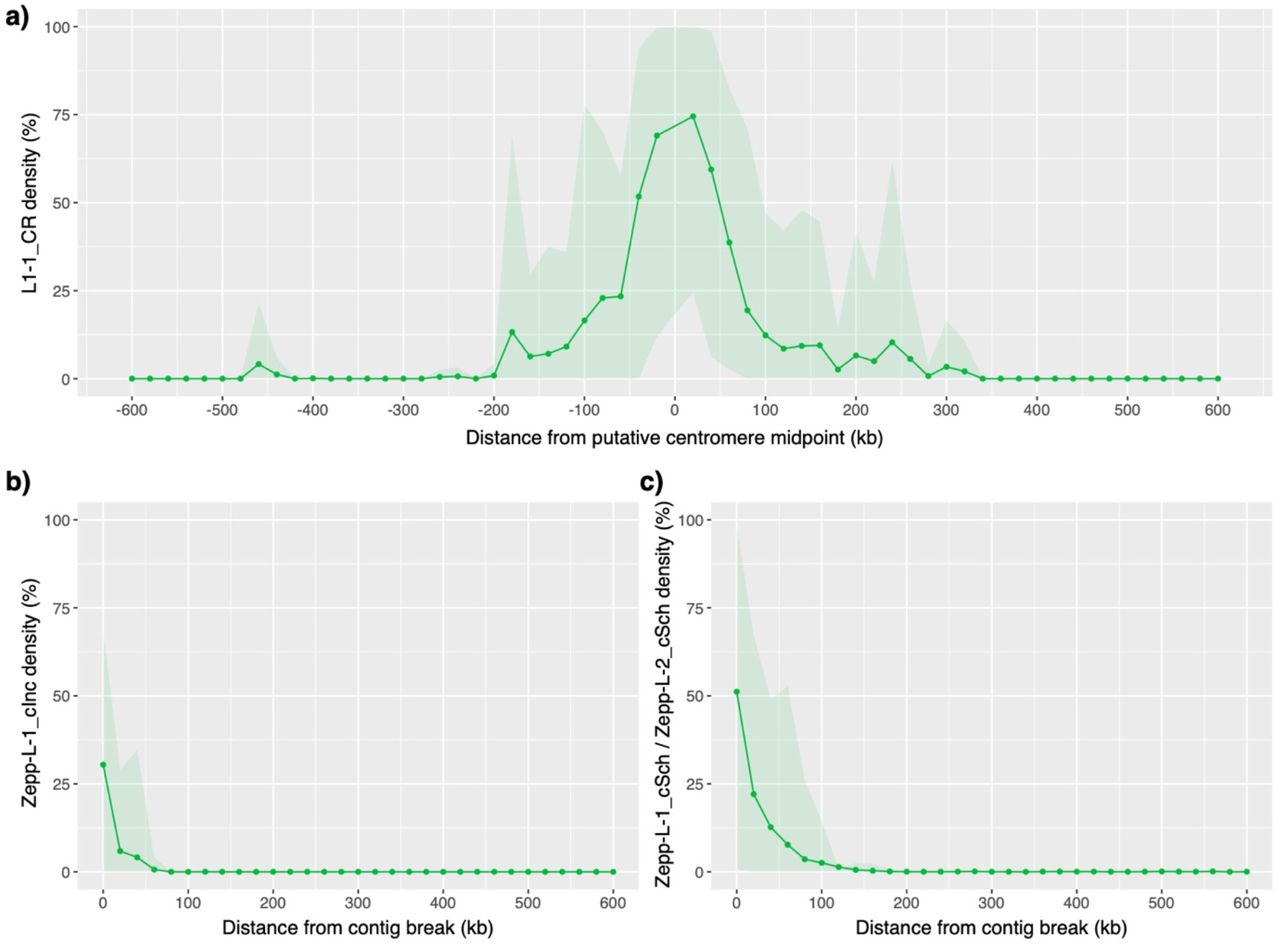{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
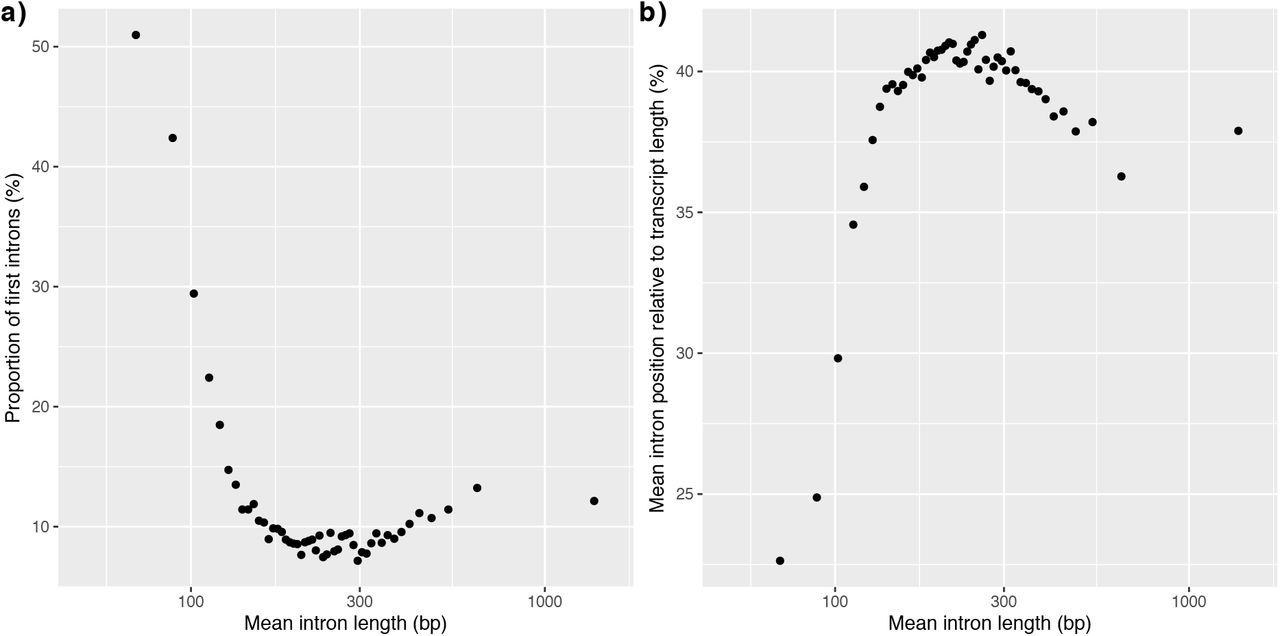{kind=link}