

Abstract
Recently, a novel isolate of the SARS-CoV-2 virus carrying a point mutation in the Spike protein (D614G) has emerged and rapidly surpassed others in prevalence, including the original SARS-CoV-2 isolate from Wuhan, China. This Spike variant is a defining feature of the most prevalent clade (A2a) of SARS-CoV-2 genomes worldwide. Using phylogenomic data, several groups have proposed that the D614G variant may confer increased transmissibility leading to positive selection, while others have claimed that currently available evidence does not support positive selection. Furthermore, in the A2a clade, this mutation is in linkage disequilibrium with a ORF1b protein variant (P314L), making it difficult to discern the functional significance of the Spike D614G mutation from population genetics alone.
Here, we perform site-directed mutagenesis on a human codon-optimized spike protein to introduce the D614G variant and produce SARS-CoV-2-pseudotyped lentiviral particles (S-Virus) with this variant and with D614 Spike. We show that in multiple cell lines, including human lung epithelial cells, that S-Virus carrying the D614G mutation is up to 8-fold more effective at transducing cells than wild-type S-Virus. This provides functional evidence that the D614G mutation in the Spike protein increases transduction of human cells. Further we show that the G614 variant is more resistant to cleavage in vitro and in human cells, which may suggest a possible mechanism for the increased transduction. Given that several vaccines in development and in clinical trials are based on the initial (D614) Spike sequence, this result has important implications for the efficacy of these vaccines in protecting against this recent and highly-prevalent SARS-CoV-2 isolate.
Recently, a novel isolate of the SARS-CoV-2 virus carrying a point mutation in the Spike protein (D614G) has emerged and rapidly surpassed others in prevalence, including the original SARS-CoV-2 isolate from Wuhan, China. This Spike variant is a defining feature of the most prevalent clade (A2a) of SARS-CoV-2 genomes worldwide1,2. Using phylogenomic data, several groups have proposed that the D614G variant may confer increased transmissibility leading to positive selection1,3, while others have claimed that currently available evidence does not support positive selection4. Furthermore, in the A2a clade, this mutation is in linkage disequilibrium with a ORF1b protein variant (P314L)1, making it difficult to discern the functional significance of the Spike D614G mutation from population genetics alone.
Here, we perform site-directed mutagenesis on a human codon-optimized spike protein to introduce the D614G variant5 and produce SARS-CoV-2-pseudotyped lentiviral particles (S-Virus) with this variant and with D614 Spike. We show that in multiple cell lines, including human lung epithelial cells, that S-Virus carrying the D614G mutation is up to 8-fold more effective at transducing cells than wild-type S-Virus. This provides functional evidence that the D614G mutation in the Spike protein increases transduction of human cells. Further we show that the G614 variant is more resistant to cleavage in vitro and in human cells, which may suggest a possible mechanism for the increased transduction. Given that several vaccines in development and in clinical trials are based on the initial (D614) Spike sequence6,7, this result has important implications for the efficacy of these vaccines in protecting against this recent and highly-prevalent SARS-CoV-2 isolate.
The first sequenced SARS-CoV-2 isolate (GenBank accession MN908947.3) and the majority of viral sequences acquired in January and February 2020 contained an aspartic acid at position 614 of the Spike protein (Fig. 1a). Beginning in February 2020, an increasing number of SARS-CoV-2 isolates with glycine at position 614 of the Spike protein were identified. We found that ∼72% of 22,103 SARS-CoV-2 genomes that we surveyed from the GISAID public repository in early June 2020 contained the G614 variant8. Previously, Cardozo and colleagues reported a correlation between the prevalence of the G614 variant and the case-fatality rate in individual localities using viral genomes available through early April 20209. Using a ∼10-fold larger dataset, we found a smaller yet significant positive correlation between the prevalence of G614 in a country with its case-fatality rate (r = 0.29, p = 0.04) (Fig. 1b). There has been little consensus on the potential function of this mutation and whether its spread may or may not be due to a founder effect1,4. Recently, two separate groups at the University of Sheffield and at the University of Washington have found that in COVID-19 patients there is a ∼3-fold increase in viral RNA during quantitative PCR-based testing for those patients with the G614 variant3,10 (Fig. 1c, d). Although there is a consistent difference in qPCR amplification between the sites (∼5 Ct) potentially due to different sampling procedures, RNA extraction methods, qRT-PCR reagents or threshold cycle settings (Fig. 1c), the difference in amplification (ΔΔCt) between G614 and D614 variants is remarkably consistent (1.6 Ct for Sheffield, 1.8 Ct for Washington), suggesting that this may be due to a biological difference between COVID-19 patients with specific Spike variants (Fig. 1d).
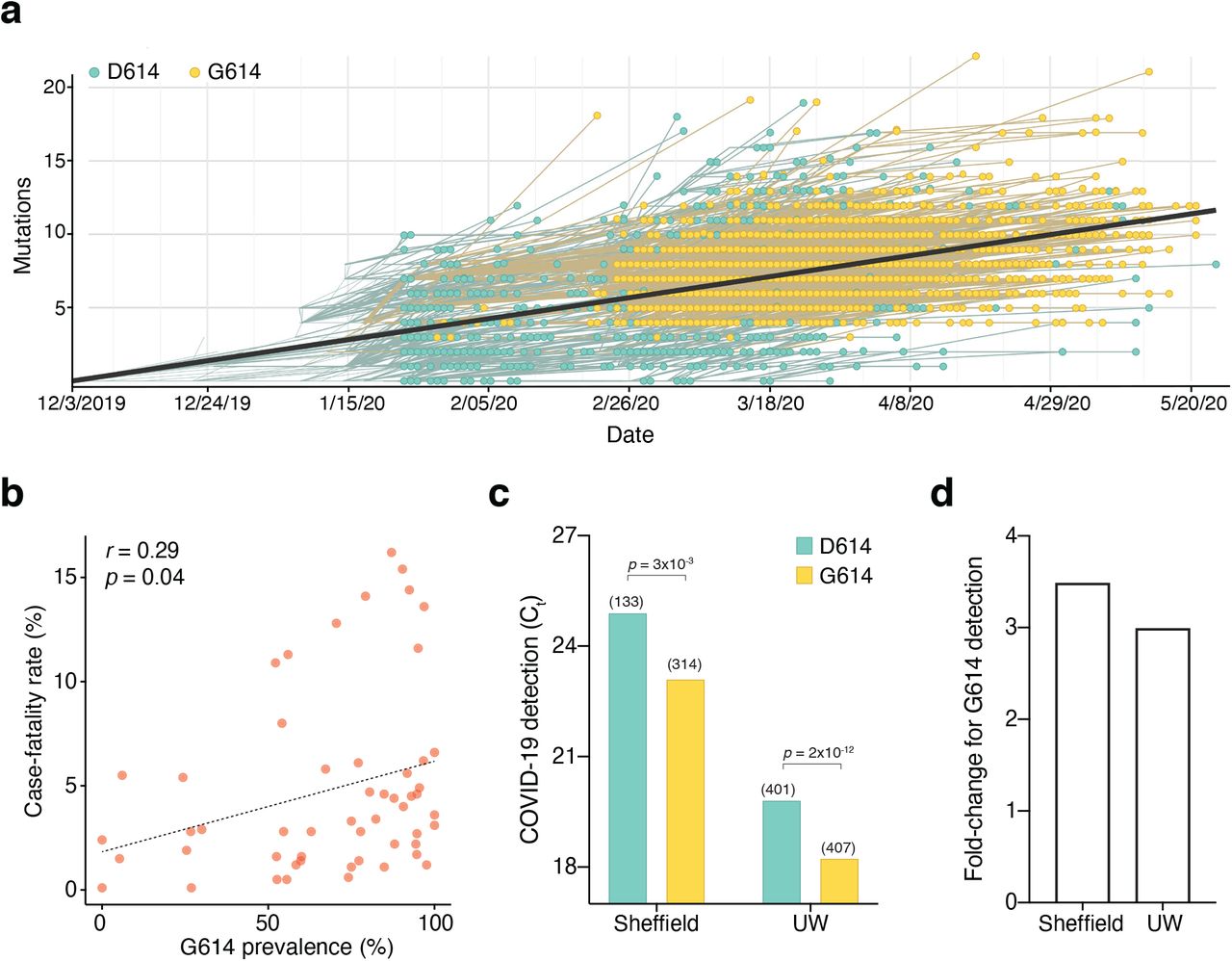
(a) Prevalence of D614G-containing SARS-CoV-2 genomes over time. This visualization was produced by the Nextstrain webtool using GISAID genomes (n = 3,866 genomes samples from January 2020 to May 2020). (b) Per-country correlation of G614 prevalence versus the case-fatality rate (n = 56 countries and 22,103 genomes). (c) Threshold cycle for quantitative polymerase chain reaction (qPCR) detection of COVID-19 from patients with D614 and G614 Spike. Numbers in parentheses indicate the number of COVID-19 patients in each group and significance testing is using the Wilcoxon rank sum test. This Sheffield data was originally presented in Korber et al. (2020). The University of Washington data was originally presented in Wagner et al. (2020). (d) Fold-change of increase in viral RNA present in COVID-19 patient samples with G614 Spike as compared to those with D614 Spike.
Given these findings, we wondered whether the G614 variant may confer some functional difference that impacts viral transmission or disease severity. To address this question, we used a pseudotyped lentiviral system similar to those developed previously for SARS-CoV-111. Using site-directed mutagenesis and a human-codon optimized SARS-CoV-2 spike coding sequence5, we constructed EGFP-expressing lentiviruses pseudotyped with no pseudotype protein or pseudotyped with D614 Spike or G614 Spike (Fig. 2a). After production and purification of these Spike-pseuodtyped viral particles (“S-virus”), we transduced human cell lines derived from lung, liver and colon. Others have observed increased S-virus transduction in cells that overexpress the angiotensin-converting enzyme 2 (ACE2) receptor11,12; we also found that S-virus is much more efficient at transducing human cell lines when the human ACE2 receptor is overexpressed (Supplementary Fig. 1). Given this, for two of the human cell lines (A549 lung and Huh7.5 liver), we overexpressed the ACE2 receptor to boost viral transduction.

(a) Schematic of EGFP lentivirus pseudotyped with SARS-CoV-2 Spike proteins (or no pseudotype) and readout of EGFP fluorescence by flow cytometry. (b) Flow cytometry of A549-ACE2 cells at 3 days post-transduction with 30 or 100 uL SARS-CoV-2 spike pseudotyped lentivirus. (c) Percent of EGFP+ cells at 3 days post-transduction with the indicated volume of virus and pseudotype in human liver Huh7.5-ACE2 cells, lung A549-ACE2 cells, and colon Caco-2 cells (n = 3 replicates, error bars are s.e.m.). (d) The maximum fold-change in viral transduction in each cell line of G614 Spike as compared to D614 Spike (error bars are s.e.m.).
After transduction with 4 different viral volumes, we waited 3 days and then performed flow cytometry to measure GFP expression (Fig. 2b). We found in all 3 cell human cell lines at all viral doses that G614 S-Virus resulted in a greater number of transduced cells than D614 S-virus (Fig. 2c). Unpseudotyped lentivirus resulted in negligible transduction (Fig. 2c). With the G614 Spike variant, the maximum increase in viral transduction over the D614 variant was 2.4-fold for Caco-2 colon, 4.6-fold for A549-ACE2 lung, and 7.7-fold for Huh7.5-ACE2 liver (Fig. 2d). To control for any potential differences in viral titer, we also measured viral RNA content by qPCR. We observed only a small difference between D614 and G614 pseudotyped viruses using 2 independent primer sets (average of 7% higher viral titer for D614), which may result in a slight underestimation of the increase in transduction efficacy of the G614 pseudotyped virus (Supplementary Fig. 2).
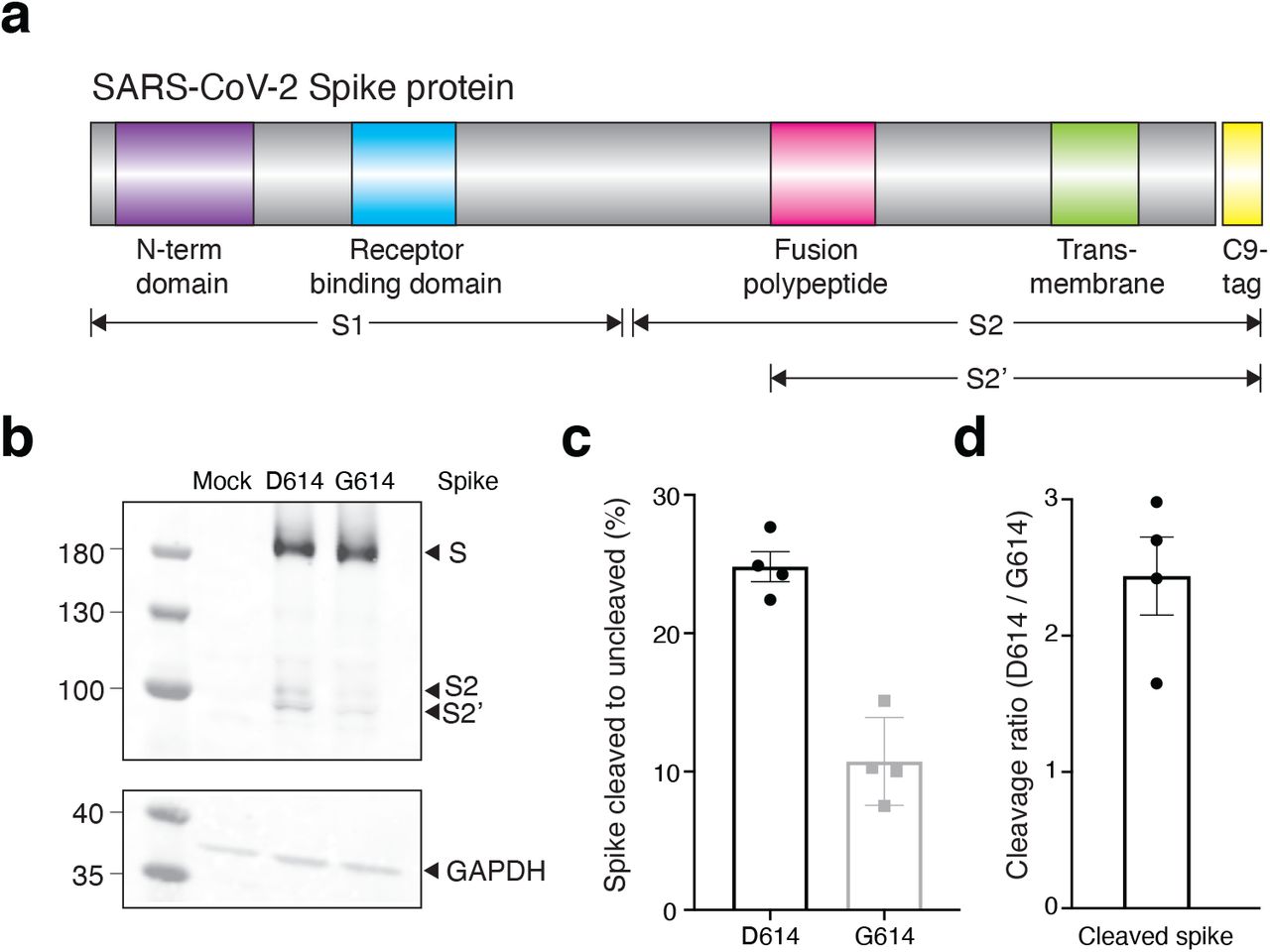
We next sought to understand the mechanism through which the G614 variant increases viral transduction of human cells. Like SARS-CoV-1, the SARS-CoV-2 Spike protein has both a receptor-binding domain and also a hydrophobic fusion polypeptide that is used after binding the receptor (e.g. ACE2) to fuse the viral and host cell membranes13 (Fig. 3a). In order for SARS-CoV-2 to enter cells, the Spike protein must be cleaved at two sites by host proteases. It is thought that Spike must first be cleaved into S1 and S2 fragments, which exposes another cleavage site14,15. The second cleavage event (creating the S2’ fragment) is thought to enable membrane fusion with the host cell. We transfected both D614 and G614 Spike variants into human HEK293FT cells to see if Spike cleavage might differ between these variants. Both constructs were tagged at their C-termini with a C9 tag to visualize full-length, S2, and S2’ fragments via western blot (Fig. 3b). To measure cleavage, we quantified the ratio of cleaved Spike (S2 + S2’) to full-length Spike (Fig. 3c). We found that the G614 variant is ∼2.5-fold more resistant to cleavage in the host cell than the D614 variant (Fig. 3d). This suggests that the 2.4-to 7.7-fold increased transduction observed with G614 S-virus (Fig. 2d) may be due to superior stability and resistance to cleavage of the G614 variant during Spike protein production and viral capsid assembly in host/producer cells.
{kind=link}
{kind=link}
{kind=link}
(a) Schematic diagram of SARS-CoV-2 Spike protein structure with the added C9 affinity tag on the C-terminus. Spike cleavage fragments S1, S2, and S2’ are also indicated. (b) Western blot of total protein lysate from HEK293FT cells after transfection with D614 Spike, G614 Spike, or mock transfection. (upper) Detection of full-length Spike and cleavage fragments using a anti-C9 (rhodopsin) antibody. (lower) Detection of GAPDH via anti-GAPDH antibody. (c) Fraction of cleaved (S2 + S2’) to uncleaved (full-length) fragments for Spike D614 and G614 (n = 4 replicates, error bars are s.e.m.). (d) Fold-change in cleavage between Spike variants (D614 / G614) (n = 4 replicates, error bars are s.e.m.).
Previous work showed that cleavage by the host protease furin at the Spike S1/S2 site in SARS-CoV-2 is essential for cell-cell fusion and viral entry14. To test for differences in furin-mediated cleavage, we performed in vitro digestion of both Spike variants after pull-down. We immunoprecipitated D614 and G614 Spike protein from HEK293FT cell lysates and then performed on-bead digestion using different concentrations of purified furin protease. Over a range of furin concentrations, we found that the G614 variant was more resistant to cleavage than the D614 variant (Supplementary Fig. 3). Importantly, the cleaved S2 and S2’ fragments might still be incorporated into new virions since they contain the required C-terminal transmembrane domain; however, they cannot functionally bind receptor due to lack of a N-terminal receptor binding domain (Fig. 3a). Thus, the greater fraction of uncleaved G614 Spike may allow each newly-assembled virion to include more receptor binding-capable Spike protein.
Given the global efforts underway to develop a COVID-19 vaccine, we also sought to understand the impact of the Spike variant on immune responses. According to the World Health Organization, there are presently 126 COVID-19 vaccine candidates in preclinical development and 10 vaccine candidates in patient-enrolling clinical trials (https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines, accessed June 10th, 2020). Despite the tremendous diversity of vaccine formulations and delivery methods, many of the them utilize Spike sequences (RNA or DNA) or peptides and were developed prior to the emergence of the G614 variant. Using epitope prediction for common HLA alleles16, we found that the G614 variant can alter predicted MHC binding (Supplementary Fig. 4). For example, the predicted binding for one high-affinity epitope decreased by nearly 4-fold (58nM for D614 versus 221nM for G614 with HLA-A*02:01). Although full-length Spike protein likely can produce many immunogenic peptides, several vaccines use only portions of Spike6,7 and thus it may be better to adapt vaccine efforts to the D614 variant given its global spread.
In summary, we have demonstrated that the recent and now dominant mutation in the SARS-CoV-2 spike glycoprotein D614G increases transduction of the virus across a broad range of human cell types, including cells from lung, liver and colon. We also found that G614 Spike is more resistant to proteolytic cleavage during production of the protein in host cells, suggesting that replicated virus produced in human cells may be more infectious due to a greater proportion of functional (uncleaved) Spike protein per virion. One important caveat of this work is that we use a pseudotyped lentivirus model, which has a different virion assembly pathway. It is unclear whether the number of Spike molecules on the pseudotyped lentivirus is comparable to that of the full SARS-CoV-2 virus. Future work with isogenic SARS-CoV-2 strains (differing only at D614G) will be needed to further bolster the functional differences seen in the pseudotyped virions. Of course, even if there is a measurable functional difference, it is still uncertain whether this will have a clinical impact on COVID-19 disease progression. Two studies that have examined potential differences in clinical severity or hospitalization rates did not see a correlation Spike mutation status3,10, although one study found a small but not significant enrichment of G614 mutations among intensive care unit (ICU) patients3.
Given its rapid rise in human isolates and enhanced transduction across a broad spectrum of human cell types, the G614 variant merits careful consideration by biomedical researchers working on candidate therapies, such as those to modulate cellular proteases, and on vaccines that deliver Spike D614 nucleic acids or peptides.
Author contributions
Z.D. and N.E.S. conceived the project and designed the study. Z.D. performed all wet lab experiments. Z.D. and N.E.S. performed analyses. X.G. analyzed SARS-CoV-2 genomes from patient isolates. All authors contributed to drafting and reviewing the manuscript, provided feedback and approved the manuscript in its final form.
Methods
SARS-CoV-2 genome analyses
For temporal tracking of D614G mutations in SARS-CoV-2 genomes, we used the Nextstrain analysis tool (https://nextstrain.org/ncov) with data obtained from GISAID (https://www.gisaid.org/)2,8. With the Nextstrain webtool, we visualized 3,866 genomes using the “clock” layout with sample coloring based on Spike 614 mutation status.
All complete SARS-CoV-2 genomes submitted before June 2nd 2020 were obtained from GISAID. We excluded genomes classified by GISAID as low coverage and downloaded the remaining 23,755 high-coverage genomes. To classify each genome as D614 or G614, we flanked the mutation site with 11-nt of surrounding sequence context on each side and identified genomes matching either mutation. For 1,652 genomes, we could not identify the mutation site and excluded these from further analysis. For the remaining 22,103 genomes, we were able to uniquely classify them as D614 or G614. Case-fatality rate data was downloaded on June 3rd 2020 from the Johns Hopkins Coronavirus Resource Center (https://coronavirus.jhu.edu/data/mortality). For accurate estimation of D614G prevalence, we only included countries with at least 9 genomes in GISAID.
COVID-19 patient quantitative PCR
Threshold cycle data and statistical test results for Sheffield quantitative PCR data from COVID-19 patients is from Korber et al. (2020)3. Threshold cycle data and statistical test results for University of Washington (UW) quantitative PCR data from COVID-19 patients is from Wagner et al. (2020) (https://github.com/blab/ncov-D614G)10. For the Sheffield study, the reported threshold cycle was the median in each group. For the UW study, the reported threshold cycle was the mean in each group. The reported p-values were computed by the respective study authors using the Wilcoxon Rank Sum test.
Cell culture
A549 cells were obtained from ATCC, HEK293FT cells were obtained from Thermo Scientific, and Huh-7.5 and Caco-2 were a kind gift of T. Jordan and B. tenOever (Mt. Sinai). All cells were cultured in D10 media: Dulbecco’s Modified Eagle Medium (Caisson Labs) supplemented with 10% Serum Plus II Medium Supplement (Sigma-Aldrich). Cells were regularly passaged and tested for presence of mycoplasma contamination (MycoAlert Plus Mycoplasma Detection Kit, Lonza).
Spike plasmid cloning and lentiviral production
To express the D614 Spike, we used an existing CMV-driven SARS-CoV-2 plasmid (pcDNA3.1-SARS2-Spike, Addgene 145032)5. To express the G614 Spike, we cloned pcDNA3.1-SARS2-SpikeD614G using the Q5 site-directed mutagenesis kit (NEB E0554S) and the following primers: 5’-CTGTACCAGGgCGTGAATTGCAC-3’ and 5’-CACGGCCACCTGGTTGCT-3’.
To make spike-pseudotyped lentivirus, we co-transfected a d2EGFP-containing transfer plasmid (Addgene 138152) with accessory plasmid psPAX2 (Addgene 12260) and the pseudotyping plasmid (or omitted the pseudotyping plasmid to produce no-pseudotype lentivirus). Briefly, for each virus, a T-225 flask of 80% confluent HEK293T cells (Thermo) was transfected in OptiMEM (Thermo) using 25 μg of the transfer plasmid, 20 μg psPAX2, 22 μg spike plasmid, and 175 μl of linear Polyethylenimine (1 mg/ml) (Polysciences). After 6 hours, media was changed to D10 media, DMEM (Caisson Labs) with 10% Serum Plus II Medium Supplement (Sigma-Aldrich), with 1 % bovine serum albumin (Sigma) added to improve virus stability. After 60 hours, viral supernatants were harvested and centrifuged at 3,000 rpm at 4 °C for 10 min to pellet cell debris debris and filtered using 45 μm PVDF filters (CellTreat). The supernatant was then ultracentrifuged for 2 hours at 100,000g (Sorvall Lynx 6000) and the pellet resuspended overnight at 4 °C in PBS with 1% BSA.
Quantitative PCR (qPCR) of Spike pseudoviruses
Viral RNA was isolated from 100 mL of 100x-concentrated Spike D614 or G614 pseudotyped lentiviruses using 500 mL Trizol (Thermo 15596026) and following the Zymo Direct-zol RNA MicroPrep kit protocol. RNA was eluted with 15 mL RNase-free water. The RNA was then diluted 1:50 and 2 mL were used to perform a one-step qPCR protocol using Luna Universal One-step qPCR kit (NEB). Two primer sets were used: 5’-CGCTATGTGGATACGCTGC-3’ and 5’-GCGAAAGTCCCGGAAAGGAG-3’ that amplify WPRE, and 5’-CGTGCAGCTCGCCGACCAC-3’ and 5’-CTTGTACAGCTCGTCCATGCC-3’ that amplify EGFP. qPCR was performed following the Luna Universal One-step qPCR kit protocol on a ViiA 384-well qPCR machine.
Spike pseudovirus transductions
We plated 50,000 cells per well of a 48-well plate. The cells were transduced the following morning using the indicated pseudotyped lentiviral amounts plus media supplemented with polybrene 8 μg/mL to a final volume of 150 μL per well. The media was changed 8 hours post-transduction. The cells were analyzed by flow cytometry 72 hours post-transduction.
ACE2 lentiviral cloning and ACE2 stable cell line overexpression
To generate pLenti-ACE2-Hygro, we amplified human ACE2 (hACE2) from pcDNA3.1-ACE2 (Addgene 1786) and cloned it into a lentiviral transfer pLEX vector carrying the hygromycin resistance gene using Gibson Assembly Master Mix (NEB E2611L). A 2A epitope tag was added to hACE2 at the C-terminus. Huh7.5-ACE2 and A549-ACE2 cell lines were generated by lentiviral transduction of ACE2. The protocol for lentiviral production was the same as above except we used the common lentiviral pseudotype (VSV-g) using plasmid pMD2.G (Addgene 12259). Transduced cells were selected with hygromycin at 50 ug/mL for Huh7.5-ACE2 and 500 ug/mL for A549-ACE2 for 10 days before use.
Flow cytometry of transduced human cells
Cells were harvested and washed with Dulbecco’s phosphate-buffered saline (Caisson Labs) twice. Cell acquisition and sorting was performed using a Sony SH800S cell sorter with a 100 μm sorting chip. We used the following gating strategy: 1) We excluded the cell debris based on the forward and reverse scatter; 2) Doublets were excluded. For all samples, we recorded at least 5000 cells that pass the gating criteria described above. Gates to determine GFP+ cells were set based on control GFP− cells, where the percent of GFP+ cells was set as <0.5% (background level). Flow cytometry analyses were performed using FloJo v10.
Protein expression of ACE2 and spike variants in human cells
HEK293FT cells were transiently transfected with equal amounts of spike or ACE2 vectors using PEI. Cells were collected 18-24 hours post-transfection with TrypLE (Thermo), washed twice with PBS (Caisson Labs) and lysed with TNE buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1mM EDTA, 1% Nonidet P-40) supplemented with protease inhibitor cocktail (Bimake B14001) for 1 hour on a rotator at 4°C. Cells lysates were spun for 10 min at 10,000 g at 4°C, and protein concentration was determined using the BCA assay (Thermo 23227). Whole cell lysates (10 μg protein per sample) were denatured in Tris-Glycine SDS sample buffer (Thermo LC2676) and loaded on a Novex 4-12% Tris-Glycine gel (Thermo XP04122BOX). PageRuler pre-stained protein ladder (Thermo 26616) was used to determine the protein size. The gel was run in 1x Tris-Glycine-SDS buffer (IBI Scientific IBI01160) for about 120 min at 120V. Protein transfer was performed using nitrocellulose membrane (BioRad 1620112) using prechilled 1x Tris-Glycine transfer buffer (Fisher LC3675) with 20% methanol for 100 min at 100V. Membranes were blocked with 5% skim milk dissolved in PBST (1x PBS + 1% Tween 20) at room temperature for 1 hour. Primary antibody incubations were performed overnight at 4°C using the following antibodies: rabbit anti-GAPDH 14C10 (0.1 μg/mL, Cell Signaling 2118S), mouse anti-rhodopsin antibody clone 1D4 (1 μg/mL, Novus NBP1-47602) which recognizes the C9-tag added to the Spike proteins. Following the primary antibody, the blots were incubated with IRDye 680RD donkey anti-rabbit (0.2 μg/mL, LI-COR 926-68073) or with IRDye 800CW donkey anti-mouse (0.2 μg/mL, LI-COR 926-32212) for 1 hour at room temperature. The blots were imaged using Odyssey CLx (LI-COR). Band intensity quantification was performed by first converting Odyssey multichannel TIFFs into 16-bit grayscale image (Fiji) and the then selecting lanes and bands in ImageLab 6.1 (BioRad). In ImageLab, background subtraction was applied uniformly across all lanes on the same gel.
On-bead Furin digestion of Spike protein
We transiently transfected 10-cm plates with 80% confluent HEK293FT with 10 μg of either spike D614 or G614 using PEI. About 24 hours later, cells were collected and lysed with 800 mL TNE buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1mM EDTA, 1% Nonidet P-40) supplemented with protease inhibitor cocktail (Bimake B14001) for 1 hour on a rotator at 4°C. Cells lysates were spun for 10 min at 10,000 g at 4°C. Spike was immunoprecipitated using 2 μg C9 antibodies (Novus NBP1-47602) per sample and incubated on a rotator at 4°C for at least 4 hours.
Recombinant Protein G Sepharose 4B beads (Thermo 101241) were washed twice with 1 mL TNE buffer and then were added to the immunoprecipitated cell lysate and incubated on a rotor at 4°C for 2 hours. Beads were then spun using a prechilled centrifuge at 4°C for 1 min at 2,000 rpm and washed 3x with 1 mL TNE. After the final spin, the beads were washed twice with 1 mL of furin reaction buffer (100 mM HEPES pH 7.5, 1 mM CaCl2, 1 mM b-Mercaptoethanol). Finally, the beads were resuspended in 150 μL and split equally in microcentrifuge tubes. The indicated amount of furin protease (NEB P8077) was added per reaction tube in a final volume of 20 μL. The reaction was incubated at 37°C for 1 hour and was occasionally mixed by gently tapping the tubes. Then the beads were denatured in Tris-Glycine SDS sample buffer (Thermo LC2676) and incubated at 95°C for 5 min. Samples were then loaded on a Novex 4-12% Tris-Glycine gel (Thermo XP04122BOX). Western blotting was performed as described above using mouse anti-rhodopsin antibody clone 1D4 (1 μg/mL, Novus NBP1-47602) which recognizes the C9-tag added to the Spike proteins. Following the primary antibody, the blots were incubated with IRDye 800CW donkey anti-mouse (0.2 μg/mL, LI-COR 926-32212) for 1 hour at room temperature. The blots were imaged using Odyssey CLx (LI-COR). Band intensity quantification was performed by first converting Odyssey multichannel TIFFs into 16-bit grayscale image (Fiji) and the then selecting lanes and bands in ImageLab 6.1 (BioRad). In ImageLab, background subtraction was applied uniformly across all lanes on the same gel.
Epitope prediction using NetMHC
Since 9mer epitopes are most commonly presented by MHC receptors17, we constructed all possible 9mers surrounding the D/G 614 site in the Spike protein. We predicted binding affinities for 5 common HLA-A alleles and 7 common HLA-B alleles using the NetMHC 4.0 prediction webserver16 (http://www.cbs.dtu.dk/services/NetMHC/). For each peptide, we computed the difference in predicted affinity between the D614 and G614 variant using R/RStudio and visualized them using the pheatmap R package.
Statistical analysis
Data analysis was performed using R/Rstudio 3.6.1 and GraphPad Prism 8 (GraphPad Software Inc.). Specific statistical analysis methods are described in the figure legends where results are presented. Values were considered statistically significant for p values below 0.05.
Reagent availability
All plasmids cloned for this study will be available on Addgene.
Acknowledgements
We thank the entire Sanjana laboratory for support and advice. We are grateful to B. tenOever, T. Jordan, T. Maniatis, M. Legut, K. McGhee, C. Lu, and M. Prober for help with this work. Z.D. is supported by an American Heart Association postdoctoral fellowship. N.E.S. is supported by New York University and New York Genome Center startup funds, National Institutes of Health (NIH)/National Human Genome Research Institute (grant nos. R00HG008171, DP2HG010099), NIH/National Cancer Institute (grant no. R01CA218668), Defense Advanced Research Projects Agency (grant no. D18AP00053), the Sidney Kimmel Foundation, the Melanoma Research Alliance, and the Brain and Behavior Foundation.
References




