

Abstract
Arbuscular mycorrhizal (AM) symbiosis is a mutually beneficial association of plants and fungi of the sub-phylum Glomeromycotina. The endosymbiotic AM fungi colonize the inner cortical cells of the roots, where they form branched hyphae called arbuscules that function in nutrient exchange with the plant. To support arbuscule development and subsequently bidirectional nutrient exchange, the root cortical cells undergo substantial transcriptional re-programming. REDUCED ARBUSCULAR MYCORRHIZA 1 (RAM1), studied in several dicot plant species, is a major regulator of this cortical cell transcriptional program. Here, we generated ram1 mutants and RAM1 overexpressors in a monocot, Brachypodium distachyon. The AM phenotypes of two ram1 lines revealed that RAM1 is only partly required to enable arbuscule development in B. distachyon. Transgenic lines constitutively overexpressing BdRAM1 showed constitutive expression of AM-inducible genes even in the shoots. Following inoculation with AM fungi, BdRAM1-overexpressing roots showed higher arbuscule densities relative to controls, indicating the potential to manipulate the relative proportion of symbiotic interfaces via modulation of RAM1. However, the overexpressors also show altered expression of hormone biosynthesis genes and aberrant growth patterns including stunted bushy shoots and poor seed set. While these phenotypes possibly provide additional clues about BdRAM1’s scope of influence, they also indicate that directed approaches to increase the density of symbiotic interfaces will require a more focused, potentially cell-type specific manipulation of transcription factor gene expression.
Introduction
The GRAS (for GA3 INSENSITIVE [GAI], REPRESSOR OF GAI [RGA], and SCARECROW [SCR]) transcription factor REDUCED ARBUSCULAR MYCORRHIZA (RAM1) has been characterized in three dicot plant species where it is a major regulator of AM symbiosis. In Medicago truncatula, Lotus japonicus, and Petunia hybrida ram1 mutants, AM fungi display limited arbuscule branching and reduced hyphal colonization of the root, which results in a non-functional symbiosis (Gobbato et al., 2013; Park et al., 2015; Rich et al., 2015; Xue et al., 2015; Pimprikar et al., 2016). RAM1 expression is induced in colonized cortical cells and is regulated by CYCLOPS, a transcription factor of the common symbiosis signaling pathway (Pimprikar et al., 2016) and also by DELLA proteins (Park et al., 2015; Pimprikar et al., 2016), negative regulators of GA signaling (Daviere and Achard, 2013). RNA sequencing of ram1 mutants (Luginbuehl et al., 2017) as well as smaller scale gene expression analyses of roots overexpressing RAM1 (Park et al., 2015; Jiang et al., 2017), indicate that RAM1 either directly or indirectly regulates expression of several symbiosis-associated transcription factors, including the GRAS transcription factor RAD1, and three AP2-domain transcription factors of the WRINKLED5 (WRI5) family. RAM1 also either directly or indirectly regulates expression of genes involved in the production and transfer of lipids to the fungal symbiont (e.g. FatM, RAM2, STR) and the phosphate transporter PT4 (Gobbato et al., 2012; Park et al., 2015; Pimprikar et al., 2016; Jiang et al., 2017; Luginbuehl et al., 2017). However, so far, only one lipid biosynthesis gene, RAM2, has been established as a direct target of RAM1 (Gobbato et al., 2012). Regulation of the other lipid biosynthesis and transport genes likely occurs indirectly through the action of the WRI5 family genes (Luginbuehl et al., 2017; Jiang et al., 2018).
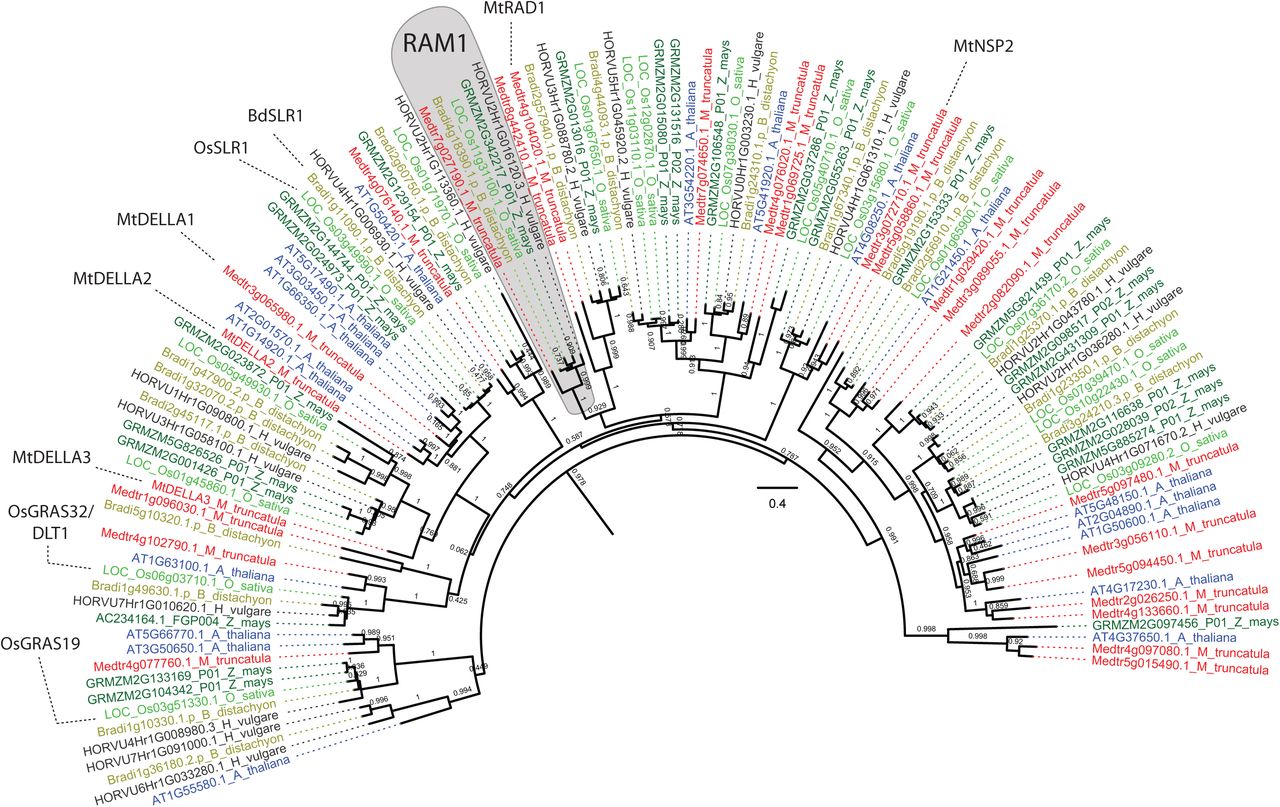
RAD1, a GRAS transcription factor very closely related to RAM1 (Supplemental Fig. 1) (Park et al., 2015; Xue et al., 2015) is also required for AM symbiosis. In L. japonicus rad1 mutants, AM fungi display defective arbuscule branching phenotypes reminiscent of those seen in ram1 (Xue et al., 2015); however, in M. truncatula rad1, AM fungi show normal arbuscule branching but reduced colonization levels (Park et al., 2015). In line with this observation, several predicted RAM1 target genes were induced in colonized L. japonicus ram1 mutants but induction was completely abolished in M. truncatula and P. hybrida ram1 (Park et al., 2015; Rich et al., 2015; Pimprikar et al., 2016; Luginbuehl et al., 2017). Thus, there are slight differences in regulation of AM symbiosis genes even between relatively closely related plant species (Pimprikar and Gutjahr, 2018).

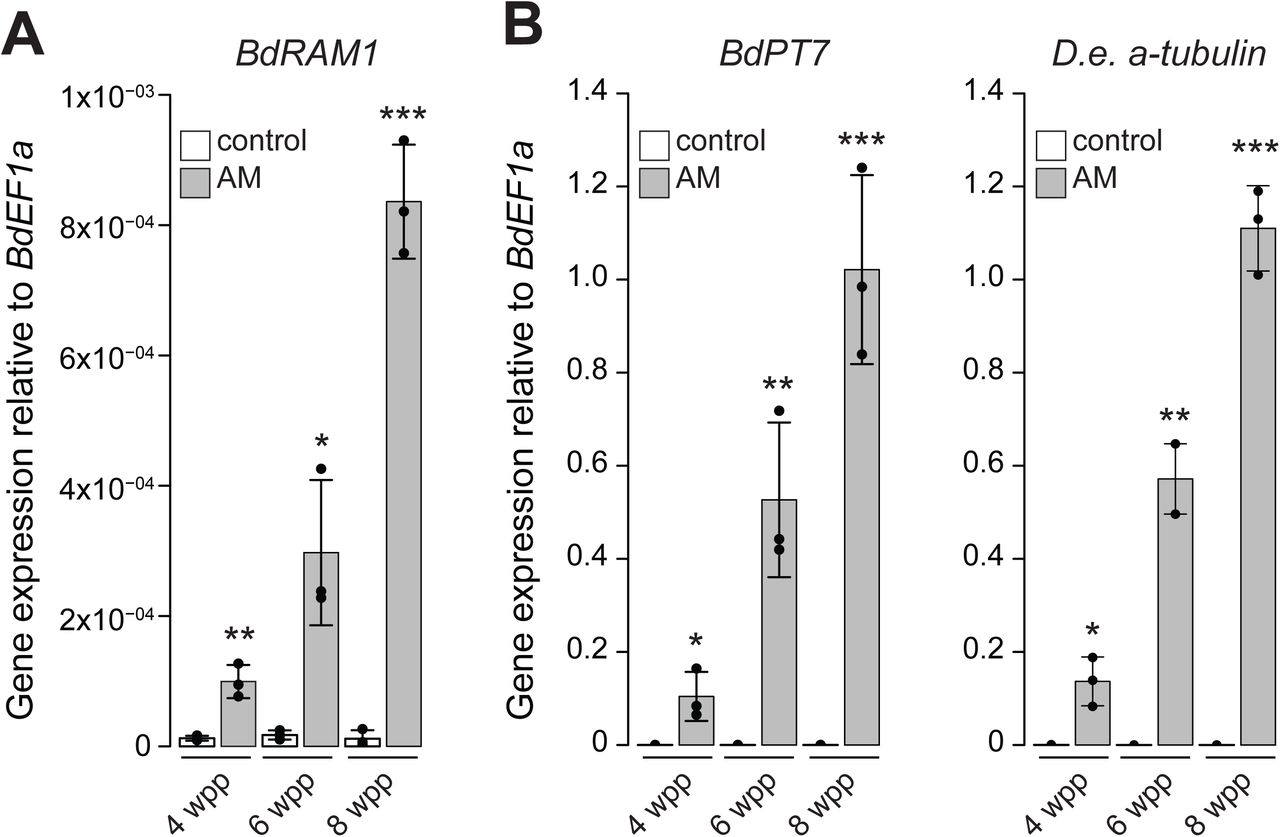
A) BdRAM1 gene expression is induced in roots colonized by the AM fungus D. epigaea (grey bars, “AM”) relative to non-mycorrhizal, mock-inoculated roots (white bars, “control”). Plants were harvested 4, 6, and 8 weeks post planting (wpp). AM-induced BdRAM1 gene expression increases over time. B) Gene expression of the AM marker genes BdPT7 and D. epigaea (D.e.) a-tubulin in D. epigaea-colonized and mock-inoculated control roots over time. A) and B) Gene expression was normalized to the B. distachyon elongation factor BdEF1a. Bar graphs show the mean, error bars the standard deviation. Single points represent individual measurements. Pairwise comparisons of gene expression in AM and control roots were analyzed separately for each time point (Student’s t-test). Significance values: ***p<0.001; **p<0.01; *p<0.05.
Several other GRAS proteins are essential for AM symbiosis including DELLA/SLR1, a negative regulator of GA signaling (Floss et al., 2013; Foo et al., 2013; Yu et al., 2014; Floss et al., 2017). In della mutants, AM fungi show a severely reduced ability to enter cortical cells, and as a result almost no arbuscules are formed (Floss et al., 2013; Foo et al., 2013; Yu et al., 2014). Arbuscules are ephemeral structures, and the few arbuscules that are formed in della mutants display an increased lifespan, indicating that DELLA not only regulates arbuscule formation but also their degradation (Floss et al., 2017). Two other GRAS transcription factors critical for hormone signaling and AM symbiosis are NSP1 and NSP2. These transcription factors regulate phosphate-dependent strigolactone (SL) biosynthesis in M. truncatula and rice (Liu et al., 2011). SLs serve as direct plant communication molecules with AM fungi at the onset of the symbiosis. Mutants impaired in NSP or enzymes required for SL biosynthesis show a reduction in fungal entry into the root and consequently reduced colonization (Gomez-Roldan et al., 2008; Liu et al., 2011; Kobae et al., 2018). Thus, there are several examples of GRAS factors that connect hormone signaling and AM symbiosis.
Many GRAS factors operate in complexes with other GRAS proteins and emerging evidence suggests that this is also true of those involved in AM symbiosis. M. truncatula and L. japonicus RAM1 were reported to interact with RAD1 and NSP2 (which also interact with each other), but not NSP1 (Gobbato et al., 2012; Park et al., 2015; Xue et al., 2015; Heck et al., 2016). In addition, rice RAM1 interacts with the GRAS transcription factor DIP1, which in turn interacts with DELLA (Yu et al., 2014). M. truncatula DELLA proteins were found to interact with numerous other GRAS transcription factors, including RAD1, MIG1, NSP1, and NSP2 (Floss et al., 2016; Fonouni-Farde et al., 2016; Heck et al., 2016; Jin et al., 2016). While their functional significance for symbiosis remains to be determined, the interactions suggest the existence of interconnected transcriptional modules regulated by multiple GRAS transcription factors.
Brachypodium distachyon is a monocot model species capable of forming AM symbiosis (Hong et al., 2012) and amenable to genetic manipulation (Bragg et al., 2015). A recent study identified 48 GRAS transcription factors in the genome of B. distachyon (Niu et al., 2019). Here we report functional analyses of the GRAS transcription factor RAM1 in a monocot and assess the potential to alter the levels of symbiotic interfaces by manipulating RAM1 expression.
Results and Discussion
We identified Bradi4g18390 as the single B. distachyon homolog of the GRAS transcription factor RAM1 (Supplemental Fig. 1), a gene that is conserved in AM host plants and missing from non-hosts (Bravo et al., 2016). Similar to orthologous RAM1 genes of M. truncatula (Gobbato et al., 2013; Park et al., 2015), L. japonicus (Xue et al., 2015; Pimprikar et al., 2016) and P. hybrida (Rich et al., 2015), B. distachyon RAM1 expression is induced in mycorrhizal roots. Following inoculation with the AM fungus Diversispora epigaea (formerly Glomus versiforme), BdRAM1 transcripts increased over time in parallel with increasing colonization of the root system as reported by D. epigaea a-tubulin transcripts and the phosphate transporter gene BdPT7 (Hong et al., 2012), a plant gene marker of AM symbiosis (Figure 1A, B). However, while the transcriptional patterns mirrors the marker genes, it is noticeable that BdRAM1 transcript levels are low, as is often the case for transcriptional regulators.
The role of RAM1 in AM has been established in at least three dicot host plants (Gobbato et al., 2013; Park et al., 2015; Rich et al., 2015; Xue et al., 2015; Pimprikar et al., 2016) where it is essential to support arbuscule development and appears to act in the upper tier of a transcription factor hierarchy (Luginbuehl et al., 2017); when ectopically over-expressed in roots, RAM1 is sufficient to induce expression of several AM-induced genes in the absence of symbiosis (Park et al., 2015; Pimprikar et al., 2016). Given its AM-inducible expression and pivotal regulatory role, we hypothesized that constitutive, high-level expression of RAM1 might increase the occurrence of arbuscules and possibly overall colonization levels, and this might provide an opportunity to evaluate the functional consequences of modifying colonization patterns. To test this hypothesis, we transformed B. distachyon with an overexpression construct, BdRAM1 under the control of two copies of the constitutively active CaMV 35S promoter (35S:BdRAM1). In addition, we generated B. distachyon ram1 loss-of-function mutants via CRISPR/Cas9 editing.
Arbuscule development in B. distachyon ram1 mutants is partly impaired
Five independent transgenic lines carrying a CRISPR/CAS9 construct targeting BdRAM1 were generated and two lines in which BdRAM1 had been edited were chosen for subsequent analysis. In both transgenic lines, the genome had been edited by both guides (Supplemental Fig. 2); editing by the upstream-most guide resulted in premature stop codons and created a truncated protein of 16 amino acids in the first line, designated ram1-1. The second line, designated ram1-2, was bi-allelic with edits resulting in premature stop codons that generated truncated protein products of 16 and 42 amino acids.

A) A root piece with “ram1-like” D. epigaea arbuscules in CRISPR ram1 mutants. “Ram1-like” infections contain solely arbuscules that are not fully developed and show only sparse branching (asterisks). B) A root piece with “wild-type-like” D. epigaea arbuscules in CRISPR ram1 mutants. “Wild-type-like” infections contain fully developed arbuscules (arrows). A) and B), D. epigaea fungal structures are visualized using WGA-Alexafluor488 (green), plant cell walls were counter-stained using Propidium Iodide (pink). C) Quantification of “ram1-like” infections relative to the total number of infections in ram1 CRISPR plants and B. distachyon plants transformed with the empty vector (E.V.). The proportion of aberrant infections is increased in two ram1 alleles (ANOVA p=2.38×10−5). D) Quantification of total D. epigaea root length colonization in CRISPR ram1 plants relative to E.V.-controls. Root length colonization is significantly decreased in two ram1 mutant alleles (ANOVA p=0.0014). Pairwise comparisons in C) and D) were performed using Tukey’s HSD post-hoc test; different letters denote significant differences. Box-and-whiskers plots show lower and upper quartiles, and minimum and maximum values. The horizontal bar represents the median, and the points individual measurements. All results presented in this figure were obtained from the T3 generation.
ram1-1 and ram1-2 were inoculated with D. epigaea and the fungal colonization patterns were examined. Some ram1 roots showed aberrant infection units, reminiscent of the typical dicot ram1 phenotype, with intraradical hyphae and only small, sparsely branched arbuscules but no fully developed arbuscules (Figure 2A). However, infection units in other roots of the same plant, or even in other parts of the same root, showed an apparent wild-type morphology with some large, well-branched arbuscules (Figure 2B). In comparison with the empty vector control (E.V.) the frequency of aberrant infections in the B. distachyon ram1 mutants was 2.5 to 3-fold higher and their overall root colonization levels were 34 to 52% lower. Similar results were obtained in several experiments across several generations (Supplemental Fig. 2). These results indicate that BdRAM1 is required to enable wild-type levels of arbuscule development similar to its orthologs in dicots; however, the B. distachyon ram1 phenotype is clearly milder than that observed in dicot ram1 mutants. The finding that B. distachyon ram1 can support some full arbuscule development suggests that other proteins or pathways have the potential to compensate for loss of BdRAM1 function. One possible candidate is the GRAS protein RAD1, which is closely related to RAM1 and induced in roots highly colonized by AM fungi (Supplemental Fig. 1, Supplemental Fig. 3). In legumes, there is evidence of a species-specific “micro-diversification” of RAM1 and RAD1, with the relative contributions of the two transcription factors to arbuscule development and symbiotic gene expression varying depending on the host species (Park et al., 2015; Xue et al., 2015; Pimprikar et al., 2016; Pimprikar and Gutjahr, 2018). It is therefore conceivable that some diversification of GRAS factor functions has occurred during the evolution of monocots, which might explain the milder arbuscule development phenotype of B. distachyon ram1 mutants relative to M. truncatula ram1. Interestingly, there are other GRAS factor examples where the converse is true, for example the DELLA proteins, where rice slr (Yu et al., 2014) shows a stronger phenotype than the M. truncatula della double or triple mutants (Floss et al., 2013; Floss et al., 2017). However, in the absence of other monocot ram1 mutants for comparison, it is also possible that the milder ram1 phenotype observed here is a feature specific to B. distachyon.

A) Gene expression levels of B. distachyon orthologs of BdRAM1 target genes in non-colonized 35S:NLS-GFP (denoted as “GFP”) and 35S:BdRAM1ox line #1 (denoted as “ox1”) roots. 35S:BdRAM1ox roots display induced expression of BdRAM1, as well as BdRAM2, BdPT7, BdFatM1, and BdFatM2 in the absence of symbiosis relative to 35S:NLS-GFP control roots. BdSTR gene expression is not affected in these roots. B) Gene expression of B. distachyon RAD1 and WRI5 orthologs. Only expression of BdWRI5.1 is induced in non-colonized roots over-expressing BdRAM1 (35S:BdRAM1ox) relative to 35S:NLS-GFP control roots. A), B), Bar graphs show the mean, error bars the standard deviation. Single points represent individual measurements. Pairwise comparisons were estimated using the Student’s t-test. Significance codes: ***p<0.001; **p<0.01; *p<0.05; n.s., not significant. C) Quantification of total root colonization in independent lines transformed with 35S:BdRAM1. There is no difference in overall root colonization between three lines ectopically over-expressing 35S:BdRAM1 (35S:BdRAM1ox, denoted as “ox1”, “ox2”, “ox3”) and control plants (35S:BdRAM1WT, which does not over-express BdRAM1 and was therefore denoted as “WT”; and 35S:NLS-GFP, labeled as “GFP”). ANOVA p=0.71. Root length colonization was quantified using the grid-line method (McGonigle et al., 1990). D) Quantification of D. epigaea arbuscules in a defined area at the fungal appressorium. Roots of 3 independent transgenic 35S:BdRAM1ox lines (denoted as “ox1”, “ox2”, ox3”) contain more arbuscules than roots transformed with the control construct 35S:NLS-GFP (“GFP”), or roots that contain 35S:BdRAM1 but do not overexpress the gene (“WT”). Arbuscule number was normalized to the volume of the confocal stack. Kruskal-Wallis test p=1.32×10−4, pairwise comparisons were conducted using the Dunn’s post-hoc test; different letters denote significant differences. C) and D), Box-and-whiskers plots show lower and upper quartiles, and minimum and maximum values. The horizontal bar represents the median, and the points individual measurements. E) Representative image of a 35S:NLS-GFP root colonized by D. epigaea. F) Representative image of a RAM1-overexpressing 35S:BdRAM1ox (line #1) root colonized by D. epigaea. E) and F), arbuscules are highlighted with arrows.
Overexpression of RAM1 alters plant morphology and results in constitutive expression of AM marker genes
The generation of transgenic B. distachyon plants overexpressing RAM1 was surprisingly challenging; from two, full-scale independent transformation experiments, only three viable independent transgenic 35S:BdRAM1 overexpressing lines (35S:BdRAM1ox) were obtained and the seed production from these lines was exceedingly poor. In addition, we obtained two lines, which carried the 35S:BdRAM1 T-DNA but displayed wild type-like BdRAM1 transcript levels (35S:BdRAM1WT). By contrast, transgenic plants carrying 35S:NLS-GFP-GUS (hereto referred to simply as 35S:NLS-GFP), were generated without difficulty. Seed production from the latter two genotypes was not impaired.
In addition to poor seed production and viability, the shoot and root phenotypes of the three lines with transcriptional up-regulation of BdRAM1 (35S:BdRAM1ox) differed from the vector controls and from the 35S:BdRAM1WT plants. The 35S:BdRAM1ox plants were characterized by a stunted bushy shoot with increased tiller formation and increased leaf angles, as well as a decreased number of node roots (Figure 4A, Supplemental Fig. 4). Thus, constitutive, ectopic, overexpression of BdRAM1, clearly influences plant development. While the cause is unknown, it might be the result of ectopic expression of BdRAM1 target genes and/or perhaps an interference of RAM1 with other GRAS transcriptional networks, many of which regulate development (reviewed in (Cenci and Rouard, 2017). Either way, the aberrant developmental phenotypes likely explain the difficulties in regenerating transgenic lines and their fecundity.

A) Photograph of 4.5 week-old B. distachyon plants transformed with 35S:NLS-GFP or 35S:BdRAM1. The three independent transformant lines overexpressing BdRAM1 (“ox1”, “ox2”, “ox3”) display a bushy stature, whereas the 35S:BdRAM1-transformant line not overexpressing BdRAM1 (“WT”) resembles the 35S:NLS-GFP control plant. B) Gene expression of BdRAM1 and of several root AM marker genes and BdWRI5.1 is strongly induced in in 4.5-week shoots of three 35S:BdRAM1ox lines (“ox1”, “ox2”, “ox3”) relative to control plants transformed with 35S:NLS-GFP (“GFP”) or the 35S:BdRAM1-transformant line not overexpressing BdRAM1 (“WT”). Bar graphs show the mean, error bars the standard deviation. Single points represent individual measurements. Significance values (ANOVA) for each gene are indicated in the figure. Pairwise comparisons were conducted using Tukey’s HSD post-hoc test; different letters denote significant differences.
The 35S:BdRAM1ox plants showed constitutive expression of B. distachyon orthologs of RAM2, STR, PT4 and FatM (Figure 3A); elevated expression of these genes would normally occur only in response to colonization by AMF (for example, Harrison et al., 2002; Paszkowski et al., 2002; Gutjahr et al., 2008; Zhang et al., 2010; Gobbato et al., 2012; Gutjahr et al., 2012; Hong et al., 2012; Bravo et al., 2017), and we observed a similar expression pattern of their orthologs in B. distachyon roots (Figure 1B, Supplementary Fig. 3). Given the prior knowledge from dicots, we had anticipated that 35S:BdRAM1ox would increase expression of these genes in roots, but it was surprising to see that expression of these genes was also induced in shoots (Figure 4B). There were some exceptions, expression of BdSTR increased in 35S:BdRAM1ox shoots, but not in roots while BdFatM2 showed the opposite expression pattern (Figure 3A, Figure 4B, Supplemental Fig. 5). Overall, these data indicate that BdRAM1 alone is sufficient to drive increased expression of these genes in the absence of AM fungi and that transcription co-factors - if required for BdRAM1 function - must be present in all tissues.

A) B. distachyon orthologs of three genes involved in the Strigolactone biosynthesis pathway (BdD27, BdD17, Bd10) are down-regulated in non-colonized roots ectopically overexpressing BdRAM1 (35S:BdRAM1ox line#1, denoted as “ox1”) relative to 35S:NLS-GFP (“GFP”) control roots. B) Two genes with a putative function in Gibberellic acid biosynthesis (BdGA3ox1, BdGA20ox1) are down-regulated in 35S:BdRAM1ox roots. C) Two B. distachyon genes orthologous to known Brassinosteroid biosynthesis genes (BdCPD, BdD2/BdCYP91D) are induced in 35S:BdRAM1ox roots. A third gene, BdDWF4, is not affected. Bar graphs show the mean, error bars the standard deviation. Single points represent individual measurements. Pairwise comparisons were estimated using the Student’s t-test. Significance codes: ***p<0.001; **p<0.01; *p<0.05; n.s., not significant.
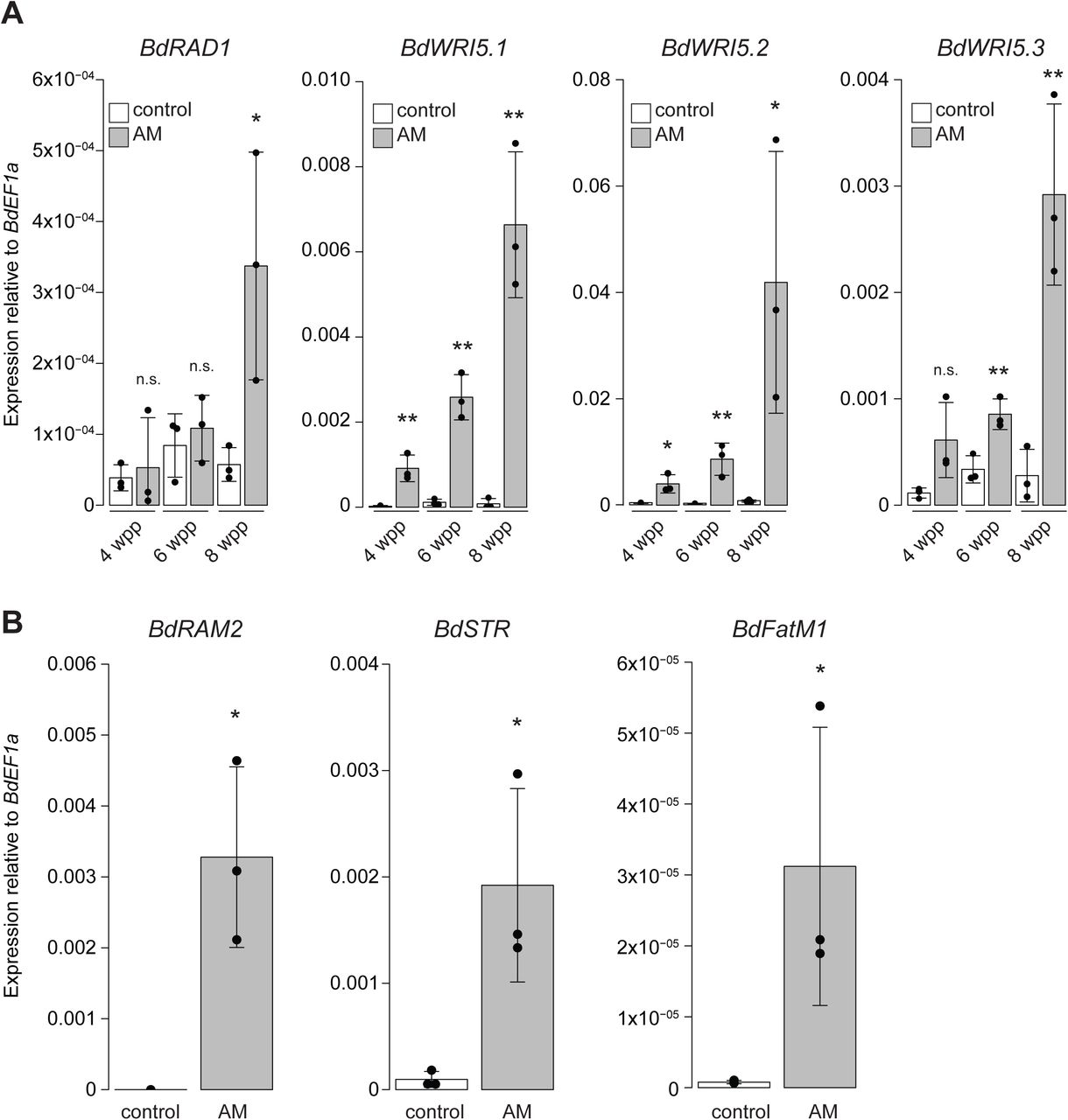
In dicots, RAM1 regulates expression of a second tier of transcription factors including RAD1 and three members of the WRINKLED family (WRI5a-c); the latter directly regulate expression of lipid genes (Park et al., 2015; Jiang et al., 2017; Luginbuehl et al., 2017). We found that BdRAD1, and the three B. distachyon AP2 family transcription factors most closely related to MtWRI5a-c (further denoted as BdWRI5.1, BdWRI5.2, and BdWRI5.3) were strongly induced in wild-type roots colonized with D. epigaea relative to mock-inoculated controls (Supplemental Fig. 3) but interestingly, only BdWRI5.1 was induced in non-colonized 35S:BdRAM1ox roots (Figure 3B). A similar pattern was observed in 35S:BdRAM1ox shoots (Figure 4B, Supplemental Fig. 5). Thus, in contrast to M. truncatula, a RAM1-independent pathway likely leads to up-regulation of BdRAD1, BdWRI5.2, and BdWRI5.3 in mycorrhizal roots. This points to functional diversification of the regulatory cascade responsible for the transcriptional reprogramming of roots during AM symbiosis in B. distachyon. In addition, it may provide an explanation for the relatively mild ram1 mutant phenotype we observed (Figure 2). Future research in other monocot species is required to determine if such a functional diversification is unique to B. distachyon or a monocot-specific phenomenon.
Arbuscule density is higher in RAM1 overexpressors relative to controls
The initial goal of this study was to test the hypothesis that constitutive overexpression of RAM1 would increase arbuscule density and/or colonization and then to use the plants to address secondary hypotheses about symbiotic performance.
To test the first hypothesis, we grew 35S:BdRAM1ox, 35S:BdRAM1WT and 35S:NLS-GFP control plants in substrate containing D. epigaea spores and evaluated colonization levels and arbuscule morphology. Colonization levels in 35S:BdRAM1ox and control plants did not differ significantly, although the variation was much greater in the 35S:BdRAM1ox plants (Figure 3C). Arbuscules in 35S:BdRAM1ox plants showed a wild-type morphology, but the number of arbuscules, which we assessed within a defined root volume below the hyphopodium, was on average 2-fold greater in the 35S:BdRAMox plants relative to controls (Figure 3D-F, Supplemental Fig. 5). Thus, 35S:BdRAM1ox plants have a higher capacity to establish and/or to maintain arbuscules relative to the control plants. As RAM1 regulates the expression of several other transcription factors, as well as genes involved in lipid biosynthesis and nutrient transport, the increased arbuscule density in the 35S:BdRAM1ox plants may result from a combination of factors including arbuscule initiation and/or regulation of arbuscule lifespan.
Unfortunately, the severe shoot growth and branching phenotype of the BdRAM1 overexpressors prevented a fair evaluation of symbiotic performance (Supplemental Fig. 5). While colonized 35S:BdRAM1ox plants and controls both showed an increase in shoot fresh weight and tiller number relative to their respective mock-inoculated controls, the differences in the developmental architecture of these lines precluded direct physiological comparisons. Consequently, it was not possible to determine whether the increased arbuscule density influenced symbiotic performance.
Hormone biosynthetic and regulatory gene expression is altered in BdRAM1 overexpressors
The shoot architecture phenotype of the 35S:BdRAM1ox plants is reminiscent of the phenotypes of several monocot hormone mutants. For example, rice and B. distachyon mutants defective in GA, SL, and BR biosynthesis or signaling display dwarf phenotypes with increased tillering (e.g. (Spielmeyer et al., 2002; Ishikawa et al., 2005; Asano et al., 2009; Lin et al., 2009; Thole et al., 2012). To obtain further clues about the BdRAM1 overexpression phenotype, we evaluated the expression of several genes associated with SL, GA, and BR signaling. B. distachyon orthologues of genes involved in SL biosynthesis (BdD27, BdD17, BdD10) (Seto and Yamaguchi, 2014) and GA biosynthesis (potential orthologs of A. thaliana GA3ox1 and GA20ox1) (Kakei et al., 2015) were down-regulated, while key BR biosynthesis genes (BdCPD, BdD2/CYP90D) but not BdDWF4 (Kakei et al., 2015) were elevated in non-colonized 35S:BdRAM1ox roots relative to the controls (Figure 5A, B). In contrast, the GA receptor GID1 and the GA-regulator DELLA/SLR1 (Daviere and Achard, 2013) as well as the regulators of SL signaling D3 and D53 (Seto and Yamaguchi, 2014), and the B. distachyon BR receptor BdBRI1 and the BR-responsive transcription factor BdBZR1 (Corvalan and Choe, 2017) were differentially regulated in 35S:BdRAM1ox roots relative to controls (Supplemental Fig. 6). Thus the transcript data indicate a disturbance in hormone biosynthetic and regulatory gene expression likely contributing to the altered shoot architecture. Because of substantial cross-talk between hormone signaling pathways (Itoh et al., 2001; Umehara et al., 2008; Unterholzner et al., 2015; Corvalan and Choe, 2017), it is not possible to predict the initial cause. As GA, SL and BR hormone pathways each involve regulation via GRAS-transcription factors (Tong et al., 2009; Liu et al., 2011; Chen et al., 2013), it is possible that ectopic overexpression of BdRAM1 disturbs GRAS-factor complexes, leading to mis-regulation of these pathways. Alternatively, one of the native functions of BdRAM1 may be to regulate aspects of hormone signaling. For example, in rice, RAM1 interacts with a DELLA interacting protein, DIP, and therefore it is possible that one of RAM1’s native functions is to influence GA signaling and that this is exacerbated in the 35S:BdRAM1ox, leading to further downstream effects on other pathways. If mis-regulation of GA biosynthesis gene expression translates to disturbed GA homeostasis in 35S:BdRAM1ox roots, an imbalance in GA-regulated arbuscule formation and degradation could result (Floss et al., 2013; Floss et al., 2017). Such a scenario might also explain the increased arbuscule numbers in 35S:BdRAM1ox roots as well as a dwarf shoot phenotype.
Conclusion
In conclusion, BdRAM1, similar to its orthologs in dicots, regulates arbuscule development and transcriptional regulation of several AM symbiosis-induced genes, although it is likely that there is some functional redundancy with other GRAS or WRI5 transcription factors. Constitutive overexpression of 35S:BdRAM1 increased the number of arbuscules relative to control plants; although the plants were unsuitable for experiments to assess the functional consequences of increasing the symbiotic interfaces, the data nevertheless indicate that it is possible to manipulate arbuscule density through expression of RAM1. Future research should focus on increasing RAM1 gene expression specifically in the root cortex. We predict such a strategy would increase arbuscule numbers without the accompanying developmental defects and would enable evaluation of the consequences of increasing the density of symbiotic interfaces and the effects on nutrient exchange during AM symbiosis.
Materials and Methods
Plant material and growth conditions
B. distachyon plants were grown in a growth chamber under a 12 h light (24°C)/12 h dark (22°C) regime. For all experiments that were conducted in the absence of an AM fungal symbiont, B. distachyon plants were grown in 20.5 cm long cones filled with sterile Terragreen (Oli-Dri) and play sand (Quikrete) in a ratio of 1:1. For all experiments involving AM symbiosis, B. distachyon plants were grown in cones filled with a sand-gravel mix, and were inoculated with 250 Diversispora epigaea spores (formerly Glomus versiforme) as previously described (Muller et al., 2019). For mock-inoculated controls, we added an appropriate volume of filtered spore wash solution instead of the spores. Unless otherwise stated, B. distachyon plants were fertilized once per week with 1/4-strength Hoagland’s fertilizer containing 20μM Pi and harvested 4-5 weeks after transplanting to cones.
To monitor AM growth responses, seedlings were planted into pots (3 seedling per 11cm diameter pot and 8 pots per genotype) containing a 1:20 mixture of autoclaved N7/N8 soil (Watts-Williams et al., 2019) to sand/gravel mix. The sand/gravel mix is a 2:2:1 mixture of play sand, fine black sand and gravel (as described in (Floss et al., 2017). 250 surface sterilized D. epigaea spores were place below each plant. Beginnning at 3 weeks post planting, the pots were fertilized weekly with 50 ml of 1/4-strength Hoaglands solution lacking phosphate and 9 ml of 0.5mM Ca3(PO4)2. Plants were harvested at 9 weeks post planting. The growth chamber conditions were as described above.
Plasmid generation
To clone the CRISPR/Cas9 construct targeting BdRAM1, we used the vector and cloning system described by Xie et al. (Xie et al., 2015). To design the primers (shown in Supplemental Table 1), gene-specific guide RNA sequences targeting Bradi4g18390 were identified using CRISPR-P (Lei et al., 2014) and CRISPR-PLANT (Xie et al., 2015) and selected based on their location in the coding sequence and low number of off-target sites. We generated a 2-guide CRISPR/Cas9 construct that targeted Bradi4g18390 at positions 32-54 bp (guide RNA1) and 280-302 bp (guide RNA2) downstream of the transcription start site (Supplemental Fig. 2). As a negative control we used the empty vector pRGEB32 (Xie et al., 2015).
To clone 35S:BdRAM1 overexpression constructs, the coding sequence of Bradi4g18390 was amplified using gene-specific primers flanked by attB1 and attB2 recombination sites (Supplemental Table 1), and cloned into pDONR221, resulting in the pENTR1-2 BdRAM1 entry clone. pENTR1-2 clones containing the coding sequence of NLS-GFP-GUS, as well as pENTR4-1 entry clones containing the CaMV35S promoter and pENTR2-3 containing the CaMV35S terminator were cloned previously (Ivanov and Harrison, 2014; Floss et al., 2017). To assemble the binary vectors for B. distachyon transformation, four vectors (pENTR4-1 containing the double CaMV35S promoter, pENTR1-2 containing BdRAM1 or NLS-GFP-GUS, pENTR2-3 containing the CaMV35S terminator and pHb7m34GW (Karimi et al., 2005)), were combined to generate 35S:BdRAM1 or 35S:NLS-GFP using the multi-site gateway cloning system (Invitrogen). All vector sequences were confirmed by Sanger sequencing.
Generation of B. distachyon transformants
The CRISPR/Cas9 constructs targeting BdRAM1 as well as the 35S:BdRAM1 and 35S:NLS-GFP constructs were transformed into B. distachyon (accession Bd21-3) following a previously established protocol(Bragg et al., 2015). Plantlets emerging from transformed calli (selectable marker: Hygromycin) were transplanted into Metro-Mix 350 and genotyped to test for the presence of the construct (see Supplemental table 1 for primer sequences). In addition, in the case of the CRISPR/Cas9 constructs, the CRISPR/Cas9 target loci were amplified using flanking primers and purified PCR products were Sanger-sequenced in order to identify gene edits.
Visualization and quantification of fungal root colonization
Fungal colonization of B. distachyon roots was visualized by staining with Wheat-Germ Agglutinin (WGA) coupled to Alexafluor488 as previously described (Hong et al., 2012). Roots were observed using a Leica M205 stereomicroscope and root colonization was quantified using the gridline-intersect method (McGonigle et al., 1990). To quantify the ram1 phenotype, roots intersecting the gridlines were scored into one of three categories: (1), not colonized; (2), colonized with wildtype-like arbuscules; (3), colonized with aberrant (sparsely branched or collapsed arbuscules) or no arbuscules. The ratio of category 3 over the overall number of intersections of colonized roots (category 2+3) x100 was used to determine the percentage of intersections without arbuscules/total colonization. Total root length colonization was calculated as the percentage of category 2+3 over the total number of intersections counted x 100. To study arbuscule morphology, WGA-Alexafluor488-stained roots were counterstained with propidium iodide to visualize plant cell walls, and observed with a Leica SP5 confocal microscope. To quantify arbuscule numbers in 35S:BdRAM1ox roots, confocal stacks from highly colonized roots were taken so that the fungal hyphopodium was in the center of the image to ensure we capture infections of similar developmental stages. The total number of arbuscules per stack was assessed manually using the Fiji Image Analysis Package (Schindelin et al., 2012). Stack depth (z-plane) was chosen to encompass the whole infection, and arbuscule numbers were normalized against the stack volume (length of x*y*z planes). To avoid potentially confounding effects caused by different B. distachyon root types, we selected only thin lateral roots with a single layer of cortical cells for analysis.
RNA isolation, cDNA synthesis, and quantitative PCR
RNA isolation, cDNA synthesis, and quantitative PCR were performed as previously described (Muller et al., 2019). Primers used to quantify expression of target genes are shown in Supplemental table 1. Ct values of the tested genes were normalized against BdEF1a {resulting in Ct} and relative expression levels were calculated with the formula 2-Ct.
Assessment of plant morphology
Plants were grown in the absence of AM fungi and whole plants were harvested 2, 4, and 6 weeks after planting. Tiller and node root numbers were counted, and maximal root system and shoot length were measured. The angle between individual leaves and the stem was measured on images of the same plants using the Fiji Image Analysis Package (Schindelin et al., 2012).
Phylogenetic analyses
B. distachyon orthologs of M. truncatula RAM1, RAD1, RAM2, PT4, FatM, STR, WRI5a-c as well as O. sativa D27, D17/CCD7, CCD8a, D3, D53, D14, D14L and SLR1 were identified using phylogenetic approaches described before (Supplemental Fig. 1, 7)(Bravo et al., 2016) B. distachyon genes putatively involved in Brassinosteroid and Gibberellic acid biosynthesis and signaling were identified previously (Kakei et al., 2015; Corvalan and Choe, 2017; Niu et al., 2019).
Statistical analyses and data representation
All experiments were performed using biological replicates. All experiments were repeated at least two times. The distribution of residuals was tested for normality using the Shapiro-Wilk test. If normality assumption was met, pairwise comparisons were analyzed using a two-sided Student’s t-test. For multiple comparisons, the raw data was subjected to a one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. If normality assumption was not met, data were analyzed using the Kruskal-Wallis test followed by Dunn’s post-hoc test (p-values adjusted after Benjamini-Hochberg). All statistical analyses were performed using R software. Quantification data for n>5 biological replicates are represented as box-and-whiskers plots, which show the lower and upper quartiles as well as the minimum and maximum values. The horizontal line in the box plots represents the median. Points represent single measurements. For datasets with less than 5 measurements per genotype, bar plots were chosen. Bars represent the mean, and error bars the standard deviation. Points represent single measurements.
Figure legends

Phylogenetic tree showing GRAS transcription factors related to BdRAM1 in the AM host species (B. distachyon, O. sativa, H. vulgare, Z. mays, M. truncatula) and the non-host species A. thaliana. The phylogeny is based on the BdRAM1 protein sequence and was constructed using a previously published pipeline(Bravo et al., 2016). MtRAD1 regulates arbuscule formation(Park et al., 2015). OsGRAS19 and OsGRAS32 function in Brassinosteroid signaling (Tong et al., 2009; Chen et al., 2013; Floss et al., 2013; Floss et al., 2016) and MtDELLA function in Gibberellic acid signaling and AM symbiosis (Floss et al., 2013; Yu et al., 2014; Floss et al., 2017). MtNSP2 regulates strigolactone biosynthesis and AM symbiosis (Liu et al., 2011).

A) Alignments of Bdram1 DNA sequences in ram1-1 and ram1-2 lines, as well as in B. distachyon plants transformed with the empty-vector control (E.V.). Target sequences of the two guide RNAs are indicated, as well as CRISPR/Cas9-induced insertions and deletions (pink, T3 generation). Numbers in blue indicate nucleotide position in the wild-type Bdram1 coding sequence. We detected a homozygous single-base pair deletion in ram1-1 at the target sequence of the upstream-most guide. In ram1-2, two edited variants (a one base-pair insertion in one allele, and a one base-pair deletion in the other; denoted as a and b, respectively) were detected at the target sequence of guideRNAl. Both ram1-1 and ram1-2 showed homozygous edits caused by guideRNA2 further downstream in the gene. B) All indels caused by guideRNAl lead to a frameshift and a premature stop codon (pink asterisk) after amino acid positions l6 or 42, respectively. C) Morphologic analysis of root colonization phenotypes in Tl ram1-1 and ram1-2 plants compared to empty vector controls. Both ram1 alleles display increased numbers of sparsely branched or collapsed arbuscules and infections without any arbuscules relative to control plants, where most arbuscules were of wildtype-like morphology. Each bar represents an individual root system; fungal colonization patterns were divided in four categories (color-coded) and counted microscopically using the grid-line method (McGonigle et al., l990). No difference in overall root colonization was observed in this experiment. D) Overall root colonization of B. distachyon ram1-1 mutants (T2 generation) is reduced relative to empty-vector controls (E.V.). Plants were harvested at two time points: 2.5 and 5 weeks post planting (wpp). Pairwise comparisons were calculated separately for each time point (Student’s t-test). Significance codes: **p<0.0l; *p<0.05. Arbuscule phenotypes were not analyzed in this experiment.

A) Gene expression of the GRAS transcription factor BdRADl and three AP2-family transcription factors related to MtWR/5. Gene expression was measured in mock-inoculated roots (“control”) and roots colonized with D. epigaea (“AM”) harvested at 4, 6, and 8 weeks after planting (wpp). Pairwise comparisons between AM and control roots were calculated separately for each time point (Student’s t-test). B) Gene expression of selected B. distachyon orthologs of the AM marker genes RAM2, STR, and FatM in 6 week-old D. epigaea-colonized (“AM”) roots relative to mock-inoculated control roots. Pairwise differences were calculated using Student’s t-test. Significance codes in A) and B): ***p<0.00l; **p<0.0l; *p<0.05; n.s., not significant.
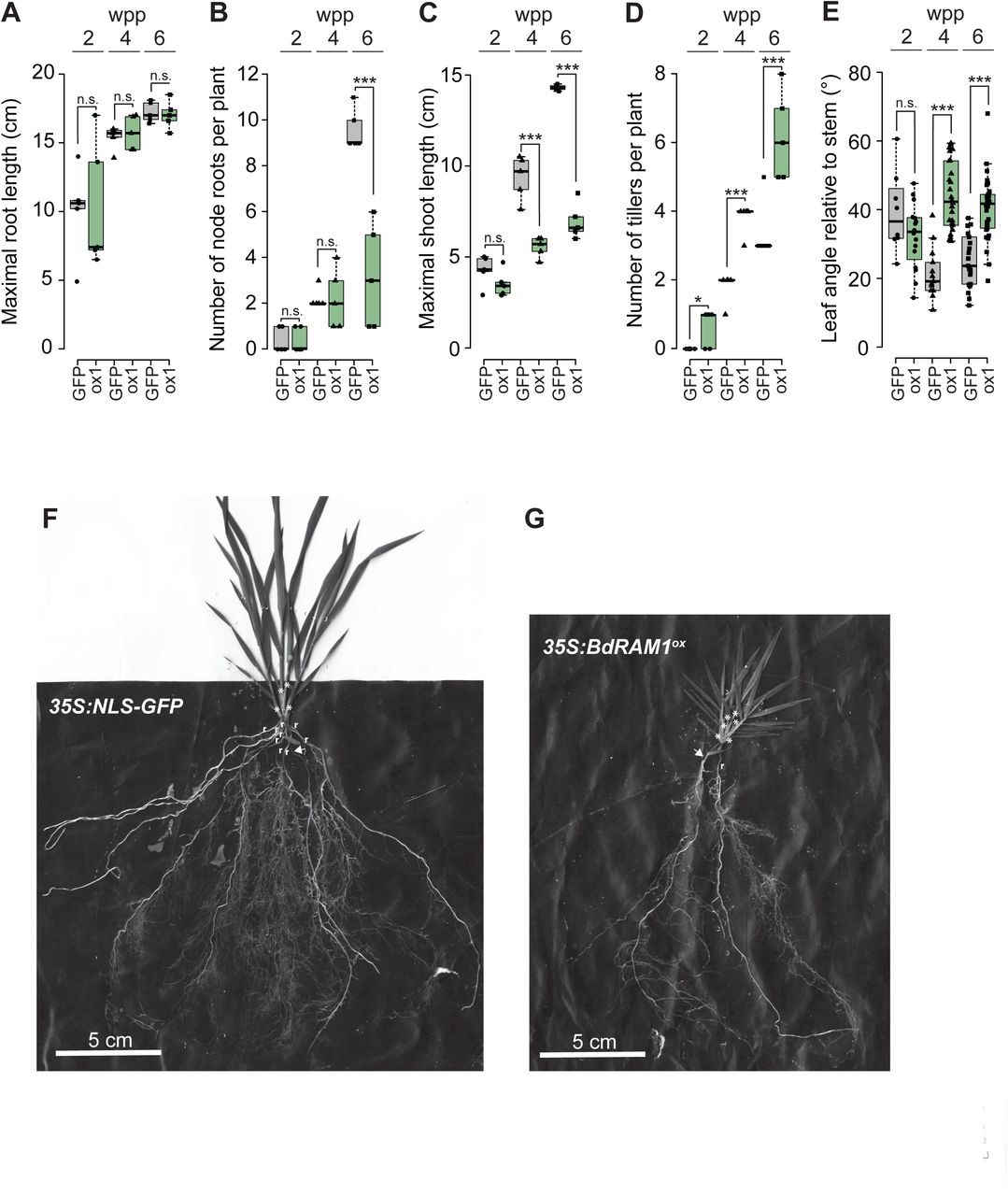
A) Maximal root system length does not differ between 355:Bdram1ox (line #1, “ox1”) and control plants (355:NL5-GFP, denoted as “GFP”). B) The collective number of coleoptile and leaf node roots is decreased in 6-week old 355:Bdram1ox relative to control plants. C) Overall shoot length is strongly decreased in 4- and 6-week old 355:Bdram1ox relative to control plants. D) Ectopic overexpression of Bdram1 results in increased tiller numbers relative to control plants. E) The leaf angle relative to the stem is increased in in 4- and 6-week old 355:Bdram1ox plants relative to the control. A)-E) Box-and-whiskers plots show lower and upper quartiles, and minimum and maximum values. The horizontal bar represents the median, and the points individual measurements. Pairwise comparisons were calculated separately for each time point (Student’s t-test). Significance codes: ***p<0.001; **p<0.01; *p<0.05; n.s., not significant. F) Representative image of a plant expressing 355:NL5-GFP. G) Representative image of a 355:Bdram1ox plant. F), G) Node roots are designated with “r”, and tillers are designated with “*”; arrow points to seminal root.

A) Arbuscule density in a defined volume below the hyphopodium. This is a second independent experiment that shows increased arbuscule density per root volume in 355:Bdram1ox roots relative to 355:NL5-GFP and 355:Bdram1WT control plants (compare Fig. 3D). Kruskal-Wallis test p=3.78×10−07. B) Gene expression levels of BdFatM2, BdRADl, BdWR/5.2, and BdWR/5.3 in shoots overexpressing Bdram1 (355:Bdram1ox lines 1-3) and control roots (355:NL5-GFP and 355:Bdram1WT). No significant differences were detected (p-value after ANOVA is shown in the figure). C)-E) B. distachyon growth response experiment: C) Shoot freshweight of 9-week old mock-inoculated (“mock”) 355:Bdram1ox (line 1) and 355:NL5-GFP and 355:Bdram1WT control plants and plants inoculated with D. epigaea (“D.e.”). Kruskal-Wallis test p= 9.78×10−11. D) Tiller numbers of the same plants. Kruskal-Wallis test p= 8.22×10−11. E) Representative images of mock-inoculated and D. epigaea-inoculated plants at harvest (9 weeks after planting). The developmental defects of 355:Bdram1ox plants were so severe that we could not draw conclusions from the growth response measurements in response to AM colonization. Note: the plant marked with an asterisk is a wild-type segregant and was not included in the analysis shown in C) and D). A), C), D) Individual measurements are displayed as single points. Box plots show median as horizontal line, and upper and lower quartiles (dashed lines). Different letters denote significant differences (p<0.05) after Dunn’s posthoc test.

A) B. distachyon orthologs of two genes involved in the strigolactone signaling pathway (BdD3, BdD53) are differentially regulated in non-colonized roots ectopically overexpressing Bdram1 (355:Bdram1ox line#1, denoted as “ox1”) relative to 355:NL5-GFP (“GFP”) control roots. Gene expression of the putative strigolactone receptor BdDl4 is not influenced by ectopic overexpression of Bdram1. Gene expression of the putative karrikin receptor BdDl4L is reduced in Bdram1ox roots. B) Two genes with a putative function in Gibberelic acid signaling (BdG/D, Bd5LRl) are differentially regulated in 355:Bdram1ox roots relative to control roots. C) Two B. distachyon genes orthologous to known Brassinosteroid signaling genes (BdBR/l, BdBZRl) are induced in 355:Bdram1ox roots. Bar graphs show the mean, error bars the standard deviation. Single points represent individual measurements. Pairwise comparisons were estimated using the Student’s t-test. Significance codes: ***p<0.001; **p<0.01; *p<0.05; n.s., not significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Sophia Cotraccia, Cassandra Proctor, and Stephanie Roh for technical assistance and the BTI Biotechnology center for generating some of the B. distachyon transgenic lines. Financial support for the project was provided by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research (grant no. DE-SC0012460) and the TRIAD Foundation. LMM was supported by Postdoctoral Fellowships from the Swiss National Science Foundation (SNF, Early Postdoc.Mobility) and the German Research Foundation (DFG), LCS was supported by a Marie Curie Fellowship (FP7-PEOPLE-2013-IOF-624739).
Footnotes
↵* joint second co-authors
References




