

Abstract
The sudden global emergence of SARS-CoV-2 urgently requires an in-depth understanding of molecular functions of viral proteins and their interactions with the host proteome. Several omics studies have extended our knowledge of COVID-19 pathophysiology, including some focused on proteomic aspects1–3. To understand how SARS-CoV-2 and related coronaviruses manipulate the host we here characterized interactome, proteome and signaling processes in a systems-wide manner. This identified connections between the corresponding cellular events, revealed functional effects of the individual viral proteins and put these findings into the context of host signaling pathways. We investigated the closely related SARS-CoV-2 and SARS-CoV viruses as well as the influence of SARS-CoV-2 on transcriptome, proteome, ubiquitinome and phosphoproteome of a lung-derived human cell line. Projecting these data onto the global network of cellular interactions revealed relationships between the perturbations taking place upon SARS-CoV-2 infection at different layers and identified unique and common molecular mechanisms of SARS coronaviruses. The results highlight the functionality of individual proteins as well as vulnerability hotspots of SARS-CoV-2, which we targeted with clinically approved drugs. We exemplify this by identification of kinase inhibitors as well as MMPase inhibitors with significant antiviral effects against SARS-CoV-2.
Main text
To identify interactions of SARS-CoV-2 and SARS-CoV with cellular proteins, we transduced A549 lung carcinoma cells with lentiviruses expressing individual HA-tagged viral proteins (Figure 1a; Extended data Fig. 1a; Supplementary Table 1). Affinity purification followed by mass spectrometry analysis (AP-MS) and statistical modelling of the MS1-level quantitative data allowed identification of 1484 interactions between 1086 cellular proteins and 24 SARS-CoV-2 and 27 SARS-CoV bait proteins (Figure 1b; Extended data Fig. 1b; Supplementary Table 2). The resulting virus-host interaction network revealed a wide range of cellular activities intercepted by SARS-CoV-2 and SARS-CoV (Figure 1b; Extended data Table 1; Supplementary Table 2). In particular, we discovered that SARS-CoV-2 targets a number of key cellular regulators involved in innate immunity (ORF7b-MAVS, -UNC93B1), stress response components (N-HSPA1A) and DNA damage response mediators (ORF7a-ATM, -ATR) (Figure 1b; Extended data Fig. 1c - e). Overall, SARS-CoV-2 interacts with specific protein complexes contributing to a range of biological processes (Supplementary Table 2). To evaluate the consequences of these interactions on cellular proteostasis, we proceeded with the total proteome analysis of A549 cells expressing the 54 individual viral proteins (Figure 1a, d; Supplementary Table 3). The analysis of the proteome changes induced by each viral protein and consideration of the interactions of this respective protein provided direct insights into their functions. For instance, we confirmed that ORF9b of SARS-CoV-2 leads to a dysregulation of mitochondrial functions (Figure 1d; Supplementary Table 3), as was previously reported for SARS-CoV4, correlating with the binding of ORF9b of both viruses to TOMM70 (Figure 1b; Supplementary Table 2)1, a known regulator of mitophagy5, which was not yet known for SARS-CoV ORF9b. Importantly, this approach identified novel SARS-CoV-2 activities, such as the regulation of proteins involved in cholesterol metabolism by NSP6 (Figure 1d; Supplementary Table 3). Despite the high similarity of SARS-CoV-2 and SARS-CoV6, these datasets allow to discriminate the commonalities and differences of both viruses, which may in part explain the characteristics in pathogenicity and transmission capabilities. By comparing the AP-MS data of homologous SARS-CoV-2 and SARS-CoV proteins, we identified significant differences in the enrichment of individual host targets, highlighting potential virus-specific interactions (Figure 1b (edge color); Figure 1c; Extended data Fig. 1f - h; Supplementary Table 2). For instance, we could recapitulate the known interaction between SARS-CoV NSP2 and prohibitins (PHB, PHB2)7, whereas their enrichment was not observed for NSP2 of SARS-CoV-2 (Extended data Fig. 1g). Alternatively, we found that ORF8 of SARS-CoV-2, but not its SARS-CoV homolog, binds specifically to the TGFB1-LTBP1 complex (Extended data Fig. 1f, h).


(a) Expression of HA-tagged viral proteins, in stably transduced A549 cells, used in AP-MS and proteome expression measurements. (b) The extended version of the virus-host protein-protein interaction network with 24 SARS-CoV-2 and 27 SARS-CoV proteins, as well as ORF3 of HCoV-NL63 and ORF4 and 4a of HCoV-229E, used as baits. Host targets regulated upon viral protein overexpression or SARS-CoV-2 infection (based on the analysis of all data of this study) are highlighted (see the in-plot legend). (c-f) Co-precipitation experiments in HEK 293T cells showing a specific enrichment of (c) endogenous MAVS co-precipitated with c-term HA-tagged ORF7b of SARS-CoV-2 and SARS-CoV (negative controls: SARS-CoV-2 ORF6-HA, ORF7a-HA), (d) ORF7b-HA of SARS-CoV-2 and SARS-CoV co-precipitated with SII-HA-UNC93B1 (control precipitation: SII-HA-RSAD2), (e) endogenous HSPA1A co-precipitated with N-HA of SARS-CoV-2 and SARS-CoV (control: SARS-CoV-2 ORF6-HA) and (f) endogenous TGFβ with ORF8-HA of SARS-CoV-2 vs ORF8-HA, ORF8a-HA, ORF8b-HA of SARS-CoV or ORF9b-HA of SARS-CoV-2. (g, h) Differential enrichment of proteins in (g) NSP2 and (h) ORF8 of SARS-CoV-2 (x-axis) vs SARS-CoV (y-axis) AP-MS experiments.
Proteins identified as SARS-CoV-2 and/or SARS-CoV host binders via AP-MS (Figure 1b) were assemble in functional groups based on functional enrichment analysis of GOBP, GPCC, GPMF and Reactome terms (Supplementary table 2)

(a) Experimental design to systematically compare the AP-MS interactomes and induced host proteome changes of the homologous SARS-CoV-2 and SARS-CoV viral proteins, with ORF3 homologs of HCoV-NL63 and HCoV-229E as reference for pan coronavirus specificity. (b) Combined virus-host protein interaction network of SARS-CoV-2 and SARS-CoV measured by affinity-purification coupled to mass spectrometry. Homolog viral proteins are displayed as one node. Shared and virus-specific interactions are denoted by the edge color. (c) The numbers of unique and shared host interactions between the homologous proteins of SARS-CoV-2 and SARS-CoV. (d) Gene Ontology Biological Processes enriched among the cellular proteins that are up- (red arrow) or down- (blue arrow) regulated upon overexpression of individual viral proteins.
To obtain information on the concerted activity of the viral proteins during infection, we infected ACE2-expressing A549 cells (Extended data Fig. 2a-b) with SARS-CoV-2, and profiled the impact of viral infection on mRNA transcription, protein abundance, ubiquitination and phosphorylation in a time-resolved manner (Figure 2a – e; Supplementary Tables 4 - 7; Methods). In line with previous reports8,9, we did not observe major upregulation of type-I interferons and related genes at the mRNA level (e.g. IFNB, IFIT3, MX1; Extended data Fig. 2c – d; Supplementary Table 4), suggesting active viral inhibition of this system. In contrast, SARS-CoV-2 upregulated NF-κB and stress responses, as inferred from the induction of IL6, CXCL2 and JUNs and transcription factor enrichment analysis (Extended data Fig. 2c – e; Supplementary Tables 4, 8; Methods). At the proteome level, we found 1053 regulated proteins (Figure 2a - b). Most notably, SARS-CoV-2 infection failed to induce an interferon response pattern indicative for an appropriate cellular antiviral response in this dataset (Extended data Fig. 2f; Supplementary Table 5). We complemented these data with global MS analysis of protein ubiquitination, which revealed 884 sites that were differentially regulated after SARS-CoV-2 infection (Figure 2a and c, Extended data Fig. 2f; Supplementary Table 6). A number of proteins displayed both differential abundance and dynamic ubiquitination patterns in an infectiondependent manner (e.g. mediators of caveolar-mediated endocytosis signaling) (Figure 2c; Extended data Fig. 2f; Supplementary Tables 5, 6, 8). Notably, EFNB1, POLR2B, TYMS and DHFR showed concomitant ubiquitination and a decrease at the protein level (Figure 2c; Extended data Fig. 2f; Supplementary Tables 5, 6). Moreover, we identified two upregulated ubiquitination sites on ACE2, including one previously unknown (K702) (Figure 2c; Extended data Fig. 2f - i), suggesting an alternative post-translational mechanism of its degradation upon SARS-CoV-2 infection besides the cleavage by matrix metalloproteinases10,11. We identified multiple yet undescribed ubiquitination sites on viral proteins, which may be tied to the interactions with several E3 ligases observed in the interactome (e.g. ORF3 and TRIM47, WWP1/2, STUB1; M and TRIM7; NSP13 and RING1) (Extended data Fig. 2j; Supplementary Table 2, 6) and likely indicative of cross-talks between ubiquitination and viral protein functions. Moreover, in the phosphoproteomic analysis, we mapped multiple novel phosphorylation sites on viral proteins (M, N, S and ORF9b), which correspond to known recognition motifs of GSK3, CSNKs, GPCR, AKT, CAMKs, and ERKs (Extended data Fig. 2k; Supplementary Table 7). Of 11,847 total quantified phosphorylation sites, 1483 showed significant changes after SARS-CoV-2 infection (Extended data Fig. 2l; Supplementary Table 7). The regulation of known phosphosites suggests an involvement of central kinases known to modulate key cellular pathways, e.g. EPHA2 – focal adhesion, RPS6Ks – cell survival, CDKs – cell cycle progression, AKT – cell growth, survival and motility, p38, JNK, ERK – stress responses, ATM, and CHEK1/2 – DNA damage response, during virus infection (Extended data Fig. 2l, m; Supplementary Tables 7, 8). Intriguingly, we could also observe an interplay of phosphorylation and ubiquitination on YAP1, a downstream regulatory target of Hippo signaling (Figure 2d), underlining the value of testing different post-translational modifications simultaneously. Combining these datasets, describing different aspects of SARS-CoV-2 infection, allowed us to determine the key pathways perturbed during the infection, such as stress and DNA damage response, regulation of transcription and cell junction organization, at various levels (Figure 2e, see Methods).

(a) Western blot showing ACE2-HA expression levels in A549 cells untransduced (wt) or transduced with ACE2-HA encoding lentivirus. (b) mRNA expression levels of SARS-CoV-2 N relative to RPLP0 as measured by qRT-PCR upon infection of wt A549 and A549-ACE2 cells at the indicated MOIs. Mean +/- standard deviation of three biological replicates are shown. (c) Volcano plot of mRNA expression changes of A549-ACE2 cells, infected with SARS-CoV-2 at an MOI of 3, shown as a fold change versus mock at 24 h.p.i.. Selected significant hits are marked in black (Wald test, n=3). (d) Expression levels, as measured by qRT-PCR, of SARS-CoV-2 N and host transcripts relative to RPLP0 after infection of A549-ACE2 cells at MOI of 3 at indicated time points after infection with indicated mean +/- standard deviation (n=3). ND: not detectable. (e) Transcription factor enrichment analysis of up- (red arrow) and down- (blue arrow) regulated genes in A549-ACE2 cells infected with SARS-CoV-2 for indicated time periods (Fisher’s exact test, unadjusted). (f) Volcano plot of protein abundance changes at 24 h.p.i. in comparison to mock measured by proteome profiling (DDA MS). Viral proteins are highlighted in orange, selected significant hits are marked in black (Student’s t-test, two-tailed, permutation-based FDR < 0.05, S0 = 0.1, n = 4). (g) Total levels of ACE2 protein at 6 and 24 hours (top) and its ubiquitination at indicated sites (bottom) at 24 hours after infection with SARS-CoV-2 as measured by proteome and diGly proteome profiling (DDA MS). (h) Western blot showing the expression levels of SARS-CoV-2 proteins, ACE2-HA and ACTB in A549-ACE2 cells at indicated time points post-infection with SARS-CoV-2 compared to mock. (i) Stable expression of ACE2 mRNA transcript relative to RPLP0, as measured by qRT-PCR, after SARS-CoV-2 infection (MOI 3) of A549-ACE2 cells at indicated time points post-infection with indicated mean +/- standard deviation of three biological replicates. (j) Mapping the ubiquitination sites of SARS-CoV-2 proteins and measuring their regulation (DDA MS of diGly PTMs) in SARS-CoV-2-infected A549-ACE2 cells (MOI 3) at 24 h.p.i., highlighting the binding of TRIM47 to SARS-CoV-2 ORF3 and TRIM7 to SARS-CoV-2 M as examples of potential E3 ubiquitin ligases driving ubiquitination and associated with the shown proteins. (k) Mapping the phosphorylation sites of SARS-CoV-2 N and measuring their regulation (DDA MS) in SARS-CoV-2-infected A549-ACE2 cells (MOI 3) at 24 h.p.i.. The host kinases potentially driving the phosphorylation on the shown sites are indicated. (l) Volcano plot of phosphorylation sites regulation indicated as a log2-fold change compared to mock as measured by phosphoproteome profiling (DDA MS). Viral proteins are marked in orange. Selected significant hits (Student’s t-test, two-tailed, permutation-based FDR < 0.05, S0 = 0.5, n = 3) are marked in black. (m) The enrichment of host kinases known to regulate the phosphorylation sites identified by phosphoproteome profiling (DIA and DDA MS) of infected A549-ACE2 cells (MOI 3) at the indicated time points after infection (Fisher’s exact test, unadjusted). DIA measurements are marked in grey, DDA in black.

(a) Time-resolved profiling of SARS-CoV-2 infection by multiple-omics methods. The plot shows normalized MS intensities of three SARS-CoV-2 viral proteins over time. (b) Numbers of distinct transcripts, proteins, ubiquitination and phosphorylation sites, up- or down-regulated at the indicated time points after infection, as identified using data independent (DIA) or dependent (DDA) acquisition methods. (c) Volcano plot showing ubiquitination sites regulated at 24h after SARS-CoV-2 infection. Viral proteins are marked in orange. Selected significant ubiquitination sites (Student’s t-test, two-tailed, permutation-based FDR < 0.05, S0 = 0.1, n = 4) are marked in black. (d) Scatter plot of phosphorylation and ubiquitination sites on Yes-associated protein (YAP1) regulated upon SARS-CoV-2 infection. Presented are fold changes compared to mock at 6, 24, and 30 hours after infection. S61 and S127 dephosphorylation lead to nuclear translocation, S131 phosphorylation regulates protein stability. Phosphorylation of S289 and S367 is involved in cell cycle regulation. K321 deubiquitination leads to a decrease in YAP1 activation. (e) Reactome pathways enriched in up- (red arrow) or downregulated (blue arrow) transcripts, proteins, ubiquitination- and phosphorylation sites (Fisher’s exact test, unadjusted). DIA MS measurements are marked in grey, DDA in black.
The systematic interactome and proteome profiling of individual viral proteins provided us with the opportunity to gain deeper understanding of their molecular mechanisms. For each viral protein, we mapped the collected data onto the global network of cellular interactions12 and applied a network diffusion approach13. Such analysis identifies short links of known protein-protein interactions, signaling and regulation events that connect the interactors of the viral protein with the proteins affected by its expression (Figure 3a, Extended data Fig. 3a, b; Supplementary Data 1).The connections predicted using the real data were significantly shorter than for the randomized data, confirming both relevance of the approach and the data quality (Extended data Fig. 3a, b). Amongst many other findings, this approach pointed towards the potential mechanisms of autophagy regulation by ORF3 and NSP6; the modulation of innate immunity by M, ORF3 and ORF7b; and the Integrin-TGFβ-EGFR-RTK signaling perturbation by ORF8 of SARS-CoV-2 (Figure 3b - d, Supplementary Data 1). Enriching these subnetworks with the SARS-CoV-2 infection-dependent mRNA abundance, protein abundance, phosphorylation and ubiquitination (Figure 3a) allowed us to gain unprecedented insights into the regulatory mechanisms employed by SARS-CoV-2 (Figure 3e, Extended data Fig. 3c, e). For instance, this analysis confirmed a role of NSP6 in autophagy14 and revealed a significant inhibition of autophagic flux by ORF3 leading to the accumulation of autophagy receptors (SQSTM1, GABARAPL2, NBR1, CALCOCO2, MAP1LC3B, TAX1BP1), also observed in virus-infected cells (SQSTM1, MAP1LC3B) (Figure 3e - i). This inhibition may be explained by the interaction of ORF3 with the HOPS complex (VPS11, −16, −18, −39, −41), which is essential for autophagosome-lysosome fusion, as well as by the differential phosphorylation of regulatory sites of key components (AKT1, AKT1S1, SQSTM1, RPS6). The inhibition of the interferon response observed at transcriptional and proteome levels was similarly explained by the network diffusion analysis (Extended data Fig. 3c), demonstrating that multiple proteins of SARS-CoV-2 are employed in the disruption of antiviral immunity. Additional functional experiments corroborated the inhibition of interferon induction or signaling by ORF3, ORF6, ORF7a, ORF7b, ORF9b (Extended data Fig. 3d). Upon virus infection, we observed upregulation of TGFβ and EGFR pathways, which modulate cell survival, motility and innate immune responses (Extended data Fig. 3e - g). Besides promoting virus replication, activation of these pathways has been implicated in fibrosis15,16, one of the hallmarks of COVID-1917. Specifically, our network diffusion analysis revealed the connection between the binding of ORF8 and ORF3 to TGFβ-associated factors (TGFB1, TGFB2, LTBP1, TGFBR2, FURIN, BAMBI) and the virus-induced upregulation of fibrinogens, fibronectin, SERPINE1 and integrin(s) (Extended data Fig. 3e, h)18. The phosphorylation of SMAD1/5, and ERK, JNK, p38 cascade activation, as well as an increased expression of MMPs, DUSPs, JUN, and EGR1 are indicative of TGFβ and EGFR pathway regulation. In turn, they are known to be potentiated by the increased integrin signaling and activation of YAP-dependent transcription19, which we observed upon SARS-CoV-2 infection (Extended data Fig. 3e).

(a) The host subnetwork perturbed by SARS-CoV-2 ORF7a, as predicted by the network diffusion approach. (b) Selection of the optimal cutting threshold for the network diffusion graph of SARS-CoV-2 ORF7a-induced proteome changes. The plot shows the correlation between the minimal allowed edge weight (X axis), and the average path length from the regulated proteins to the host targets of the viral protein along the edges of the filtered subnetwork (Y axis). The red curve represents the path length for the network diffusion analysis of the actual data. The grey band shows 50% confidence interval, and dashed lines correspond to 95% confidence interval for the path lengths of 1000 randomised datasets. Optimal edge weight threshold that maximises the difference between the median path length in randomised data and the path length in the real data is highlighted by the blue vertical line. (c) Overview of perturbations to host-cell innate immunity related pathways, induced by distinct proteins of SARS-CoV-2, derived from the network diffusion model and overlaid with transcriptional, protein abundance, ubiquitination and phosphorylation changes upon SARS-CoV-2 infection. (d) Heatmap showing effects of the indicated SARS-CoV-2 proteins on type-I IFN expression levels, ISRE and GAS promoter activation in HEK293-R1. Accumulation of type-I IFN in the supernatant was evaluated by testing supernatants of PPP-RNA (IVT4) stimulated cells on MX1-luciferase reporter cells, ISRE promoter activation - by luciferase assay after IFN-α stimulation and GAS promoter activation - by luciferase assay after INF-γ stimulation in cells expressing SARS-CoV-2 proteins as compared to the controls (ZIKV NS5 and SMN1). Average of three independent experiments is shown. (e) Overview of perturbations to host-cell Integrin-TGFβ-EGFR-RTK signaling, induced by distinct proteins of SARS-CoV-2, derived from the network diffusion model and overlaid with transcriptional, protein abundance, ubiquitination and phosphorylation changes upon SARS-CoV-2 infection. (f) Profile plots showing intensities of indicated phosphosites and total protein levels of EGFR, EPHA2 and AKAP12 in SARS-CoV-2 infected A549-ACE2 cells at the indicated time points post infection. Points are normalized intensities of individual replicates, solid line is median, filled area corresponds to 25–75 percentiles, dashed lines mark 2.5–97.5 percentiles of the posterior distribution. n = 3 independent experiments; Bayesian statistical modelling. (g) Western blot showing phosphospecies and total protein levels of p38 (T180/Y182, MAPK14) and JNK (T183/Y185, MAPK8) in SARS-CoV-2 infected A549-ACE2 cells. (h) Profile plots of total protein levels of ITGA3, SERPINE1 and FN1 in SARS-CoV-2 infected A549-ACE2 cells at 6 and 24 hours post infection with indicated median and confidence intervals. n = 4 independent experiments.

(a) Network diffusion approach to identify functional connections between the host targets of a viral protein and downstream proteome changes followed by the integration of RNA expression, protein abundance, ubiquitination and phosphorylation changes upon SARS-CoV-2 infection to streamline the identification of affected host pathways. (b-d) Subnetworks of the network diffusion predictions linking host targets of (b) SARS-CoV-2 ORF3 to the accumulation of factors involved in autophagy, (c) ORF7b to the factors involved in innate immunity and (d) ORF8 to the factors involved in TGFβ signaling. (e) Overview of perturbations to host-cell autophagy, induced by distinct proteins of SARS-CoV-2, derived from the network diffusion model and overlaid with the changes in protein levels, ubiquitination and phosphorylation induced by SARS-CoV-2 infection. (f-i) Western blot of autophagy-associated factors MAP1LC3B-II and SQSTM1 accumulation upon SARS-CoV-2 ORF3 expression in (f) HEK293R1 and (g-i) SARS-CoV-2 infection of A549-ACE2 cells. (h) Profile plot of SQSTM1 MS intensity and (i) line diagram showing SQSTM1 mRNA level relative to RPLP0 tested by qRT-PCR upon SARS-CoV-2 infection.
Taken together, the viral-host protein-protein interactions and pathway regulations observed at multiple levels identify potential vulnerability points of SARS-CoV-2 that could be targeted by well-characterized selective drugs for antiviral therapies. To test their antiviral efficacy, we established time-lapse fluorescent microscopy of SARS-CoV-2 GFP-reporter virus infection20. Inhibition of virus replication by type-I interferon treatment corroborated the necessity for SARS-CoV-2 to block this pathway and confirmed the reliability of this screening approach (Figure 4a)9,21. We tested a panel of 48 drugs modulating the pathways perturbed by the virus for their effects on SARS-CoV-2 replication (Figure 4b, Supplementary Table 9). Notably, B-RAF (Sorafenib, Regorafenib, Dabrafenib), JAK1/2 (Baricitinib) and MAPK (SB 239063) inhibitors, among others, led to a significant increase of virus growth in our in vitro infection setting (Figure 4b, Extended data Fig. 4, Supplementary Table 9). In contrast, inducers of DNA damage (Tirapazamine, Rabusertib) or the mTOR inhibitor (Rapamycin) led to suppression of the virus. The highest antiviral effect was seen for Gilteritinib (a designated FLT3/AXL inhibitor), Ipatasertib (AKT inhibitor), Prinomastat and Marimastat (matrix metalloproteinases inhibitors) (Figure 4b - e, Extended data Fig. 4, Supplementary Table 9). Remarkably, these compounds profoundly inhibited replication of SARS-CoV-2 while having no or minor influence on cell growth (Extended data Fig. 4, Supplementary Table 9). These inhibitors may perturb host pathways required by the virus or influence viral protein activity through post-translational modifications. Notably, we identified AKT as a potential kinase phosphorylating SARS-CoV-2 protein N (Extended data Fig. 2k), indicating the possibility of a direct influence of Ipatasertib on the viral protein.
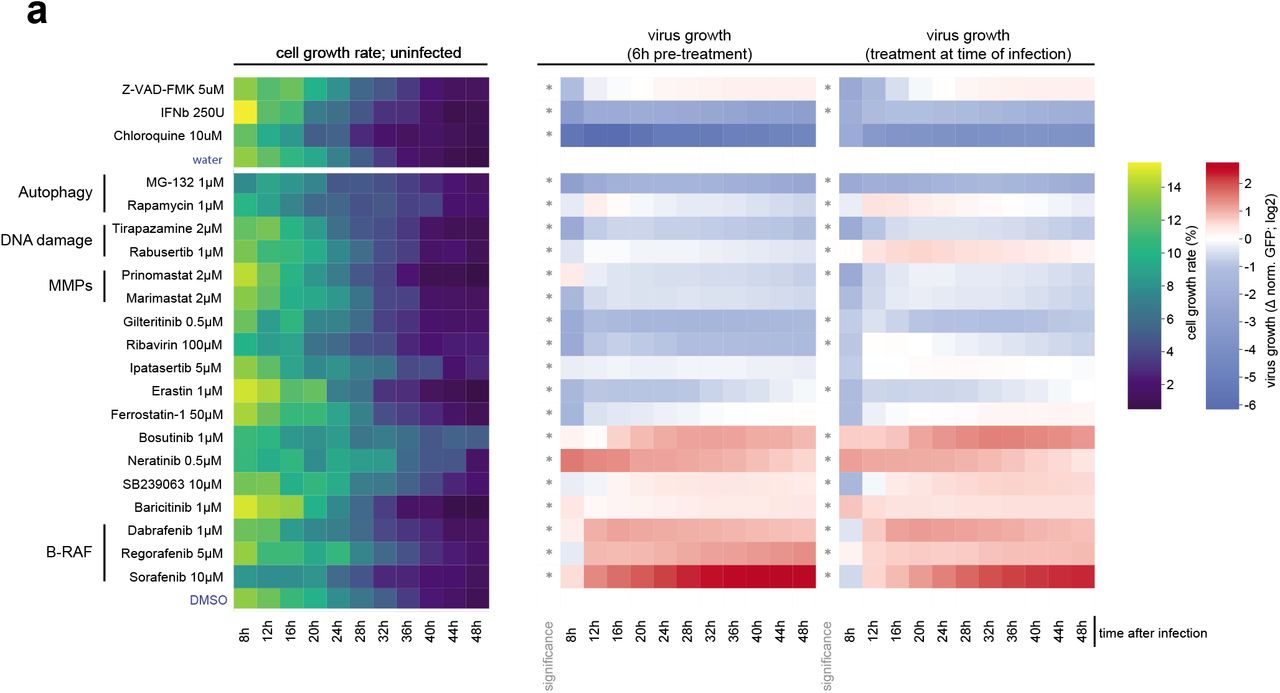
(a) A549-ACE2 cells were pre-treated for 6h or treated at the time of infection with SARS-CoV-2-GFP reporter virus (MOI 3). GFP signal and cell growth were evaluated for 48h by live cell imaging using an Incucyte S3 platform. Heatmap show the cell growth rate over time in uninfected conditions, and GFP signal vs total cell confluency and normalized to the signal measured in control treatment (water, DMSO), over time. Only treatments with significant effects on SARS-CoV-2-GFP are shown. Asterisks indicate significance to control treatment (Wilcoxon test; p-value ≤ 0.05).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) A549-ACE2 cells, exposed for 6h to the specified concentrations of interferon alpha and infected with SARS-CoV-2-GFP reporter virus (MOI 3). GFP signal and cell confluency were analysed by live-cell imaging for 48h. Line diagrams show virus growth over time of GFP-positive vs total cell area with indicated mean of four biological replicates. (b) A549-ACE2 cells were treated with the indicated drugs 6h prior to infection with SARS-CoV-2-GFP (MOI 3). Scatter plot represents GFP vs total cell area signal (y-axis) versus cell confluency in uninfected control treatments (x-axis) at 48h after infection. A confluence cutoff of −0.2 log2 fold change was applied to remove cytotoxic compounds. (c-e) as (a) but line diagrams showing virus replication after (c) Prinomastat, (d) Ipatasertib and (e) Gilteritinib pre-treatment. Asterisks indicate significance to control treatment (Wilcoxon test; p-value ≤ 0.01).
This drug screen demonstrates the value of our combined dataset that profiles the infection of SARS-CoV-2 at multiple levels. Further exploration of these rich data by the scientific community and investigating the interplay between different-omics levels will substantially advance our knowledge of coronavirus biology, in particular on the pathogenicity caused by highly virulent strains such as SARS-CoV-2 and SARS-CoV. Moreover, this resource may streamline the search for antiviral compounds and serve as a base for intelligent design of combination therapies that aim at targeting the virus from multiple, synergistic angles, thus potentiating the effect of individual drugs while minimizing side-effects on healthy tissues.
Material and Methods
Cell lines and reagents
HEK293T, A549, Vero E6 and HEK293R1 cells and their respective culturing conditions were described previously22. All cell lines were tested to be mycoplasma-free. Expression constructs for C-terminal HA tagged viral ORFs were synthesised (Twist Bioscience and BioCat) and cloned into pWPI vector as described previously23 with the following modifications: starting ATG codon was added, internal canonical splicing sites were replaced with synonymous mutations and C-terminal HA-tag, followed by amber stop codon, was added to individual viral open reading frames. C-terminally hemagglutinin(HA)-tagged ACE2 sequence was amplified from an ACE2 expression vector (kindly provided by Stefan Pöhlmann)24 into the lentiviral vector pWPI-puro. A549 cells were transduced twice, and ACE2-expressing A549 (ACE2-A549) cells were selected with puromycin. Lentiviruses production, transduction of cells and antibiotic selection were performed as described previously23. RNA-isolation (Macherey-Nagel NucleoSpin RNA plus), reverse transcription (TaKaRa Bio PrimeScript RT with gDNA eraser) and RT-qPCR (Thermo-Fisher Scientific PowerUp SYBR green) were performed as described previously25. RNA-isolation for NGS applications was performed according to manufacturer’s protocol (Qiagen RNeasy mini kit, RNase free DNase set). For detection of protein abundance by western blotting, HA-HRP (Sigma-Aldrich), ACTB-HRP (Santa Cruz), ATM, MAP1LC3B, MAVS, HSPA1A, TGFβ and SQSTM1, phospho-JNK (T183/Y185), JNK, phospho-p38 (T180/Y182), p38 (Cell Signaling), SARS-CoV-2 (Sino Biological) antibodies were used. For AP-MS and AP-WB applications, HA-beads (Sigma-Aldrich and Thermo Fisher Scientific) and Streptactin II beads (IBA Lifesciences) were used. Secondary Abs: HRP and WB imaging was performed as described previously25. For the stimulation of cells in the reporter assay, recombinant human interferon-α (IFN-α) was a kind gift from Peter Stäheli, recombinant human IFN-γ were purchased from PeproTech and IVT4 was produced as described before26. All compounds tested during the viral inhibitor assay are listed in Suppl. Table 9.
Virus strains, stock preparation, plaque assay and in vitro infection
SARS-CoV-2-MUC-IMB-1 and SARS-CoV-2-GFP strains20 were produced by infecting Vero E6 cells cultured in DMEM medium (10% FCS, 100 ug/ml Streptomycin, 100 IU/ml Penicillin) for 2 days (MOI 0,01). Viral stock was harvested and spun twice (1000g/10min) before storage at −80°C.Titer of viral stock was determined by plaque assay. Confluent monolayers of VeroE6 cells were infected with serial five-fold dilutions of virus supernatants for 1 h at 37□°C. The inoculum was removed and replaced with serum-free MEM (Gibco, Life Technologies) containing 0.5% carboxymethylcellulose (Sigma-Aldrich). Two days post-infection, cells were fixed for 20 minutes at room temperature with formaldehyde directly added to the medium to a final concentration of 5%. Fixed cells were washed extensively with PBS before staining with H2O containing 1% crystal violet and 10% ethanol for 20 minutes. After rinsing with PBS, the number of plaques was counted and the virus titer was calculated.
A549-ACE2 cells were infected with SARS-CoV-2-MUC-IMB-1 strain (MOI 3) for the subsequent experiments. At each time point, the samples were washed once with 1x TBS buffer and harvested in SDC lysis buffer (100 mM Tris HCl pH 8.5; 4% SDC) or 1x SSB lysis buffer (62.5 mM Tris HCl pH 6.8; 2% SDS; 10% glycerol; 50 mM DTT; 0.01% bromophenol blue) or RLT (Qiagen) for proteome-phosphoproteome-ubiquitinome,western blot, and transcriptome analyses, respectively. The samples were heat-inactivated and frozen at −80°C until further processing, as described in the following sections.
Affinity purification mass spectrometric analyses of SARS-COV-2, SARS-COV and HCoV protein expressing A549 cells
For the determination of SARS-COV-2, SARS-COV and partial HCoV interactomes, four replicate affinity purifications were performed for each HA-tagged viral protein. A549 cells (6×106 cells per 15cm dish) were transduced with HA-tagged SARS-COV-2, SARS-COV or HCoV protein coding lentivirus and harvested three days post transduction. Cell pellets of two 15-cm dishes were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1.5 mM MgCl2, 0.2% (v/v) NP-40, 5% (v/v) glycerol, cOmplete protease inhibitor cocktail (Roche), 0.5% (v/v) 750 U/μl Sm DNAse) and sonicated (5 min, 4°C, 30 sec on, 30 sec off, low settings; Bioruptor, Diagenode SA). Following normalization of protein concentrations cleared lysates, virus protein-bound host proteins were enriched by adding 50 μl anti-HA-agarose slurry (Sigma-Aldrich, A2095) with constant agitation for 3h at 4°C. Non-specifically bound proteins were removed by four subsequent washes with lysis buffer followed by three detergent-removal steps with washing buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1.5 mM MgCl2, 5% (v/v) glycerol). Enriched proteins were denatured, reduced, alkylated and digested by addition of 200 μl digestion buffer (0.6 M GdmCl, 1 mM TCEP, 4 mM CAA, 100 mM Tris-HCl pH 8, 0.5 μg LysC (WAKO Chemicals), 0.5 μg trypsin (Promega) at 30°C overnight. Peptide purification on StageTips with three layers of C18 Empore filter discs (3M) and subsequent mass spectrometry analysis was performed as described previously22,23. Briefly, purified peptides were loaded onto a 20□cm reverse-phase analytical column (75□μm diameter; ReproSil-Pur C18-AQ 1.Mμm resin; Dr. Maisch) and separated using an EASY-nLC 1200 system (Thermo Fisher Scientific) with a 90 min gradient (80% acetonitrile, 0.1% formic acid; 5% (80% acetonitrile) to 30% for 65 min, 30% to 95% for 10 min, wash out at 95% for 5 min, readjustment to 5% in 10 min) at a flow rate of 300 nl per min. Eluting peptides were directly analyzed on a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) with data-dependent acquisition including repeating cycles of one MS1 full scan (150-2,000 m/z, R□=□60,000 at 200□m/z) followed by 15 MS2 scans of the highest abundant isolated and higher-energy collisional dissociation fragmented peptide precursors.
Proteome analyses of SARS-COV-2, SARS-COV and HCoV protein expressing cells
For the determination of proteome changes in A549 cells expressing SARS-COV-2, SARS-COV or HCoV proteins, a fraction of 1×106 lentivirus-transduced cells from the affinity purification samples were lysed in guanidinium chloride buffer (6 M GdmCl, 10 mM TCEP, 40 mM CAA, 100 mM Tris-HCl pH 8), boiled at 95°C for 8 min and sonicated (10 min, 4°C, 30 sec on, 30 sec off, high settings). Protein concentrations of cleared lysates were normalized to 50 μg and proteins were pre-digested with 1 μg LysC at 37°C for 1h followed by a 1:10 dilution (100 mM Tris-HCl pH 8) and overnight digestion with 1 μg trypsin at 30°C. Peptide purification on StageTips with three layers of C18 Empore filter discs (3M) and subsequent mass spectrometry analysis was performed as described previously22,23. Briefly, 300 ng of purified peptides were loaded onto a 50 cm reversed phase column (75 μm inner diameter, packed in house with ReproSil-Pur C18-AQ 1.9 μm resin [Dr. Maisch GmbH]). The column temperature was maintained at 60°C using a homemade column oven. A binary buffer system, consisting of buffer A (0.1% formic acid (FA)) and buffer B (80% ACN, 0.1% FA), was used for peptide separation, at a flow rate of 300 nl/min. An EASY-nLC 1200 system (Thermo Fisher Scientific), directly coupled online with the mass spectrometer (Q Exactive HF-X, Thermo Fisher Scientific) via a nano-electrospray source, was employed for nano-flow liquid chromatography. Peptides were eluted by a linear 80 min gradient from 5% to 30% buffer B (0.1% v/v formic acid, 80% v/v acetonitrile), followed by a 4 min increase to 60% B, a further 4 min increase to 95% B, a 4 min plateau phase at 95% B, a 4 min decrease to 5% B and a 4 min wash phase of 5% B. To acquire MS data, the data-independent acquisition (DIA) scan mode operated by the XCalibur software (Thermo Fisher) was used. DIA was performed with one full MS event followed by 33 MS/MS windows in one cycle resulting in a cycle time of 2.7 seconds. The full MS settings included an ion target value of 3 x 106 charges in the 300 – 1650 m/z range with a maximum injection time of 60 ms and a resolution of 120,000 at m/z 200. DIA precursor windows ranged from 300.5 m/z (lower boundary of first window) to 1649.5 m/z (upper boundary of 33rd window). MS/MS settings included an ion target value of 3 x 106 charges for the precursor window with an Xcalibur-automated maximum injection time and a resolution of 30,000 at m/z 200.
To generate the proteome library for DIA measurements purified peptides from the first replicates and the fourth replicates of all samples were pooled separately and 25 μg of peptides from each pool were fractionated into 24 fractions by high pH reversed-phase chromatography as described earlier27. During each separation, fractions were concatenated automatically by shifting the collection tube every 120 seconds. In total 48 fractions were dried in a vacuum centrifuge, resuspended in buffer A* (0.3% TFA/ 2% ACN) and subsequently analyzed by a top12 data-dependent acquisition (DDA) scan mode using the same LC gradient and settings. The mass spectrometer was operated by the XCalibur software (Thermo Fisher). DDA scan settings on full MS level included an ion target value of 3 x 106 charges in the 300 – 1650 m/z range with a maximum injection time of 20 ms and a resolution of 60,000 at m/z 200. At the MS/MS level the target value was 105 charges with a maximum injection time of 60 ms and a resolution of 15,000 at m/z 200. For MS/MS events only, precursor ions with 2-5 charges that were not on the 20 s dynamic exclusion list were isolated in a 1.4 m/z window. Fragmentation was performed by higher-energy C-trap dissociation (HCD) with a normalized collision energy of 27eV.
Infected proteome-phosphoproteome time-course
Frozen cell lysates of infected A549-ACE2 harvested at 3, 6, 12, 18, 24 and 30h post infection were thawed on ice and boiled for 5 min at 95 degrees. Lysates were transferred to a 96-well plate (Covaris) and sonicated for 5 min. Protein concentrations were estimated by tryptophan assay28 and protein material was equalized to 200 μg per sample. CAA (10 mM) and TCEP (40 mM) along with trypsin (1:100 w/w, Sigma-Aldrich) and LysC (1/100 w/w, Wako) were added to the samples that were digested at 37°C overnight. Peptides were desalted using SDB-RPS cartridges (PreOmics).
Briefly, samples were mixed with 300 μl 1% TFA in isopropanol, loaded onto cartridges and washed with 200 μl 1% TFA in isopropanol and 200 μl 0.2% TFA. Peptides were eluted with 150 μl of 1.25% Ammonium hydroxide (NH4OH)/ 80% ACN and 10 μl aliquots were taken and dried separately for global proteome analysis. The rest was dried using a SpeedVac centrifuge (Eppendorf, Concentrator plus) and resuspended in 105 μl of equilibration buffer (1% TFA/ 80% ACN) for phosphopeptide enrichment. The AssayMAP Bravo robot (Agilent) performed the enrichment for phosphopeptides by priming AssayMAP cartridges (packed with 5 μl Fe(III)-NTA) with 1 % TFA in 99 % ACN followed by equilibration in equilibration buffer and loading of peptides. Enriched phosphopeptides were eluted with 1 % Ammonium hydroxide, dried in a vacuum centrifuge and resuspended in 1 % FA buffer. Evotips were activated by wetting in 40 ml 1-propanol in Evotipbox and subsequently washed with 100 μl of 0.1% FA. Peptides were loaded onto tips which were subsequently washed with 100 μl of 0.1 % FA. The tips were then loaded with 100 μl 0.1 % FA and centrifuged very shortly.
To generate the proteome library for DIA measurements cells were lysed in 4 % SDC and 100 mM Tris pH 8.4, followed by sonication, protein quantification, reduction, and alkylation and desalting using SDB-RPS cartridges (see above). 100 μg of peptides were fractionated into 24 fractions by high pH reversed-phase chromatography as described earlier27. Fractions were concatenated automatically by shifting the collection tube every 120 seconds and subsequently dried in a vacuum centrifuge and resuspended in buffer A* (0.3% TFA/ 2% ACN).
To generate the library for phosphoproteome DIA measurements A549 cells were treated with 100 ng/ml Calyculin and 2 mM Sodium orthovanadate for 20 min. Cells were lysed and treated as for the proteome library generation. After overnight digestion, peptides were desalted using Sepax Extraction columns. 5.5 mg of desalted peptides were fractionated into 84 fractions on a C18 reversed-phase column (4.6 x 150 mm, 3.5 μm bead size) under basic conditions using a Shimadzu UFLC operating at 1 ml/minute. Buffer A (2.5 mM Ammoniumbicarbonat in MQ) and Buffer B (2.5 mM ABC in 80% CAN) were used to separate peptides on a linear gradient of 2.5% B to 44% B for 64 minutes and 44% B to 75% B for 5 minutes before a rapid increase to 100% B which was kept for 5 minutes. Fractions were subsequently concatenated into 24 fractions and lyophilized. Phosphopeptides of these 24 fractions were enriched using the AssayMAP Bravo robot and loaded on Evotips. Another 5.5 mg of desalted peptides were split into 24 samples and enriched for phosphopeptides by the AssayMAP Bravo robot. Eluted phoshopeptides were combined, dried and fractionated into 24 fractions by neutral pH reversed-phase chromatography27. Fractions were dried and loaded on Evotips as described above.
Infected proteome-phosphoproteome-diGly-proteome (6 and 24hr)
Frozen cell lysates of infected A549-ACE2 harvested at 6 and 24h post infection were thawed on ice and sonicated for 1 min (Branson Sonifierer). Protein concentrations were estimated by tryptophan assay28. To reduce and alkylate proteins, samples were incubated for 5 min at 45°C with CAA and TCEP, final concentrations of 10 mM and 40 mM, respectively. Samples were digested overnight at 37°C using trypsin (1:100 w/w, Sigma-Aldrich) and LysC (1/100 w/w, Wako).
For proteome analysis, 10 μg of peptide material were desalted using SDB-RPS StageTips (Empore) (2). Briefly, samples were diluted with 1% TFA in isopropanol to a final volume of 200 μl and loaded onto StageTips, subsequently washed with 200 μl of 1% TFA in isopropanol and 200 μl 0.2% TFA/ 2% ACN. Peptides were eluted with 60 μl of 1.25% Ammonium hydroxide (NH4OH)/ 80% ACN and dried using a SpeedVac centrifuge (Eppendorf, Concentrator plus). They were resuspended in buffer A* prior to LC-MS/MS analysis. Peptide concentrations were measured optically at 280nm (Nanodrop 2000, Thermo Scientific) and subsequently equalized using buffer A*. 500ng peptide was subjected to LC-MS/MS analysis.
To generate phosphoproteome data, the EasyPhos protocol was used for the enrichment of phosphopeptides29. Briefly, samples were adjusted with the lysis buffer to a volume of 300 μl, transferred into a 96-deep-well plate and mixed with 100 μl 48% TFA, 8 mM KH2PO4. Phosphopeptides were captured by 5 min incubation at 40°C with 5 mg TiO2 beads. Thereafter, beads were washed 5 times with 5% TFA/ 60% isopropanol, followed by a transfer in 0.1% TFA/ 60% isopropanol into C8 StageTips. Phosphopeptides were eluted twice with 30 μl 20% NH4OH/ 40% ACN and concentrated for 30 min at 45°C using a SpeedVac centrifuge. Concentrated samples were immediately diluted with 100 μl 1% TFA in isopropanol and transferred into SDB-RPS StageTips. Peptides were washed, eluted and dried as described above. Dried peptides were resuspended in 6 μl buffer A* and 5 μl was subjected to LC-MS/MS analysis.
For diGly peptide enrichment, samples were four-fold diluted with 1% TFA in isopropanol and loaded onto SDB-RPS cartridges (Strata™-X-C, 30 mg/ 3 ml, Phenomenex Inc), pre-equilibrated with 4 ml 30% MeOH/1% TFA and washed with 4 ml 0.2% TFA. Samples were washed twice with 4 ml 1% TFA in isopropanol, once with 0.2% TFA/ 2% ACN and eluted twice with 2 ml 1.25% NH4OH/ 80% ACN. Eluted peptides were diluted with ddH2O to a final ACN concentration of 35%, snap frozen and lyophilized. Lyophilized peptides were reconstituted in IAP buffer (50 mM MOPS, pH 7.2, 10 mM Na2HPO4, 50 mM NaCl) and the peptide concentration was estimated by tryptophan assay. K--GG remnant containing peptides were enriched using the PTMScan® Ubiquitin Remnant Motif (K--GG) Kit (Cell Signaling Technology). Crosslinking of antibodies to beads and subsequent immunopurification was performed with slight modifications as previously described30. Briefly, two vials of crosslinked beads were combined and equally split into 16 tubes (~31 μg of antibody per tube). Equal peptide amounts (800 μg) were added to crosslinked beads and the volume was adjusted with IAP buffer to 1 ml. After 1h of incubation at 4°C and gentle agitation, beads were washed twice with cold IAP and 5 times with cold ddH2O. Thereafter, peptides were eluted twice with 50 μl 0.15% TFA. Eluted peptides were desalted and dried as described for proteome analysis with the difference that 0.2% TFA instead of 1%TFA in isopropanol was used for the first wash. Eluted peptides were resuspended in 9 μl buffer A* and 4 μl was subjected to LC-MS/MS analysis.
DDA Measurements
Samples were loaded onto a 50 cm reversed phase column (75 μm inner diameter, packed in house with ReproSil-Pur C18-AQ 1.9 μm resin [Dr. Maisch GmbH]). The column temperature was maintained at 60°C using a homemade column oven. A binary buffer system, consisting of buffer A (0.1% formic acid (FA)) and buffer B (80% ACN plus 0.1% FA), was used for peptide separation, at a flow rate of 300 nl/min. An EASY-nLC 1200 system (Thermo Fisher Scientific), directly coupled online with the mass spectrometer (Q Exactive HF-X, Thermo Fisher Scientific) via a nanoelectrospray source, was employed for nano-flow liquid chromatography.
For proteome measurements, we used a gradient starting at 5% buffer B and stepwise increasing to 30% in 95 min, 60% in 5 min and 95% in 5 min. The mass spectrometer was operated in Top12 data-dependent mode (DDA) with a full scan range of 300-1650 m/z at 60,000 resolution with an automatic gain control (AGC) target of 3e6 and a maximum fill time of 20ms. Precursor ions were isolated with a width of 1.4 m/z and fragmented by higher-energy collisional dissociation (HCD) (NCE 27%). Fragment scans were performed at a resolution of 15,000, an AGC of 1e5 and a maximum injection time of 60 ms. Dynamic exclusion was enabled and set to 30 s.
For phosphopeptide samples 5μl were loaded and eluted with a gradient starting at 3% buffer B and stepwise increased to 19% in 60 min, 41% in 30 min, 36% in 39 min and 95% in 5 min. The mass spectrometer was operated in Top10 DDA with a full scan range of 300-1600 m/z at 60,000 resolution with an AGC target of 3e6 and a maximum fill time of 120 ms. Precursor ions were isolated with a width of 1.4 m/z and fragmented by HCD (NCE 28%). Fragment scans were performed at a resolution of 15,000, an AGC of 1e5 and a maximum injection time of 50 ms. Dynamic exclusion was enabled and set to 30 s.
For the analysis of K--GG peptide samples, we use a gradient starting at 3% buffer B and stepwise increased to 7% in 6 min, 20% in 49 min, 36% in 39 min, 45% in 10 min and 95% in 4 min. The mass spectrometer was operated in Top12 DDA with a full scan range of 300-1350 m/z at 60,000 resolution with an AGC target of 3e6 and a maximum fill time of 20ms. Precursor ions were isolated with a width of 1.4 m/z and fragmented by HCD (NCE 28%). Fragment scans were performed at a resolution of 30,000, an AGC of 1e5 and a maximum injection time of 110 ms. Dynamic exclusion was enabled and set to 15 s.
DIA Measurements
Samples were loaded onto a 15 cm reversed phase column (150 μm inner diameter, packed in house with ReproSil-Pur C18-AQ 1.9 μm resin [Dr. Maisch GmbH]), which was kept in a homemade column oven at 60°C. Peptides were separated by the Evosep One LC system using the preprogrammed 44 minutes gradient for proteome samples and the 21 minutes gradient for phosphoproteome samples. The same gradients were used for the acquisition of proteome and phosphoproteome library fractions. The Evosep One system was coupled to a Q Exactive HF-X Orbitrap (Thermo Fisher Scientific) via a nano-electrospray source.
The proteome and phosphoproteome fractions we used to build the libraries were measured in DDA mode. To acquire proteome fractions the mass spectrometer was operated in Top15 data-dependent mode with a full scan range of 300-1650 m/z at 60,000 resolution, an automatic gain control (AGC) target of 3e6 and a maximum fill time of 20ms. For the generation of the phosphoproteome library the mass spectrometer was operated in Top12 data-dependent mode (DDA) with a full scan range of 300-1650 m/z at 60,000 resolution, an automatic gain control (AGC) target of 3e6 and a maximum fill time of 25ms. For both libraries precursor ions were isolated with a width of 1.4 m/z and fragmented by higher-energy collisional dissociation (HCD) (NCE 27%). Fragment scans were performed at a resolution of 15,000, an AGC of 1e5 and a maximum fill time of 28 ms. Dynamic exclusion was enabled and set to 30 s for the proteome library and 20 s for the phosphoproteome library.
For proteome and phosphoproteome DIA measurements, full MS resolution was set to 60,000 with a full scan range of 300-1650 m/z, a maximum fill time of 60 ms and an automatic gain control (AGC) target of 3e6. One full scan was followed by 32 windows with a resolution of 30,000 and a maximum fill time of 54 ms for proteome measurements and 40 windows for phosphoproteome measurements with a resolution of 15,000 and maximum fill time of 28 ms in profile mode. Precursor ions were fragmented by higher-energy collisional dissociation (HCD) (NCE 27%).
Data-processing of affinity purification, (phospho)proteome and ubiquitinome LC-MS/MS analyses
Raw MS data files of experiments conducted in DDA mode were processed with MaxQuant (version 1.6.14) using the standard settings and label-free quantification enabled (LFQ min ratio count 1, normalization type none, stabilize large LFQ ratios disabled). For profiling of post-translational modifications, additional variable modifications for ubiquitination (GlyGly(K)) and phosphorylation (Phospho(STY)) were added. Spectra were searched against forward and reverse sequences of the reviewed human proteome including isoforms (UniprotKB, release 10.2019) and SARS-COV-2, SARS-COV and HCoV proteins by the built-in Andromeda search engine31.
For AP-MS data, the alternative protein group definition was used: only the peptides identified in AP-MS samples could be regarded as protein group-specific, protein groups that differed by the single specific peptide or had less than 25% different specific peptides were merged to extend the set of peptides used for protein group quantitation and reduce the number of protein isoform-specific interactions.
For the experiments conducted in DIA mode, Spectronaut version 13 (Biognosys) was used to generate the proteome and phosphoproteome libraries from DDA runs by combining files of respective fractionations using the human fasta file (Uniprot, 2019, 42,431 entries). For the generation of the proteome library default settings were left unchanged. For the phosphoproteome library generation 2 x 24 files received by both fractionation strategies were combined and phosphorylation at Serine/Threonine/Tyrosine was added as variable modification to default settings. Maximum number of fragment ions per peptide was increased from 6 to 25. Proteome DIA files were analyzed using the proteome library with default settings and disabled cross run normalization. Phospho DIA files were analyzed using the phosphoproteome library using default settings with disabled PTM localization filter and cross run normalization. To search for viral proteins, we also generated the “hybrid” spectral library by merging DDA proteome library with a direct-DIA library generated from the DIA analysis of DIA proteome samples. For this search, the sequences of viral proteins were added to the human fasta file.
Bioinformatic analysis
Unless otherwise specified, the bioinformatic analysis was done in R (version 3.6), Julia (version 1.4) and Python (version 3.8) using a collection of in-house scripts (available upon request).
Statistical analysis of MS data
For all MS datasets, except DDA phosphoproteome and ubiquitinome data, the Bayesian linear random effects models were used to define how the abundances of proteins change between the conditions. To specify and fit the models we employed msglm R package (https://github.com/innatelab/msglm), which depends on rstan package (version 2.19)32 for inferring the posterior distribution of the model parameters. In all the models, the effects corresponding to the experimental conditions have regularized horseshoe+ priors33, while the batch effects have normally distributed priors. Laplacian distribution was used to model the instrumental error of MS intensities. For each MS instrument used, the heteroscedastic intensities noise model was calibrated with the technical replicate MS data of the instrument. These data were also used to calibrate the logit-based model of missing MS data (the probability that the MS instrument will fail to identify the protein given its expected abundance in the sample). The model was fit using unnormalized MS intensities data. Instead of transforming the data by normalization, the inferred protein abundances were scaled by the normalization multiplier of each individual MS sample to match the expected MS intensity of that sample. This allows taking the signal-to-noise variation between the samples into account when fitting the model. Due to high computational intensity, the model was applied to each protein group separately. For all the models, 4000 iterations (2000 warmup + 2000 sampling) of the No-U-Turn Markov Chain Monte Carlo were performed in 7 or 8 independent chains, every 4th sample was collected for posterior distribution of the model parameters. For estimating the statistical significance of protein abundance changes between the two experimental conditions, the P-value was defined as the probability that a random sample from the posterior distribution of the first condition would be smaller (larger) than a random sample drawn from the second condition. No multiple hypothesis testing corrections were applied, since this is handled by the choice of the model priors.
Statistical analysis of AP-MS data and filtering for specific interactions
Given the sparsity of the AP-MS data (each peptide is quantified in a small fraction of experiments), to take advantage of the missing data modeling by msglm, the statistical model was applied directly to the MS1 intensities of protein group-specific LC peaks (evidence.txt table of MaxQuant output). In R GLM formula language, the model could be specified as
 where APMS effect models the average shift of intensities in AP-MS data in comparison to full proteome samples, Bait is the average enrichment of a protein in AP-MS experiments of homologous proteins of both SARS-CoV and SARS-CoV-2, and Bait: Virus corresponds to the virus-specific changes in protein enrichment. MS1peak is the log-ratio between the intensity of a given peak and the total protein abundance (the peak is defined by its peptide sequence, PTMs and the charge; it is assumed that the peak ratios do not depend on experimental conditions34), and MSbatch accounts for batch-specific variations of protein intensity. APMS, Bait and Bait: Virus effects were used to reconstruct the batch effect-free abundance of the protein in AP-MS samples.
where APMS effect models the average shift of intensities in AP-MS data in comparison to full proteome samples, Bait is the average enrichment of a protein in AP-MS experiments of homologous proteins of both SARS-CoV and SARS-CoV-2, and Bait: Virus corresponds to the virus-specific changes in protein enrichment. MS1peak is the log-ratio between the intensity of a given peak and the total protein abundance (the peak is defined by its peptide sequence, PTMs and the charge; it is assumed that the peak ratios do not depend on experimental conditions34), and MSbatch accounts for batch-specific variations of protein intensity. APMS, Bait and Bait: Virus effects were used to reconstruct the batch effect-free abundance of the protein in AP-MS samples.
The modeling provided the enrichment estimates for each protein in each AP experiment. Specific APMS interactions had to pass the two tests. In the first test, the enrichment of the candidate protein in a given bait AP was compared against the background, which was dynamically defined for each interaction to contain the data from all other baits, where the abundance of the candidate was within 50%-90% percentile range (excluding top 10% baits from the background allowed the protein to be shared by a few baits in the resulting AP-MS network). The non-targeting control and Gaussia luciferase baits were always preserved in the background. Similarly, to filter out any potential sideeffects of very high bait protein expression, the ORF3 homologs were always present in the background of M interactors and vice versa. To rule out the influence of the batch effects, the second test was applied. It was defined similarly to the first one, but the background was constrained to the baits of the same batch, and 40%-80% percentile range was used. In both tests, the protein has to be 4 times enriched against the background (16 times for highly expressed baits: ORF3, M, NSP13, NSP5, NSP6, ORF3a, ORF7b, ORF8b, HCoV ORF4a) with the P-value ≤ 1E-3.
Additionally, we excluded the proteins that, in the viral protein expression data, have shown upregulation, and their enrichment in AP-MS data was less than 16 times stronger than observed upregulation effects. Finally, to exclude the carryover of material between the samples sequentially analyzed by MS, we removed the putative interactors, which were also enriched at higher levels in the samples of the preceding bait, or the one before it.
For the analysis of interaction specificity between the homologous viral proteins, we estimated the significance of interaction enrichment difference (corrected by the average difference between the enrichment of the shared interactors to adjust for the bait expression variation). Specific interactions have to be 4 times enriched in comparison to the homolog with P-value ≤ 1E-3.
Statistical analysis of DIA proteome effects upon viral protein overexpression
The statistical model of the viral protein overexpression data set was similar to AP-MS data, except that protein-level intensities provided by Spectronaut were used. The PCA analysis of the protein intensities has identified that the 2nd principal component is associated with the batch-dependent variations between the samples. To exclude their influence, this principal component was added to the experimental design matrix as an additional batch effect.
As with AP-MS data, the two statistical tests were used to identify the significantly regulated proteins. First, the absolute value of log2-fold change of protein abundance upon overexpression of a given viral protein in comparison to the control samples had to be above 0.25 with P-value ≤ 1E-3. Second, the protein had to be significantly regulated (same log2-fold change and P-value applied) against the background distribution of its abundance in the selected experiments of the same batch (experiments, where the tested protein abundance was within the 20%-80% percentile range of the whole batch, were dynamically selected for each protein).
Statistical analysis of DDA proteome data of virus infection
For DDA proteome data the following linear model was used:
 where after(24h) effect corresponds to the protein abundance changes in mock-infected samples that happened between 6h and 24h, and treatment:after(ti) (ti=6,24) is the effect of interaction between the SARS-CoV-2 infection and the timepoint, and corresponds to the changes in the virus-infected cells (in comparison to the mock-infected samples) within the first 6h hours after infection, and between 6h and 24h after infection, respectively. The absolute value of log2-fold change between the conditions below 0.25 and the corresponding P-value ≤ 1E-3 criteria were used to define the significant changes.
where after(24h) effect corresponds to the protein abundance changes in mock-infected samples that happened between 6h and 24h, and treatment:after(ti) (ti=6,24) is the effect of interaction between the SARS-CoV-2 infection and the timepoint, and corresponds to the changes in the virus-infected cells (in comparison to the mock-infected samples) within the first 6h hours after infection, and between 6h and 24h after infection, respectively. The absolute value of log2-fold change between the conditions below 0.25 and the corresponding P-value ≤ 1E-3 criteria were used to define the significant changes.
Statistical analysis of DIA proteome and phosphoproteome data of virus infection
For DIA phosphoproteome data, to convert peptide-level output of Spectronaut into PTM site-level report, the Peptide Collapse Perseus plugin was used (https://github.com/AlexHgO/Perseus_Plugin_Peptide_Collapse)35. Phosphosites with less than 0.75 localization probability were excluded.
For DIA proteome and phosphoproteome datasets, the following linear model was used:
 where after(ti) effect corresponds to the protein abundance changes in mock-infected samples that happened between ti-1 and ti (ti=6,12,18,24,30), and treatment:after(ti) (ti=3,6,12,18,24,30) is the effect of interaction between the SARS-CoV-2 infection and the timepoint. The absolute value of log2-fold change between the conditions below 0.25 and the corresponding P-value ≤ 1E-3 criteria were used to define the significant changes for proteome data, and |log2-fold change| ≤ 0.5, P-value ≤ 1E-2 for phosphoproteome data.
where after(ti) effect corresponds to the protein abundance changes in mock-infected samples that happened between ti-1 and ti (ti=6,12,18,24,30), and treatment:after(ti) (ti=3,6,12,18,24,30) is the effect of interaction between the SARS-CoV-2 infection and the timepoint. The absolute value of log2-fold change between the conditions below 0.25 and the corresponding P-value ≤ 1E-3 criteria were used to define the significant changes for proteome data, and |log2-fold change| ≤ 0.5, P-value ≤ 1E-2 for phosphoproteome data.
Statistical analysis of DDA total proteome, phosphoproteome and ubiquitinome data 6 and 24 hours post SARS-CoV-2 infection of A549-ACE2 cells
The output of MaxQuant was analyzed with Perseus (version 1.6.14.0)36 and visualized with R (version 3.6.0) and RStudio (version 1.2.1335). For total proteome analysis, detected protein groups within the proteinGroups output table identified as known contaminants, reverse sequence matches, only identified by site or quantified in less than 3 out of 4 replicates in at least one condition were excluded. Following log2 transformation, missing values were imputed for each replicate individually by sampling values from a normal distribution calculated from the original data distribution (width = 0.3×s.d., downshift = −1.8×s.d.). Differentially regulated protein groups between mock and SARS-CoV-2 infection at 6 and 24 h.p.i. were identified via two-sided Student’s T-tests corrected for multiple hypothesis testing applying a permutation-based FDR (S0 = 0.1; FDR < 0.05, 250 randomizations). Protein groups were further removed for statistical testing if not at least one T-test condition contained a minimum of three non-imputed values. For phosphoproteome analysis, phosphosites within the Phospho (STY)Sites output table identified as known contaminants, reverse sequence matches or less than 0.75 localization probability were excluded. Following log2 transformation, phosphosite intensities were normalized based on sites that were quantified in at least 90% of all samples to account for technical variations. In detail, the median of phosphosite-specific intensities across samples was subtracted from individual intensities to normalize for different phosphosite abundances (row-wise normalization). Next, the median of normalized phosphosite intensities per sample was used as final normalization factor and subtracted from individual nonnormalized phosphosite intensities (column-wise normalization). Phosphosites were further filtered for quantification in at least 3 replicates and missing values were imputed for each replicate individually by sampling values from a normal distribution calculated from the original data distribution (width = 0.3 × s.d., downshift = −1.8 × s.d.). Differentially regulated phosphosites between mock and SARS-CoV-2 infection at 6 and 24 h.p.i. were identified via two-sided Student’s T-tests corrected for multiple hypothesis testing applying a permutation-based FDR (S0 = 0.5; FDR < 0.05, 250 randomizations). Phosphosites were further removed from statistical testing if not at least one T-test condition contained a minimum of 3 non-imputed values. For ubiquitinome analysis, ubiquitination sites within the GlyGly (K)Sites output table were processed as mention for phosphosites, but normalization for technical variation was based on ubiquitination sites quantified in more than 60% of all samples. Differentially regulated ubiquitination sites between mock and SARS-CoV-2 infection at 6 and 24 h.p.i. were identified via two-sided Student’s T-tests corrected for multiple hypothesis testing applying a permutation-based FDR (S0 = 0.1; FDR < 0.05, 250 randomizations) and removed from statistical testing if not at least one T-test condition contained a minimum of 3 non-imputed values. Annotation of detected protein groups, phosphosites and ubiquitination sites with GOBP, -MF, -CC, KEGG, Pfam, GSEA, Keywords and Corum as well as PhosphoSitePlus kinase-substrate relations and regulatory sites (version May 1st 2020)37 was performed in Perseus.
Transcriptomic analysis of SARS-COV-2 infected A549-ACE2 cells
A549-ACE2 cells used for transcriptional profiling of SARS-CoV-2 infection were cultured and infected as described above. RNA isolation was performed using RNeasy Mini kit (Qiagen) according to the manufacturer’s protocol with addition of a heat inactivation step after cell lysis. Library preparation for bulk 3’-sequencing of poly(A)-RNA was done as described previously38. Briefly, barcoded cDNA of each sample was generated with a Maxima RT polymerase (Thermo Fisher) using oligo-dT primer containing barcodes, unique molecular identifiers (UMIs) and an adapter. 5’ ends of the cDNAs were extended by a template switch oligo (TSO) and after pooling of all samples fulllength cDNA was amplified with primers binding to the TSO-site and the adapter. cDNA was fragmented and TruSeq-Adapters ligated with the NEBNext® Ultra™ II FS DNA Library Prep Kit for Illumina® (NEB) and 3’-end-fragments were finally amplified using primers with Illumina P5 and P7 overhangs. In comparison to Parekh et al.38 the P5 and P7 sites were exchanged to allow sequencing of the cDNA in read1 and barcodes and UMIs in read2 to achieve better cluster recognition. The library was sequenced on a NextSeq 500 (Illumina) with 75 cycles for the cDNA in read1 and 16 cycles for the barcodes and UMIs in read2.
As for the analysis of the transcriptome data, Gencode gene annotations v28 and the human reference genome GRCh38 were derived from the Gencode homepage (EMBL-EBI). Dropseq tool v1.1239 was used for mapping raw sequencing data to the reference genome. The resulting UMI filtered count matrix was imported into R v3.4.4. CPM (counts per million) values were calculated for the raw data and genes having a mean cpm value less than 1 were removed from the dataset. Prior differential expression analysis with DESeq2 v1.18.140, dispersion of the data was estimated with a parametric fit using a multiplicative model where infection status (MOCK, virus infected) and time were incorporated as covariates in the model matrix. The Wald test was used for determining differentially regulated genes across timepoints in individual infection states and shrunk log2 fold changes were calculated afterwards. Transcripts with low mean normalized count that were flagged by the independent filtering procedure of DESeq2 were removed and those with absolute apeglm shrunk log2 fold change > 0.5 and the p-value < 0.05 were considered differentially expressed in distinct conditions.
The data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB38744 (https://www.ebi.ac.uk/ena/data/view/PRJEB38744).
qRT-PCR analysis
RNA isolation from SARS-CoV-2 infected A549-ACE2 cells was performed as described above (Qiagen). 500 ng total RNA was used for reverse transcription with PrimeScript RT with gDNA eraser (Takara). For relative transcript quantification PowerUp SYBR Green (Applied Biosystems) was used. Primer sequences can be provided upon request.
Gene Set Enrichment Analysis
We have used Gene Ontology, Reactome and other EnrichmentMap gene sets of human proteins41 as well as protein complexes annotations from IntAct Complex Portal (version 2019.11)42 and CORUM (version 2019)43.
To find the nonredundant collection of annotations describing the unique and shared features of multiple experiments in a dataset, we have used Julia package OptEnrichedSetCoverjl (https://github.com/alyst/OptEnrichedSetCover.jl), which employs evolutionary multi-objective optimization technique to find a collection of annotation terms that have both significant enrichments in the individual experiments and minimal pairwise overlaps.
For transcription factor enrichment analysis the significantly regulated transcripts were submitted to ChEA3 web-based application44. Transcription factor – target gene set libraries from ENCODE were used45. Transcriptome, proteome, ubiquitinome and phosphoproteome changes along with unchanged transcripts/proteins/sites were submitted to the core ingenuity pathway analysis (IPA) (www.ingenuity.com).
The following cut-offs were used for differentially expressed transcripts: the absolute values of apeglm-shrunk log2 fold change > 0.5, the p-value < 0.05. Transcripts with low mean normalized count that were flagged by the independent filtering procedure of DESeq2 were removed prior pathway analysis. The following cut-offs were used for differentially expressed proteins or regulated sites: p-value <0.05 and absolute log2 fold change ≥ 0.5. Ingenuity knowledge base was used as a reference dataset, only experimentally observed findings were used for confidence filtering, additionally human species and A549-ATCC cell line filters were set. Input datasets were used to identify the most significant canonical pathways and upstream regulators (in case of transcriptome). Right-tailed Fisher’s exact test with Benjamini-Hochberg’s correction was used to calculate p-values, which are presented in Supplementary Table 3.
Prediction of Functional Links between AP-MS and viral protein overexpression data
To systematically detect functional interactions, which may connect the cellular targets of each viral protein with the downstream changes it induces on proteome level, we have used the network diffusion-based HierarchicalHotNet method13 as implemented in Julia package HierarchicalHotNet.jl (https://github.com/alyst/HierarchicalHotNet.jl). Specifically, for network diffusion with restart, we used the ReactomeFI network (version 2019)12 of cellular functional interactions, reversing the direction of functional interaction (e.g. replacing kinase→substrate interaction with substrate→kinase). The proteins with significant abundance changes upon bait overexpression (|median(log2-fold change)| ≥ 0.25, P-value ≤ 1E-3 both in the comparison against the controls and against the baits of the same batch) were used as the sources of signal diffusion with weights set to  , and the restart probability was set to 0.4. To find the optimal cutting threshold of the resulting hierarchical tree of strongly connected components (SCCs) of the weighted graph corresponding to the stationary distribution of signal diffusion and to confirm the relevance of predicted functional connections, the same procedure was applied to 1000 random permutations of vertex weights as described in Reyna et al.13 (vertex weights are randomly shuffled between the vertices with similar in- and out-degrees). Since cutting the tree of SCCs at any threshold t (keeping only the edges with weights above t) and collapsing each resulting SCC into a single node produces the directed acyclic graph of connections between SCCs, it allowed efficient enumeration of the paths from the “source” nodes (proteins perturbed by viral protein expression with vertex weight w, w ≥ 1.5) to the “sink” nodes (interactors of the viral protein). We have used this property of the tree to calculate the average source-to-sink path length at each cutting threshold of the network diffusion weighted graph. At each threshold t, the average path from source to sink nodes was calculated as:
, and the restart probability was set to 0.4. To find the optimal cutting threshold of the resulting hierarchical tree of strongly connected components (SCCs) of the weighted graph corresponding to the stationary distribution of signal diffusion and to confirm the relevance of predicted functional connections, the same procedure was applied to 1000 random permutations of vertex weights as described in Reyna et al.13 (vertex weights are randomly shuffled between the vertices with similar in- and out-degrees). Since cutting the tree of SCCs at any threshold t (keeping only the edges with weights above t) and collapsing each resulting SCC into a single node produces the directed acyclic graph of connections between SCCs, it allowed efficient enumeration of the paths from the “source” nodes (proteins perturbed by viral protein expression with vertex weight w, w ≥ 1.5) to the “sink” nodes (interactors of the viral protein). We have used this property of the tree to calculate the average source-to-sink path length at each cutting threshold of the network diffusion weighted graph. At each threshold t, the average path from source to sink nodes was calculated as:
 where Nsrc is the number of “sources”, Nsink is the number of “sinks”, Ndis is the number of disconnected pairs of sources and sinks, NSCC is the number of SCC at given threshold, LSCC(p) is the number of SCCs that the given path p from source to sink goes through, and the sum is for all paths from sources to sinks. For the generation of the diffusion network we have selected the topt threshold that maximized the difference between the median of Lavg(t) for randomly shuffled data and Lavg(t) for the real data.
where Nsrc is the number of “sources”, Nsink is the number of “sinks”, Ndis is the number of disconnected pairs of sources and sinks, NSCC is the number of SCC at given threshold, LSCC(p) is the number of SCCs that the given path p from source to sink goes through, and the sum is for all paths from sources to sinks. For the generation of the diffusion network we have selected the topt threshold that maximized the difference between the median of Lavg(t) for randomly shuffled data and Lavg(t) for the real data.
Co-immunoprecipitation and western blot analysis
HEK293T cells were transfected with pWPI plasmid encoding single HA-tagged viral proteins, alone or together with pTO-SII-HA expressing host factor of interest. 48 hours after transfection, cells were washed in PBS, flash frozen in liquid nitrogen and kept at −80°C until further processing. Coimmunoprecipitation experiments were performed as described previously22,23. Briefly, cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1.5 mM MgCl2, 0.2% (v/v) NP-40, 5% (v/v) glycerol, cOmplete protease inhibitor cocktail (Roche), 0.5% (v/v) 750 U/μl Sm DNAse) and sonicated (5 min, 4°C, 30 sec on, 30 sec off, low settings; Bioruptor, Diagenode SA). HA or Streptactin beads were added to cleared lysates and samples were incubated for 3h at 4°C under constant rotation. Beads were washed six times in the lysis buffer and resuspended in 1x SDS sample buffer 62,5 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT, 0.01% bromophenol blue). After boiling for 5 minutes at 95°C, a fraction of the input lysate and elution were loaded on NuPAGE™ Novex™ 4-12% Bis-Tris (Invitrogen), and further submitted to western blotting using Amersham Protran nitrocellulose membranes. Imaging was performed by HRP luminescence (ECL, Perkin Elmer).
SARS-CoV-2 infected A549-ACE2 cell lysates were sonicated (10 min, 4°C, 30 sec on, 30 sec off, low settings; Bioruptor, Diagenode SA). Protein concentration was adjusted based on Pierce660 assay supplemented with ionic detergent compatibility reagent. After boiling for 5 min at 95°C and brief max g centrifugation, the samples were loaded on NuPAGE™ Novex™ 4-12% Bis-Tris (Invitrogen), and blotted onto 0,22 μm Amersham™ Protran® nitrocellulose membranes (Merck). Primary and secondary antibody stainings were performed according to the manufacturer’s recommendations. Imaging was performed by HRP luminescence using Femto kit (ThermoFischer Scientific) or Western Lightning PlusECL kit (Perkin Elmer).
Reporter Assay and IFN Bioassay
The following reporter constructs were used in this study: pISRE-luc was purchased from Stratagene, EF1-α-ren from Engin Gürlevik, pCAGGS-Flag-RIG-I from Chris Basler, pIRF1-GAS-ff-luc, pWPI-SMN1-flag and pWPI-NS5 (ZIKV)-HA was described previously23,46.
For the reporter assay, HEK293RI cells were plated in 24-well plates 24 hours prior to transfection. Firefly reporter and Renilla transfection control were transfected together with plasmids expressing viral proteins using polyethylenimine (PEI, Polysciences) for untreated and treated conditions. In 18 hours cells were stimulated for 8 hours with a corresponding inducer and harvested in the passive lysis buffer (Promega). Luminescence of Firefly and Renilla luciferases was measured using dual-luciferase-reporter assay (Promega) according to the manufacturer’s instructions in a microplate reader (Tecan).
Total amounts of IFN-α/β in cell supernatants were measured by using 293T cells stably expressing the firefly luciferase gene under the control of the mouse Mx1 promoter (Mx1-luc reporter cells)47. Briefly, HEK293RI cells were seeded, transfected with pCAGGS-flag-RIG-I plus viral protein constructs and stimulated as described above. Cell supernatants were harvested in 8 hour. Mx1-luc reporter cells were seeded into 96-well plates in triplicates and were treated 24 hours later with supernatants. At 16 hours post incubation, cells were lysed in the passive lysis buffer (Promega), and luminescence was measured with a microplate reader (Tecan). The assay sensitivity was determined by a standard curve.
Viral inhibitors assay
A549-ACE2 cells were seeded into 96-well plates in DMEM medium (10% FCS, 100 ug/ml Streptomycin, 100 IU/ml Penicillin) one day before infection. Six hours before infection, or at the time of infection, the medium was replaced with 100ul of DMEM medium containing either the compounds of interest or DMSO as a control. Infection was performed by adding 10ul of SARS-CoV-2-GFP (MOI 3) per well and plates were placed in the IncuCyte S3 Live-Cell Analysis System where whole well real-time images of mock (Phase channel) and infected (GFP and Phase channel) cells were captured every 4h for 48h. Cell viability (mock) and virus growth (mock and infected) were assessed as the cell confluence per well (Phase area) and GFP area normalized on cell confluence per well (GFP area/Phase area) respectively using IncuCyte S3 Software (Essen Bioscience; version 2019B Rev2).
Acknowledgements
We thank Stefan Pöhlmann for sharing ACE2 plasmids and Robert Baier for technical assistance, Juan Pancorbo and Johannes Albert-von der Gönna from the Leibniz Supercomputing Centre (www.lrz.de) for technical assistance. We further thank the Karl Max von Bauernfeind - Verein for support for the screening microscope. Work in the author’s laboratories was supported by an ERC consolidator grant (ERC-CoG ProDAP, 817798), the German Research Foundation (PI 1084/3, PI 1084/4) and the German Federal Ministry of Education and Research (COVINET) to A.P. This work was also supported by the German Federal Ministry of Education and Research (CLINSPECT-M) to BK. AW was supported by the China Scholarship Council (CSC). The work of J.Z. was supported by the German Research Foundation (SFB1021, A01 and B01; KFO309, P3), the State of Hessen through the LOEWE Program (DRUID, B02) and the German Ministry for Education and Research (COVINET).
References
References




