

Abstract
Preeclampsia (PE) is a life-threatening disease of the pregnant women, and has a profound influence on fetal development. Mitochondrial-mediated placental oxidative stress plays key role in the etiology of PE. However, the underlying mechanism remains to be revealed. Here, we identify Rnd3, a small Rho GTPase, participating in the regulation of placental mitochondrial reactive oxygen species (ROS). We showed that Rnd3 is down-regulated in primary trophoblasts isolated from PE patients. Loss of Rnd3 in trophoblasts resulted in excessive ROS generation, cell apoptosis, mitochondrial injury and proton leakage from respiratory chain. Moreover, Rnd3 overexpression partially rescues the mitochondrial defects and oxidative stress in human PE primary trophoblasts. Mechanistically, Rnd3 physically interacts with the peroxisome proliferators-activated receptor γ (PPARγ) and promotes PPARγ-mitochondrial uncoupling protein 2 (UCP2) cascade. Forced expression of PPARγ rescues deficiency of Rnd3-mediated mitochondrial dysfunction. We conclude that Rnd3 acts as a novel protective factor in placental mitochondria through PPARγ-UCP2 signaling and highlight that downregulation of Rnd3 is a potential factor involved in the pathogenesis of PE.
Introduction
Preeclampsia (PE) is a pregnancy complication that occurs after the 20th gestational week and is characterized by maternal hypertension, proteinuria and multi-organ injuries. It affects 5-7% of pregnancies and is the leading cause of perinatal morbidity and mortality(Rana et al., 2019). Although the mechanisms responsible for the pathogenesis of PE have not been entirely clarified, placental oxidative stress along with poor vascularization is thought to be its main cause(Hladunewich et al., 2007; Steegers et al., 2010). Oxidative stress is generated by the imbalance of reactive oxygen species (ROS) and the endogenous oxidant scavenging system(Birben et al., 2012). Excessive ROS accumulation is identified in PE pathogenesis as the contributor to endothelial dysfunction and placental inflammation response(Myatt, 2010; San Juan-Reyes et al., 2020; Sanchez-Aranguren et al., 2014). Although shallow invasion of trophoblasts to decidua and subsequent failed spiral artery remodeling participate in placental ischemia and oxidative stress, accumulating evidence has shown that mitochondrial dysfunction during PE also can induce an increase in the level of ROS and cause placental oxidative stress(D’Souza et al., 2016; Vishnyakova et al., 2016; Wang and Walsh, 1998). Abnormalities in the morphology of mitochondria, including swelling mass and disappearance of cristae, have been observed in trophoblasts of PE placenta(Salgado and Salgado, 2011). However, a limited number of studies have investigated the molecular machinery of the mitochondrial ROS during PE.
Peroxisome proliferators-activated receptor γ (PPARγ) is a ligand-inducible transcription factor that plays key roles in mitochondrial biogenesis, dynamics and metabolism(Corona and Duchen, 2016; Janani and Ranjitha Kumari, 2015). Loss of PPARγ in a mouse model caused embryonic lethality with major placental defects(Waite et al., 2005), due to its key roles in placental vasculature and the differentiation of complicated trophoblast lineages(Barak et al., 2002; Barak et al., 1999; Schaiff et al., 2000). Recent studies have revealed that the levels of the circulating agonists of PPARγ were significantly reduced in PE patients(Waite et al., 2005; Waite et al., 2000), while administration of rosiglitazone, a selective PPARγ agonist, could largely ameliorate PE in a RUPP-induced PE rat model(McCarthy et al., 2011). Nevertheless, the regulatory mechanism of the downregulated PPARγ signaling in PE remains unknown.
Rnd3 is a small GTPase, also called RhoE that exhibits multiple regulatory functions in carcinogenesis, cardiovascular diseases and neural development(Dai et al., 2019; Lin et al., 2013; Liu et al., 2015; Yang et al., 2015; Yue et al., 2016; Yue et al., 2014). Rnd3 has been proposed to be a PE candidate gene based on a population study. A specific Rnd3 SNP (rs115015150) was associated with PE and several quantitative cardiovascular risk traits(Moses et al., 2012). Several research works illustrated that Rnd3 was involved with PE and regulating the proliferation, migration and invasion of trophoblast cells(Fang et al., 2018; Xu et al., 2017; Xu et al., 2018). In the present study, we identified decreases of Rnd3 expression in placentae and primary trophoblasts from PE patients, and indicated a novel protective role of Rnd3 in PE via maintaining mitochondrial function. The possible molecular mechanism is that RND3 protein physically interacts with and stabilizes PPARγ. Transcriptionally regulated by PPARγ, uncoupling protein 2 (UCP2) was involved in the regulation of mitochondrial membrane potential and ROS generation. The downregulated PPARγ-UCP2 cascade was identified and could be reversed by Rnd3 overexpression along with rescued mitochondrial dysfunction in human PE primary placental trophoblasts. The findings revealed a new function of Rnd3 in PE, and provided new insights into PE aetiology.
Methods
Human placental tissues
Human placental tissues were obtained and used for the present study with written patient-informed consent and approval by the Ethics Committee of Southern Hospital, Southern Medical University. PE placental tissues were obtained from 24 patients with a terminated pregnancy with the average gestational ages 36.1 ± 2.8 weeks. The healthy placental tissues were obtained from 30 spontaneous term pregnancies with the average gestational ages 37.0 ± 1.2 weeks. Freshly harvested tissues were immediately frozen by liquid nitrogen or processed by paraformaldehyde fixation and frozen embedding. Women with a history of smoking and drinking or with a diagnosis of chronic hypertension or gestational diabetes mellitus were excluded from the present study. There was no any difference in medications or management of preeclampsia.
Electron microscopy analysis
Freshly harvested placental tissues were fixed with a buffer containing 2.5% glutaraldehyde (diluted from 25% glutaraldehyde, G5882, Sigma-Aldrich, USA) and were subsequently processed and embedded in LX-112 medium. The ultrathin sections were stained with uranyl acetate and lead citrate and examined in a H-7650 transmission electron microscope (HITACH, Japan).
Isolation of human primary trophoblasts
The isolation of human primary trophoblasts was performed as described previously(Yue et al., 2018). Briefly, the fresh placental tissues were dissected into small pieces and were washed with PBS to remove blood cells. Digestion with the media containing 0.125% trypsin (diluted from 0.25% Trypsin-EDTA, Gibco 25200072, USA), 0.03% DNase (D4263, Sigma-Aldrich, USA) and 1% Penicillin-Streptomycin was performed at 37°C. Floating cells with trypsin supernatant were neutralized by 5% fetal bovine serum (FBS). Following 5 times of digestion, the total number of cells was harvested by centrifugation at 1,200 x g for 15 min and resuspended in DMEM medium. The trophoblast cells were purified with 5-65% Percoll density gradients (P1644-500 ml, Sigma, USA). The cell surface biomarker cytokeratin 7 (CK7) was used to identify the trophoblasts. A total of 20 immunofluorescent staining images were acquired in different fields by fluorescence microscopy. The numbers of CK7 positive cells and DAPI-labeled nuclei in each image were counted by the LAS V4.0 software. The purity of the trophoblasts was determined by the ratio of the number of CK7 positive cells over that of the total cells.
Cell culture and hypoxia treatment
Human primary trophoblasts were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin. BeWo cells (ATCC CCL-98, USA) were cultured in F-12K medium containing 2 mM Glutamine, 10% FBS and 1% Penicillin-Streptomycin. The cells were maintained at 37°C with 5% CO2. Hypoxic cell culture was performed in a hypoxic chamber (MIC-101, Billups-Rothenberg Inc, CA, USA) with 1% O2 for 16 h.
Expression vectors and adenoviral expression vectors
The subcloning of human Rnd3 cDNA was performed to generate the expression vectors GFP-RND3 and Myc-RND3 as described previously(Dai et al., 2019; Yue et al., 2016). Human PPARγ cDNA was subcloned into GV141 backbone (GeneChem, Shanghai, China) to generate the expression vector Flag-PPARγ. The AdMax™ system was used for the generation of recombinant adenovirus carrying human Rnd3 cDNA. Briefly, CMV-EGFP-Rnd3 and viral backbone plasmid pBHG were co-transfected into HEK293 cells and subsequently the recombinant adenovirus was harvested and amplified in HEK293 cells.
Mitochondrial respiratory function measurement
Mitochondrial respiration was measured at 37°C in a Seahorse XF24 Extracellular Flux Analyzer (Agilent, USA). The XF Cell Mito Stress Test Kit (103015-100, Agilent, USA) and XF24 FluxPak mini (102342-100, Agilent, USA) were used to determine mitochondrial oxygen consumption rates (OCR) in viable cells. The mitochondrial assay medium consisted of XF Base Medium (102353-100, Agilent, USA), 10 mM glucose, 5 mM sodium pyruvate and 2 mM L-glutamine at pH 7.4. The OCRs were measured by subsequent addition of 2 μM oligomycin, 1 μM FCCP and 1 μM antimycin A/1 μM rotenone.
Reverse transcription and quantitative PCR
The mRNA transcripts were quantified by quantitative PCR analysis as described previously(Yue et al., 2016). Total RNA was prepared by TRIzol extraction. The forward and reverse PCR primers (5’ to 3’) were as follows: RND3 (human): CCAGCCAGAAATTATCCAGCA/GAGAACCCGAAGTGTCCCA; GAPDH (human): GAGTCAACGGATTTGGTCGT/TTGATTTTGGAGGGATCTCG; PPARγ (human): TCCACATTACGAAGACATTCCA/CGACATTCAATTGCCATGAG; UCP2 (human): TGGGTTCAAGGCCACAGATG/CCATTGTAGAGGCTTCGGGG; PGC1α (human): AGCACTTCGGTCATCCCAG/CAGTTTATCACTTTCATCTTCGC; NRF1 (human): ATGGAGGAACACGGAGTGAC/TCATCAGCTGCTGTGGAGTT; TFAM (human): CCGAGGTGGTTTTCATCTGT/CCGCCCTATAAGCATCTTGA; PPARα (human): CTGTCTGCTCTGTGGACTCA/AGAACTATCCTCGCCGATGG; SOD1 (human): TGAAGGTGTGGGGAAGCATT/GTCACATTGCCCAAGTCTCC; SOD2 (human): TTTTGGGGTATCTGGGCTCC/TCAAAGGAACCAAAGTCACGT. GAPDH expression levels were used for qPCR normalization. The expression levels were determined by the 2-ΔΔCt threshold cycle method.
Luciferase assay
The luciferase reporter vector with the 1,000 bp promoter of the human UCP2 gene was generated with a GV238 backbone (GeneChem, Shanghai, China). The luciferase assay was conducted as described previously(Yue et al., 2016). Each measurement was repeated three times. All results were normalized according to the co-transfected Renilla luciferase enzyme activity (E1910, Promega).
Western immunoblotting, immunoprecipitation and ELISA
The protein samples for western blot analysis were prepared as described previously(Yue et al., 2016) and the immunoblotting densitometry was quantified by the ImageJ Software (NIH, USA). For immunoprecipitation, 293T cells were co-transfected with myc-Rnd3 and flag-PPARγ expression vectors. The cells were lysed in RIPA lysis buffer containing protease inhibitors. Cell lysates were incubated with protein A/G magnetic beads (88802, Thermo Fisher Scientific, USA) and either mouse IgG (5415S, Cell Signaling Technology, USA) or anti-Myc-Tag antibody at 4°C overnight. The beads were washed with lysis buffer for 4 times and boiled with 4X SDS loading buffer. The samples were analyzed by immunoblotting, and identified with an anti-PPARγ primary antibody. We used the conformation specific secondary antibody to recognize only the primary antibody but not the heavy and light chain of the antibody used for immunoprecipitation.
The antibodies used for the present study were from the following sources: anti-Cytokeratin 7 (ab9021, Abcam, USA); anti-PPARγ (2443S), anti-UCP2 (89326S), anti-Rnd3 (3664S), anti-Lamin B1 (12586S), anti-Myc-Tag (2276S), and mouse anti-rabbit IgG (Conformation Specific, 5127S) from Cell Signaling Technology, USA. Equal protein loading for immunoblotting was verified by the intensity of the β-actin blot (ab8226, Abcam, USA). 8-Isoprostane concentration was assessed by the 8-isoprostane ELISA Kit (516351, Cayman Chemical, USA).
Fluorescence staining
For histological analysis, fresh placental tissues were embedded in the Tissue-Tek O.C.T. Compound (4583, Sakura, USA) and frozen sections were used for dihydroethidium (DHE) staining (D1168, Thermo Fisher Scientific, USA). Cellular JC-1 (T3168, Thermo Fisher Scientific, USA) staining and DHE staining were performed with viable BeWo cells, respectively. TUNEL staining (11684795910, Roche, Germany) was performed in fixed cells for apoptosis detection.
Statistical analysis
The data was expressed as the mean ± standard deviation (SD). An unpaired, two-tailed Student’s t test was used for two-group comparison. The one-way ANOVA followed by the Student-Newman-Keuls method was used for multiple-group comparison. All analyses were conducted using GraphPad Prism 8.0. A value of P<0.05 was considered for significant difference.
Results
The expression of Rnd3 is downregulated in human placentae with PE
To investigate the role of Rnd3 in PE, its expression levels were measured in 24 human placentae from PE patients and 30 healthy controls. Women with a history of smoking and drinking or with a diagnosis of chronic hypertension or gestational diabetes mellitus were excluded from the present study. The clinical characteristics of all pregnant women were presented in Table 1. In the placentae with PE, a 62.9% decrease was noted in the RND3 protein levels (Fig. 1A) and a 46.0% decrease was noted in the Rnd3 mRNA level (Fig. 1B). This clinical observation suggested that Rnd3 may act as a potential regulator in the pathogenesis of PE.
Clinical characteristics of the PE group and of the healthy control group.
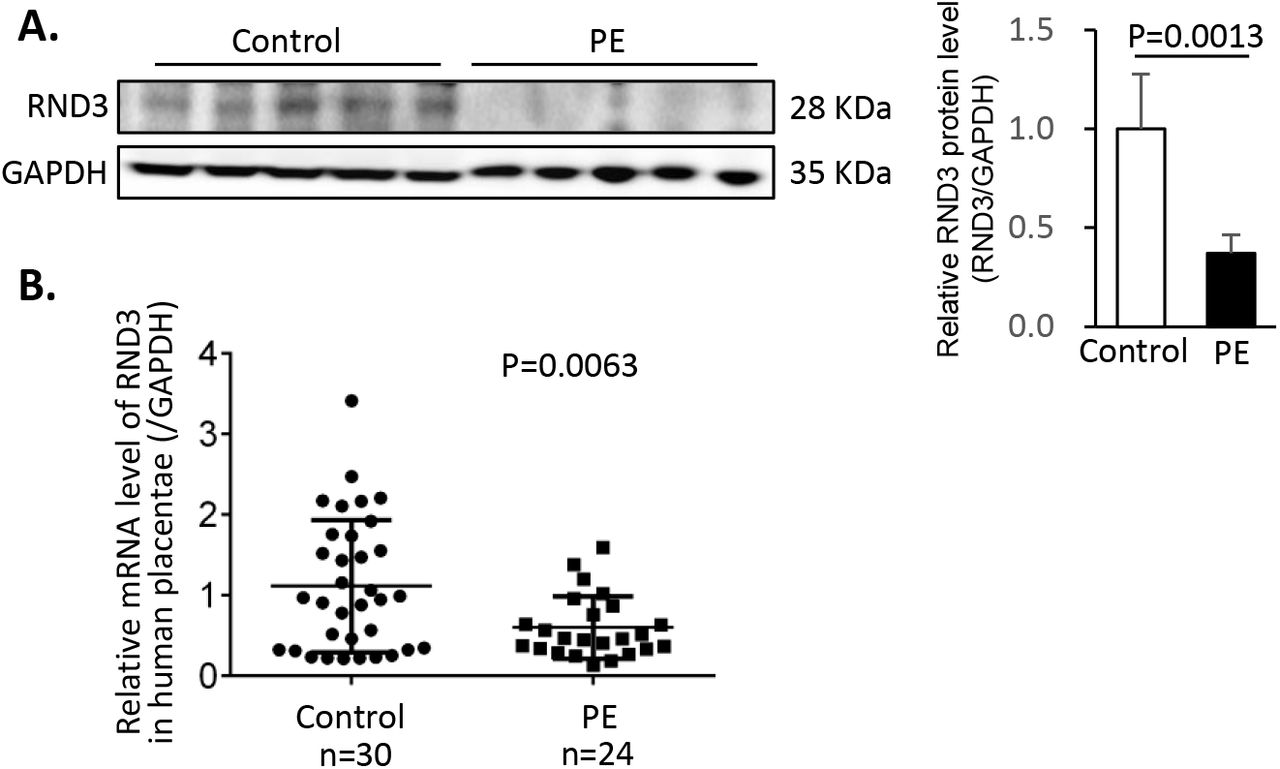
(A) Representative RND3 protein expression by Western blot analysis. Densitometry of bands was performed by Image J. (B) Relative levels of Rnd3 mRNA from 30 human healthy placentae and 24 human placentae with PE.
RND3 protein expression is downregulated in human PE primary trophoblasts and is associated with severe oxidative stress and apoptosis
Oxidative stress is a main regulator of PE development. In the placental tissue sections, severe oxidative stress was noted in the human placentae with PE (Fig. 2A). Systemic experiments based on human primary trophoblasts (PTBs) were conducted for the understanding of the underlying mechanisms involved in this process. Human PTBs were isolated from PE and healthy placentas. The PTBs were identified by immunostaining of the surface marker cytokeratin 7 (CK7) (Fig. 2B). The purity of the isolated PTBs was 97% according to nuclear counterstaining (Fig. 2B). The protein level of RND3 in the PTBs was detected by immunostaining analysis. Rnd3 was universally expressed in the cytoplasm and nuclei of the trophoblasts (Fig 2C). Reduced Rnd3 expression was associated with increased ROS accumulation and was detected in the PE patient-derived PTBs (Fig 2C). To mimic the pathological PE placental environment, hypoxic cell culture of PTBs was established. Following induction of hypoxia, additional ROS accumulation and apoptosis were observed in the PE PTBs compared with those noted in the control PTBs (Fig 2D). This suggested that PE PTBs were more sensitive to hypoxic stress. Rnd3 may participate in the development of PE via the regulation of ROS generation.
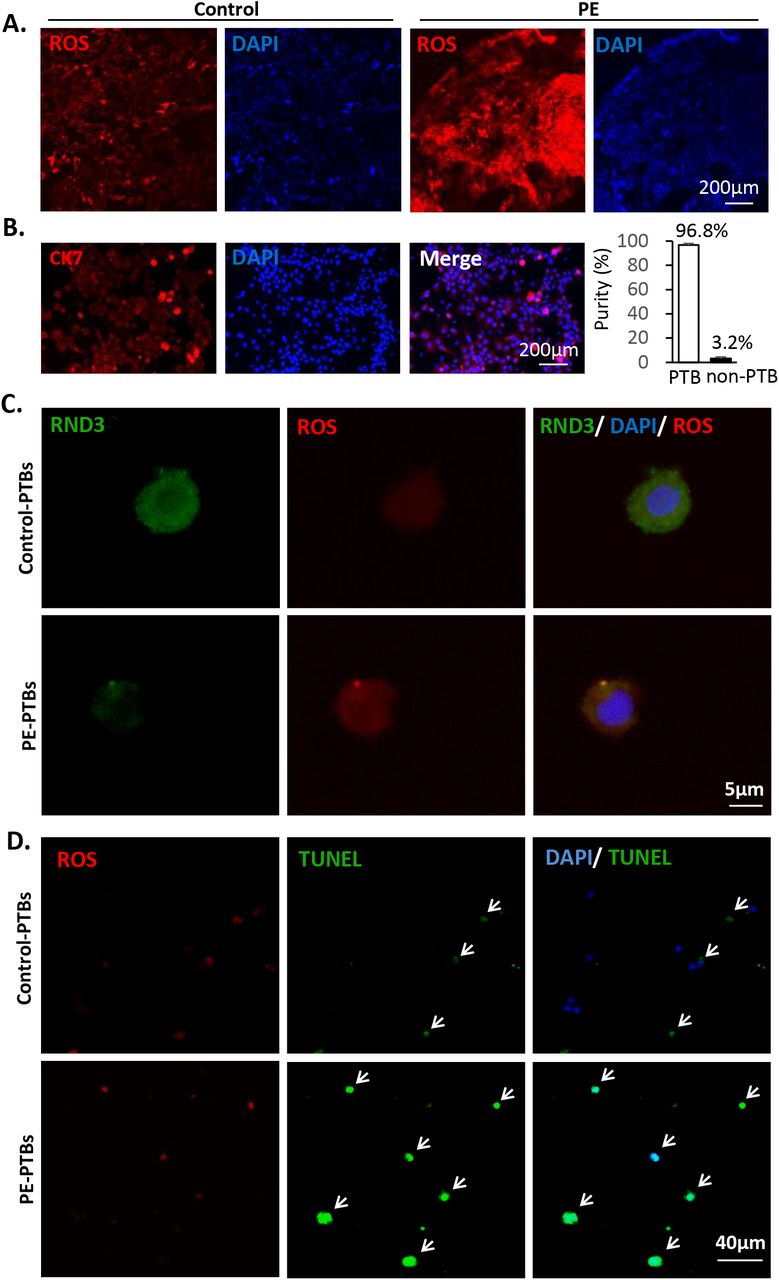
(A) Severe oxidative stress was observed in human placental tissue sections from PE patients by DHE staining. The nuclei were visualized by blue DAPI staining. Scale bar represents 200 μm. (B) Isolated human primary trophoblast cells (PTBs) were identified by the cell surface marker CK7 shown in red and the nuclei were visualized by DAPI. The purity of primary trophoblasts was calculated by the ratio of CK7-positive cells over nuclei. The scale bar represents 50 μm. (C) RND3 protein expression in PTBs was labeled by immunostaining. Decreased Rnd3 expression along with increased ROS levels were observed in the PTBs from PE patients. The scale bar represents 5 μm. (D) Elevated ROS levels and severe apoptosis were observed in the PE PTBs compared with those of the PTBs from healthy control subjects. ROS was labeled by DHE staining shown in red. The arrows point at the TUNEL-positive cells (green), which overlapped with nuclear counter-staining (blue). The scale bar represents 40 μm.
Human PE primary placental trophoblasts are predisposed to mitochondrial damage
To explore the underlying mechanism of Rnd3-associated oxidative stress in PE, trophoblastic mitochondria, which are the main source of ROS, were analyzed by electron microscopy. In the ultrathin sections of the placental tissues, a borderline between the layers of syncytiotrophoblast (STB) and cytotrophoblast (CTB) could be clearly identified. In the CTB of the PE placentae, swelling mitochondria with loss of cristae were observed, which were the typical characteristics of mitochondrial damage (arrows pointed, Fig 3A). We further analyzed the mitochondrial membrane potential in PTBs. Using JC1 staining, we compared the mitochondrial membrane potential between the control PTBs and the PE PTBs with or without hypoxic stimuli. The ratios of JC1 red/green fluorescence between the two groups indicated no difference at the baseline (Fig 3B). The mitochondrial membrane potential was reduced in response to hypoxic challenge. However, the red/green fluorescence emission ratio was collapsed in PE PTBs compared with that noted in the control PTBs (Fig 3B), indicating that PE PTBs were predisposed to hypoxia-induced mitochondrial injury.

(A) Placental tissues from PE patients and healthy control subjects were analyzed by transmission electron microscopy and viewed at a low magnification (upper panel) and a high magnification (lower panel), respectively. The arrows point at the damaged mitochondria. The scale bars represent 1 μm and 0.5 μm, respectively. (B) Mitochondrial membrane potentials were shown by the JC1 staining of the primary trophoblast cells isolated from PE patients and healthy control subjects with or without hypoxic cell culture. JC1 is a cell permeable dye that accumulates in mitochondria and yields green fluorescence. Driven by high mitochondrial membrane potential, JC1 is able to enter the mitochondrial inner membrane and yield red fluorescence. The ratios of the red/green fluorescence intensities were quantified by the image J software. The numbers in the columns represent the numbers of cells in each group. C-PTBs indicates control-primary trophoblasts, and P-PTBs indicates PE-primary trophoblasts.
Overexpression of Rnd3 protects the trophoblastic cells from ROS generation and induction of apoptosis
To investigate the potential role of Rnd3 deficiency in inducing oxidative stress in PE PTBs, we manipulated Rnd3 expression in the trophoblastic BeWo cell line. Hypoxic cell culture was applied to induce ROS generation. ROS levels were determined by DHE fluorescence labeling and the detection of cellular 8-isoprostane levels. The GFP-Rnd3 expressing BeWo cells displayed significantly decreased ROS levels compared with those of the surrounding non-GFP-Rnd3 cells (Fig 4A). Consistent with these observations, myc-Rnd3 overexpression led to decreased 8-isoprostane levels (Fig 4B) and ameliorated cell apoptosis (Fig 4C-D), indicating that Rnd3 modulated the production of ROS in trophoblasts. Rnd3 deficiency was observed in human PE PTBs and may be the cause of severe oxidative stress and apoptosis in PE.

(A) Reduced ROS levels were observed in hypoxic conditions and were challenged with GFP-Rnd3 overexpressing BeWo cells. ROS was labeled by DHE staining shown in red. The arrows point at the GFP-Rnd3 expressing cells. The scale bar represents 80 μm. (B) A decrease in 8-isoprostane levels was detected in the cell lysates of myc-Rnd3 overexpressing BeWo cells, compared with those noted in the myc control group. The experiments were repeated 3 times. (C) The comparison of the TUNEL staining in myc and myc-Rnd3 overexpressing BeWo cells following 16 h of hypoxic cell culture. The arrows indicate TUNEL-positive cells (green) overlapping with nuclear counter-staining. The scale bar represents 200 μm. (D) Quantification of TUNEL-positive cells. The experiments were repeated 3 times.
Rnd3 deficiency leads to mitochondrial damage in BeWo trophoblastic cells
To investigate if mitochondria contributed to Rnd3-mediated ROS generation, we knocked down Rnd3 in BeWo cells. Electron microscopy of Rnd3 knockdown BeWo cells revealed mitochondrial injury. The compromised mitochondria displayed a structure with damaged cristae and swelling mass (arrow pointed, Fig 5A). Consistent with the morphological changes, the mitochondrial transmembrane potential was also depolarized following Rnd3 knockdown (Fig 5B). Subsequently, the effects of Rnd3 on mitochondrial function were assessed by measuring the oxygen consumption rate (OCR) in different respiratory states. The comparison of the Rnd3 knockdown group and the control group indicated no significant differences in the OCR of the basal respiration, maximal respiration and space capacity (Fig 5C). However, a 2.4-fold increase in the proton leak associated OCR was detected in the Rnd3 knockdown BeWo cells (Fig 5C), indicating the uncoupling of ATP synthesis and substrate oxidation. Consistent with this result, the mitochondrial coupling efficiency was reduced in the siRnd3 group (Fig 5D).

(A) By transmission electron microscope analysis, mitochondrial damage was observed in siRnd3 transiently transfected BeWo cells. The arrow heads indicate the loss of crista in the damaged mitochondria. The images of a series of magnifications are displayed. (B) The depolarized mitochondrial membrane potential was detected in siRnd3 transfected BeWo cells compared with that noted in the siCtrl group. The mitochondrial membrane potential was quantified by the ratios of red/green fluorescence intensities of mitochondria-specific JC1 dye. (C) The Oxygen consumption rate (OCR) of treated BeWo cells was measured prior to and following the injections of oligomycin, FCCP, rotenone and antimycin A (Rot/AA), respectively. Increased proton leak was detected in the siRnd3 group. (D) Reduced coupler efficiency was detected in siRnd3 transfected BeWo cells compared with that noted in the siCtrl group as determined by mitochondrial OCR determination. The scale bar represents 80 μm. The numbers in the columns represent the number of cells in each group.
Rnd3 facilitates the protein accumulation of PPARγ and stimulates the expression of UCP2 in trophoblasts
To explore the underlying mechanisms of Rnd3-mediated mitochondrial dysfunction, the mRNA expression levels of the mitochondrial regulatory factors and ROS scavengers were evaluated in siRnd3 transiently transfected BeWo cells. The analysis indicated no significant difference between siRnd3 and sicontrol groups with regard to the mRNA levels of the transcription factors PPARγ, PPARγ coactivator 1-α (PGC1-α), peroxisome proliferator-activated receptor α (PPARα), nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (TFAM) (Fig S1). No significant differences were also noted with regard to the levels of the endogenous ROS scavenger superoxide dismutase 1 (SOD1) and superoxide dismutase 2 (SOD2) (Fig S1).

RND3 promoted UCP2 mRNA level. Manipulation of RND3 expression resulted in no change in the mRNA levels of PPARγ, PGC1α, NRF1, TFAM, PPARα, SOD1 and SOD2.
Mitochondrial uncoupling protein 2 (UCP2) is critical for mitochondrial respiratory coupling. Rnd3 promoted the protein and the mRNA expression levels of UCP2, whereas knockdown of Rnd3 resulted in UCP2 deficiency (Fig 6A–6C), which was consistent with the mitochondrial respiratory coupling defect in siRnd3-transfected cells. As PPARγ has been identified as a transcriptional regulator of UCP2(Medvedev et al., 2001), we next assessed its expression levels. It is interesting to note that although the mRNA transcripts of PPARγ did not change following manipulation of Rnd3 (Fig S1), the PPARγ protein expression was stimulated by myc-Rnd3 overexpression and was repressed by Rnd3 knockdown (Fig 6A–6B), suggesting a possible post-translational regulation of PPARγ by Rnd3.
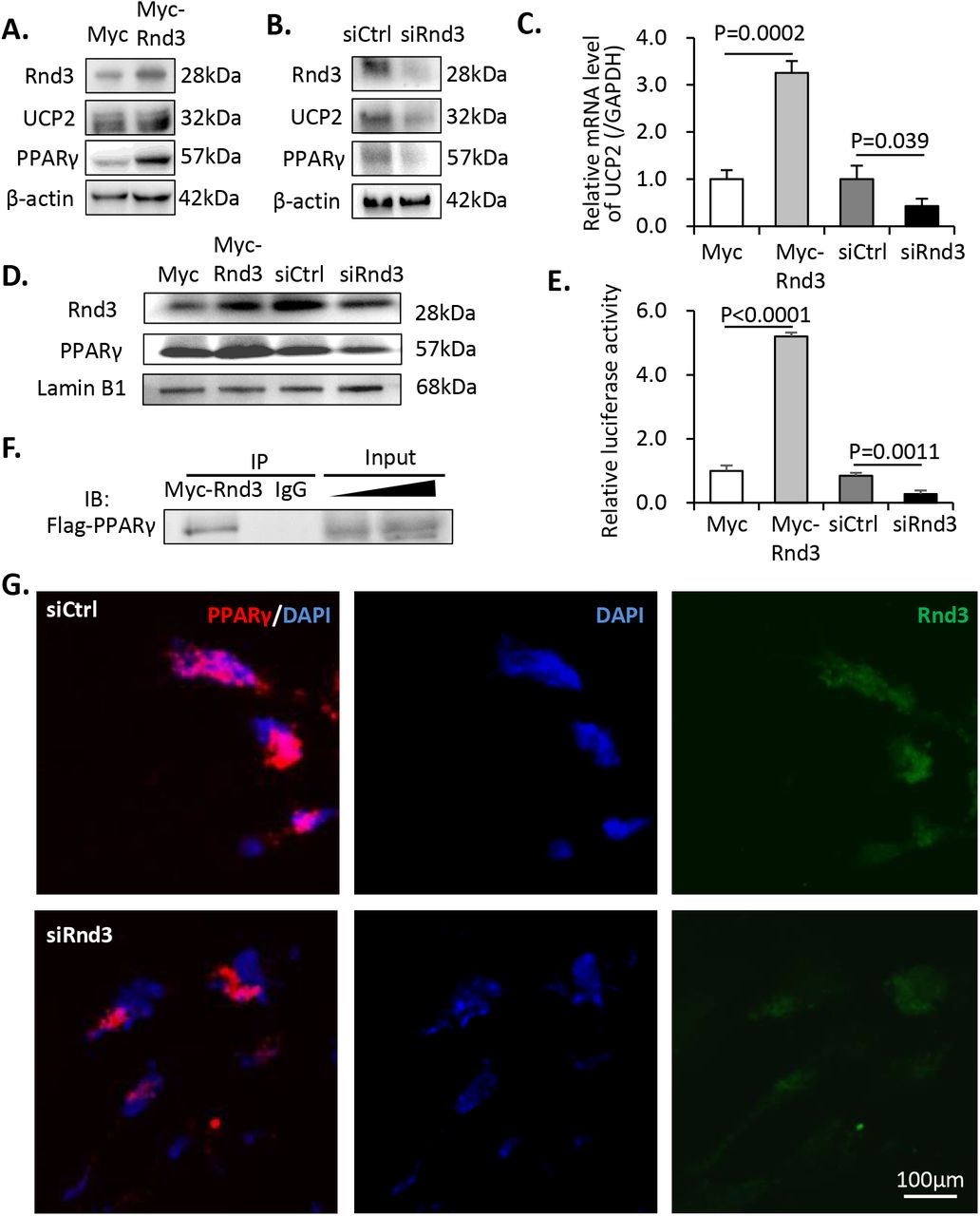
(A) Increased expression levels of UCP2 and PPARγ were observed following Rnd3 overexpression. (B) Knockdown of Rnd3 results in lower UCP2 and PPARγ protein expression levels. (C) Q-PCR analysis indicated that UCP2 mRNA levels were increased in myc-Rnd3 expressed cells, whereas they were reduced in Rnd3 deficient cells. (D) Nuclear protein levels of RND3 and PPARγ were increased in the myc-Rnd3 overexpressed cells and were decreased in the Rnd3 deficient cells. (E) Rnd3 regulated PPARγ transcriptional activity as demonstrated by luciferase assays. (F) Myc-RND3 physically interacted with flag-PPARγ in vivo as determined by coimmunoprecipitation. (G) Immunofluorescence staining indicated the co-localization of PPARγ and RND3 proteins. Knockdown of Rnd3 reduced the nuclear accumulation of the PPARγ protein. The scale bar represents 100 μm.
RND3 is physically bound to PPARγ and results in the accumulation of PPARγ in the nuclei
To investigate if PPARγ protein could be functionally regulated by Rnd3, we knocked down and overexpressed the latter in BeWo cell cultures. As a transcription factor, the nuclear distribution of PPARγ is critical for maintaining its biological function. We detected apparent nuclear accumulation of PPARγ protein corresponding to the increase in Rnd3 expression. The opposite trend was observed when Rnd3 was knocked down (Fig 6D). Subsequently, a luciferase experiment was conducted in order to determine if Rnd3 caused any effect on its transcriptional activity. The luciferase reporter was driven by a UCP2 promoter, which was responsible for the PPARγ-dependent transcriptional activity. Myc-Rnd3 overexpression resulted in a 5.2-fold increase in luciferase activity compared with that of the control, while downregulation of Rnd3 weakened PPARγ transcriptional activity and resulted in 68% declined luciferase signal (Fig 6E). To further explore the potential mechanism of Rnd3-induced PPARγ protein accumulation, we performed an in vivo coimmunoprecipitation assay with 293T cells. As shown in Fig 6F, physical interaction between Myc-RND3 and Flag-PPARγ proteins was detected. Moreover, we performed co-immunostaining of these two proteins in BeWo cells and confirmed the co-localization of RND3 and PPARγ protein molecules and the reduced nuclear PPARγ protein levels in Rnd3 knockdown cells (Fig 6G).
Mitochondrial dysfunction mediated by Rnd3 deficiency is attenuated by PPARγ overexpression
To assess whether PPARγ downregulation is responsible for Rnd3 deficiency-induced mitochondrial defects, Flag-PPARγ was overexpressed in Rnd3 knockdown BeWo cells. The improvement in mitochondrial membrane potential and mitochondrial morphological integrity was observed following PPARγ overexpression (Fig 7A-C). The ROS levels were detected by DHE labeling. We also detected 8-isopropane levels. PPARγ overexpression further ameliorated Rnd3 deficiency-mediated oxidative stress (Fig 7D-F). We analyzed the expression levels of UCP2 in Rnd3 knockdown BeWo cells with or without administration of Flag-PPARγ. As expected, PPARγ attenuated Rnd3 deficiency-induced UCP2 downregulation both at the mRNA and protein levels (Fig 7G-H). Finally, we assessed the mitochondrial respiratory functions of the different groups of BeWo cells. Overexpression of PPARγ significantly protected the mitochondrial function with improved proton leakage and coupler efficiency (Fig 7I-J).

(A) JC1 staining was performed in BeWo cells among the three following groups: siCtrl, siRnd3 and siRnd3 plus flag-PPARγ. The depolarization of the mitochondrial membrane potential caused by Rnd3 knockdown was attenuated by flag-PPARγ overexpression. The scale bar represents 80 μm. (B) The quantification of the ratios of red/green fluorescence intensities. The experiments were repeated 3 times. (C) Transmission electron microscope analysis indicates that the Rnd3 deficiency-induced mitochondrial damage was recovered by flag-PPARγ overexpression. The arrow heads display damaged mitochondrial structure in the siRnd3 group. The scale bar represents 0.5 μm. (D) DHE staining indicates ROS levels among the three groups. The scale bar represents 100 μm. (E) The ROS levels were quantified by the densitometry of DHE staining. The experiments were repeated 3 times. (F) 8-isoprostane levels were detected in the cell lysates of the three groups. (G) Q-PCR analysis of UCP2 mRNA levels among the three groups. The experiments were repeated 3 times. (H) Immunoblots revealed increased UCP2 protein expression levels in the flag-PPARγ rescue group. (I) Mitochondrial respiratory function was measured. The proton leak associated OCR was improved by PPARγ. (J) Rnd3 deficiency-mediated defective coupler efficiency was ameliorated by PPARγ.
Adenoviral-mediated Rnd3 overexpression in human PE primary trophoblasts rescues oxidative stress and mitochondrial defect
Rnd3 downregulation was demonstrated in human PE-PTBs along with severe oxidative stress and mitochondrial injury. To further reveal the critical role of Rnd3 in the clinical pathology of PE, we overexpressed hRnd3 in human PTBs by adenoviral-mediated gene delivery. Elevated RND3 protein levels in Ad-GFP-hRnd3 treated PTBs were confirmed by western blot analysis (Fig 8D). In the absence of Ad-GFP-hRnd3 infection, the baseline PE-PTBs exhibited higher ROS levels compared with those of the control-PTBs (Fig 8A, cells displaying no GFP, pointed by long arrows). Following infection with Ad-GFP-hRnd3, the ROS levels of the PE-PTBs were significantly reduced (Fig 8A, cells displaying GFP, pointed by arrow heads). The levels of 8-isopropane were decreased in PE-PTBs following Ad-GFP-hRnd3 application (Fig 8B), which was consistent with the previous observations. Increased expression levels of PPARγ and UCP2 were observed, as expected, in the PTBs with Ad-GFP-hRnd3 (Fig 8C-D). In addition to regulating the PPARγ/UCP2 pathway, Rnd3 further attenuated the mitochondrial function in PE-PTBs (Fig 8E-G).

(A) Human PTBs from PE patient and healthy control subjects were infected with Ad-GFP-hRnd3. Hypoxic cell culture was applied to induce oxidative stress. The arrow heads represent the GFP-Rnd3 expressing PTBs (green fluorescence). The long arrows represent the PTBs with non-GFP-Rnd3. ROS levels were significantly reduced in PE-PTBs following GFP-Rnd3 overexpression as determined by DHE staining. NS indicates non-specific staining. The scale bar represents 25 μm. (B) 8-isoprostane level detection in the C-PTBs and P-PTBs following treatment of Ad-GFP or Ad-GFP-Rnd3. (C) Ad-GFP-hRnd3 improved UCP2 mRNA transcripts in the PTBs. (D) Rnd3 overexpression rescued PPARγ-UCP2 signaling in PE-PTBs. Mitochondrial dysfunction in PE-PTBs was partially rescued by Ad-GFP-hRnd3, as determined by the increases in proton leak associated OCR (E) and respiratory control ratio (F). C-PTBs indicates control-primary trophoblasts; P-PTBs, PE-primary trophoblasts.
Downregulation of the expression levels of PPARγ and UCP2 were observed in human placentae with PE
The expression levels of PPARγ and UCP2 were evaluated in human PE placental tissues. The experiments aimed to offer additional insight in the clinical relevance of the regulation of the PPARγ/UCP2 pathway by Rnd3 in PE. PPARγ protein levels were downregulated in PE placentae. Consistent with the post-transcriptional regulation of PPARγ by Rnd3, the corresponding mRNA levels of PPARγ indicated no significant changes between the control and PE groups (Fig 9A-B). The mRNA transcripts of UCP2 and its protein levels were reduced in human PE placentae (Fig 9A and C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Representative protein expression levels of PPARγ and UCP2 as determined by western blot analysis. The levels of the two proteins were downregulated in the placentas with PE. (B) Relative mRNA levels of PPARγ from 30 healthy placentae and 24 placentae with PE. (C) Relative mRNA levels of UCP2 from 30 healthy placentae and 24 placentae with PE.
Discussion
PE has been suggested as a mitochondrial disorder since the late 1980s(Torbergsen et al., 1989). Proteome analysis of PE placentae revealed that the abnormal expression levels of the respiratory complex proteins were associated with excessive generation of ROS in the mitochondrial respiratory chain(Shi et al., 2013). Given that mitochondrial ROS is an important contributor to oxidative stress and inflammation, the reason causing placental mitochondrial ROS is critical and remains to be revealed. This study uncovers a novel understanding of placental oxidative stress in PE that Rnd3-mediated PPARγ/UCP2 cascade participating in the regulation of mitochondrial dysfunction.
In the pathological condition of PE, Rnd3 promotes the proliferation and migration of trophoblasts under the regulations by lncRNAs HOXA11-AS and TUG1(Xu et al., 2017; Xu et al., 2018). In contrast to these observations, Rnd3 has also been proposed as a stimulator of trophoblast cell invasion in PE, under the repression caused by miR-182-5p(Fang et al., 2018). However, the expression levels of Rnd3 in PE patients were not detected in these studies. Our study confirmed the regulatory role of Rnd3 in PE, and revealed the novel function of Rnd3 in maintaining mitochondrial respiratory chain. The supply of Rnd3 in PE patient-derived primary trophoblasts significantly improved mitochondrial function and repressed oxidative stress (Fig 8), suggesting the potential role of Rnd3 as a pharmacological target for PE. Reductions in Rnd3 mRNA and protein levels were observed in human PE placentae, while the exact mechanism of Rnd3 downregulation in PE remains to be revealed in the future. The understanding of the pathological consequence of Rnd3 downregulation is important and has considerable clinical significance.
UCP2 exerts protective properties against oxidative damage by reducing ROS generation via decreasing mitochondrial proton gradient and local oxygen availability(Cadenas, 2018). PPARγ and its ligands directly activate UCP2 gene promoter via the E-box element(Medvedev et al., 2001). By recovering the mitochondrial membrane potential and reducing ROS generation, PPARγ improves the survival of trophoblast cells under hypoxic condition(Kohan-Ghadr et al., 2019). In the present study, reduced PPARγ/UCP2 signaling was detected in human PE placentas, contributing to mitochondrial defect and oxidative stress in PE primary trophoblasts. However, excessive stimulation of PPARγ/UCP2 has also been implicated in pathological placenta with maternal nutrient restriction, by enhancing fatty acid metabolism and limiting glucose utilization(Yiallourides et al., 2009). Therefore, the homeostasis of the PPARγ/UCP2 axis is critical in maintaining normal placental function.
It is interesting to note that Rnd3 facilitates the protein accumulation of PPARγ without causing the change of PPARγ mRNA transcripts. Therefore, immunoprecipitation was performed to investigate the underlying mechanism, and the direct interaction of protein molecules of RND3 and PPARγ was observed. Meanwhile, the co-localization of the two proteins was visualized in nuclei and cytoplasm (Fig 6G). Given that the dynamics of PPARγ depends on ligand-induced transcriptional activation and ubiquitin-proteasome dependent degradation, it is reasonable that Rnd3 may stabilize PPARγ in a post-translational manner.
The present study has several limitations. First, even though we have used two cell models, including PE primary cell and transform BeWo cell line, to mimic the Rnd3 downregulation in the placentas from PE patients. It is not precisely clear whether Rnd3 deficiency could cause PE in vivo. Future studies using Rnd3 gene knockout animal model will provide stronger supports to the role of Rnd3 in PE etiology. Second, even though we have proved the direct interaction of RND3 and PPARγ protein molecules, however, the more precise mechanism of post-translational regulation also needs further investigation.
Conclusions
We identified here downregulation of Rnd3 in placental trophoblasts in patients with PE, and proved that Rnd3 deficiency can cause mitochondrial defects and oxidative stress in trophoblasts. Supply of Rnd3 in PE primary trophoblasts attenuated mitochondrial dysfunction and oxidative stress. The possible underlying mechanism is that Rnd3 interacts with PPARγ and stabilizes PPARγ protein, causing stimulation of PPARγ/UCP2 cascade. In conclusion, our study indicates the novel role of Rnd3, providing new insights into PE aetiology.
Disclosure of interests
None.
Contribution to authorship
X.Y. designed the experiments; L.H., Y.M. and X.Y. conducted most experiments; L.C. collected data in Fig. 9; Y.S., X.C., F.S., L.X., J.C., Y.L., C.Y. and X.Y. analyzed the data; X.Y. wrote the manuscript; J.C., M.Z., Z.W. and X.Y. revised the manuscript. All authors contributed to the final manuscript.
Details of ethics approval
Human placental tissues were obtained and used for the present study with written patient-informed consent and approval by the Ethics Committee of Southern Hospital, Southern Medical University. The date of approval is July 14th, 2020. The reference number of approval is NFEC-2020-155.
Funding
This study was supported by the following funding sources: the Major Science and Technology Program of Hainan Province (ZDKJ2017007), the National Natural Science Foundation of China (81771609, 81601317, 81960283, and 81971415), the Natural Science Foundation of Guangdong Province (2017A030313584, 2019A1515010290 and 2019A1515010019), the Special Fund for Cooperative Innovation and Platform Environment Construction (2015B050501006), the Outstanding Youth Development Scheme of Nanfang Hospital Southern Medical University (2018J010).
References




