

ABSTRACT
PIWI interacting (pi)RNAs are small RNAs mostly known to protect the genomes of animal germlines against transposable elements. In Drosophila, piRNA-mediated cleavage of transposon RNA triggers the release of a responder piRNAs via the ping-pong amplification cycle. Responder piRNA 3’ end formation by the endonuclease Zucchini is coupled to the production of downstream trailer piRNAs, expanding the repertoire of piRNAs that target transposons. Intriguingly, in Aedes aegypti mosquitoes, somatic piRNAs are produced from viral RNA, implying a role of viral (v)piRNAs in antiviral immunity. Knowledge on how vpiRNA 3’ ends are formed and whether viral RNA is subjected to trailer piRNA production, however, is lacking. To address these questions, we analyzed small RNA sequencing libraries from Ae. aegypti cells. We found that virus- and transposon-derived piRNAs have sharply defined 3’ ends, and that uridine residues are enriched directly downstream of dominant piRNA sequences, both of which are characteristic features of Zucchini-like cleavage of precursor piRNAs. Next, we designed a piRNA reporter system based on Sindbis virus recombinants that harbor target sites for abundant endogenous piRNAs. These piRNAs guide cleavage of the viral RNA, which is subsequently processed into abundant responder piRNAs. Using this reporter virus system, we identified the Ae. aegypti orthologs of Zucchini, which is required for sharp 3’ end formation of responder piRNAs, and Nibbler, a 3’-5’ exonuclease involved in the trimming of a subset of piRNAs and miRNAs. Furthermore, we found that cleavage of viral RNA triggers the production of trailer piRNAs, thus expanding the piRNA sequence pool that is able to target viral RNA. Our results have important implications for understanding how autonomous piRNA production from viral RNA can be triggered by just a few cleavage events by genome-encoded piRNAs.
INTRODUCTION
Blood-feeding mosquitoes of the Aedes genus are responsible for the spread of arthropod-borne (arbo)viruses that cause severe diseases, such as dengue, Zika, chikungunya and yellow fever. Due to rising global temperatures and increased human mobility, the geographical distribution of these vector mosquitoes is continuously expanding, which has resulted in a rapid increase in the number of arbovirus epidemics in the last decennia (1). For efficient transmission to occur, arboviruses have to actively replicate in several mosquito tissues to eventually infect the salivary gland (2). Therefore, suppression of virus replication by the mosquito antiviral immune response strongly affects the efficiency of arboviral spread. The cornerstone of antiviral immunity in insects is the small interfering (si)RNA pathway, in which viral double stranded (ds)RNA is cleaved by Dicer-2 (3). The resulting cleavage products are processed into siRNAs, which provide sequence specificity to the endonuclease Argonaute 2 to direct the cleavage of single stranded viral transcripts.
Intriguingly, in Aedes mosquitoes, viral RNA is also processed by a somatically active PIWI interacting (pi)RNA pathway, suggesting that two independent small RNA pathways act in parallel to combat viral infections (4). The piRNA biogenesis machinery has been thoroughly characterized in the model organism Drosophila melanogaster, where a germline-restricted piRNA pathway defends the genome from parasitic genetic elements called transposons (5,6). piRNA biogenesis is initiated by the cleavage of genomically encoded piRNA precursors, which are rich in transposon remnants. Slicing of these precursor transcripts into pre-piRNAs is mediated either by a piRNA-guided PIWI protein or the endonuclease Zucchini (Zuc), which acts independently of small RNAs (7–9). Pre-piRNAs are loaded into the PIWI proteins Aubergine (Aub) and Piwi, where their 3’ ends may be further trimmed by the exonuclease Nibbler (Nbr) and are finally 2’-O-methylated by Hen1 to generate mature piRNAs (10–14). Whereas Piwi translocates to the nucleus to silence transposons at the transcriptional level (15,16), Aub remains in the cytoplasm where it cleaves transposon mRNA with sequence complementarity to its associated piRNA (17,18). The resulting cleavage fragments are loaded into the PIWI protein Argonaute 3 (Ago3) and processed into responder piRNAs by Nbr-mediated trimming, followed by 2’-O-methylation by Hen1 (12,14). In turn, these responder piRNAs direct Ago3-mediated cleavage of piRNA precursors, triggering the production of new initiator piRNAs, completing the so-called ping-pong loop (17–20).
In Drosophila, Zuc-mediated generation of piRNA 3’ ends often produces a downstream cleavage product which is preferentially loaded into Piwi, thus forming a new pre-piRNA. This mechanism results in phased processing of piRNA precursor transcripts into a string of piRNAs, named trailer piRNAs (8,9). Hence, while the ping-pong loop amplifies those piRNAs that initially recognized active transposons, phased piRNA production expands the piRNA repertoire to allow for more efficient repression of transposons. Historically, piRNAs derived from cluster transcripts and transposon mRNAs were termed primary and secondary piRNAs, respectively. Hereafter, we use the more intuitive names initiator or responder for ping-pong amplified piRNAs and trailer for piRNAs produced through phased biogenesis, as proposed in (5).
The Ae. aegypti piRNA pathway differs from that in Drosophila in three important ways: i) the pathway is active in somatic tissues as well as germline tissues (21,22), ii) it processes non-canonical substrates such as de novo produced viral RNA (23–25), and iii) mosquito piRNA clusters contain large numbers of endogenous viral elements (EVEs): sequences of non-retroviral RNA viruses inserted in mosquito genomes (26–28). These EVEs give rise to abundant piRNAs (26,29,30) and mediate antiviral defense (31–33).
In Ae. aegypti mosquitoes, the PIWI proteins Piwi5 and Ago3 are brought together by the TUDOR proteins Veneno and Yb and the RNA helicase Vasa to promote efficient ping-pong dependent production of piRNAs (34), yet, the mechanisms involved in specifying piRNA 3’ ends is unknown. Moreover, while the Ae. aegypti piRNA pathway was recently proposed to efficiently generate trailer piRNAs (7), the molecular machinery responsible for phased piRNA production and whether viral RNA is also subjected to phased piRNA production is unclear.
It has been proposed that endogenous piRNAs encoded in the mosquito genome can trigger the production of piRNAs from viral sources (31,32). We hypothesized that through the mechanism of piRNA phasing, a single endogenous initiator piRNA could induce the production of a pool of viral piRNAs, thereby increasing the piRNA population that targets viral RNA. Here, we uncover that this interaction between an endogenous initiator piRNAs and viral RNA substrate indeed takes place in Ae. aegypti. Specifically, we demonstrate that Ae. aegypti piRNAs, both of transposon and viral origin, display sequence features indicative of a Zucchini-like biogenesis mechanism. Hence, we use a viral piRNA reporter system to show that AAEL011385 and AAEL005527, the Ae. aegypti orthologs of Drosophila Zuc and Nibbler, respectively, cooperatively determine piRNA 3’ ends. Lastly, we show that cleavage guided by a genomically encoded initiator piRNA triggers the production of additional trailer piRNAs from the viral genome, thus increasing the pool of piRNAs targeting the newly infecting virus. This mechanism may equip the Aedes piRNA pathway with an adaptive immune response that is able to adapt to newly encountered and continuously mutating viruses.
RESULTS AND DISCUSSION
Aedes aegypti piRNAs have sharp 3’ ends
In Drosophila, piRNA 3’ end formation is largely dependent on the cleavage of pre-piRNAs by the endonuclease Zucchini (Zuc). Zuc uses a sequence motif to preferentially cleave upstream of uridine residues in vivo (35–37), hence, piRNAs generated by Zuc generally have sharp 3’ ends and the nucleotide directly downstream of the 3’ end tends to be a uridine (+1U bias) (8,9). We examined whether these characteristics were present in our previously generated small RNA deep sequencing libraries from Ae. aegypti Aag2 cells (24) infected with Sindbis virus (SINV), a positive sense RNA virus of the Togaviridae family. We first analyzed transposon-derived piRNAs and found that individual piRNAs, defined by a shared 5’ end, generally had the same length (Figure 1A). Specifically, for almost 60% of piRNAs the dominant length made up more than 75% of sequenced reads. We selected those piRNAs for which the dominant sequence length represented at least 75% of the reads and inspected the identity of the nucleotides downstream of that most abundant piRNA isoform. We found that the nucleotide position directly following the 3’ end of the piRNA was biased for uridine (Figure 1B), strongly indicating that these piRNAs were generated by a mechanism that resembles Zuc cleavage in Drosophila. Strikingly, sharp 3’ ends and +1U biases were also clearly visible for SINV-derived piRNAs, irrespective of the strand from which the piRNAs were produced (Figure 1C-D). These findings suggest that 3’ ends of both (+) strand derived vpiRNAs, which are predominantly Ago3-associated and (-) strand derived vpiRNAs, which are mostly Piwi5-bound (24,34), are generated, at least in part, by Zuc-like cleavage events. Interestingly, we also observe sharp 3’ ends and +1U biases for piRNAs generated from Phasi Charoen-like virus (Figure S1), a negative sense RNA virus from the Phenuiviridae family that persistently infects Aag2 cells (38). These findings indicate that Zuc-like piRNA biogenesis targets diverse RNA viruses.
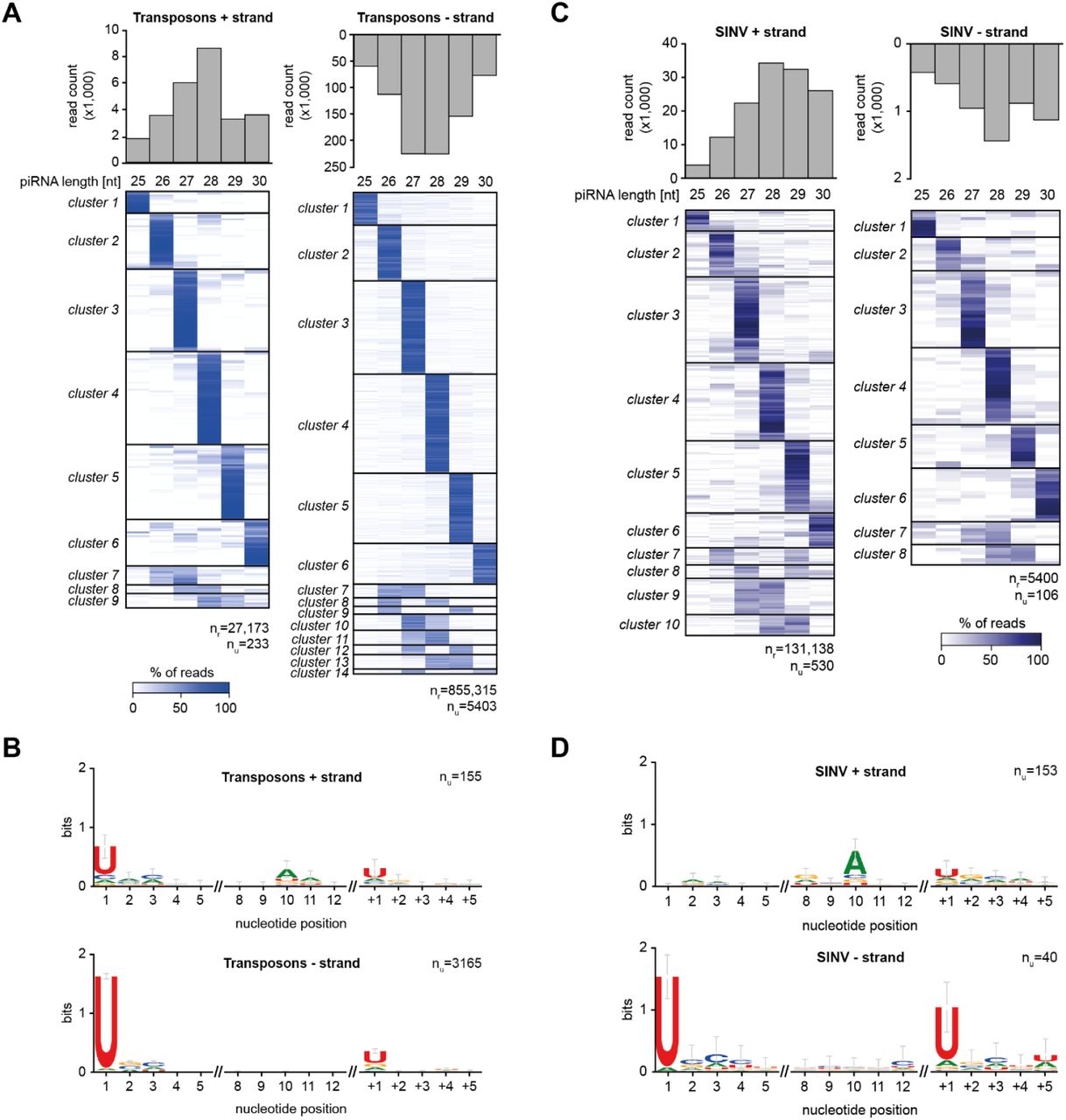
(A) Heat map showing the relative size distribution of individual transposon-derived piRNAs, defined by a shared 5’ end. Shades of blue indicate the percentage of reads contributing to the indicated read length, white represents absence of reads in the specific size class. The number of unique piRNAs (nu) and the number of reads (nr) that underlie the heat map are indicated. A minimum of 20 reads/unique piRNA was required to be included in the analysis.
(B) Nucleotide biases for the indicated nucleotide positions of transposon-derived piRNAs and the sequence at the genomic region directly downstream (+1 until +5) piRNA 3’ ends. Only piRNAs from (A) that had a dominant length (at least 75% of reads) were considered in this analysis and only unique sequences were analyzed, disregarding read count for the individual piRNAs.
(C-D) The same analysis as for A and B was applied to piRNAs mapping to SINV.
Genomically encoded piRNAs are able to trigger production of virus-derived responder piRNAs
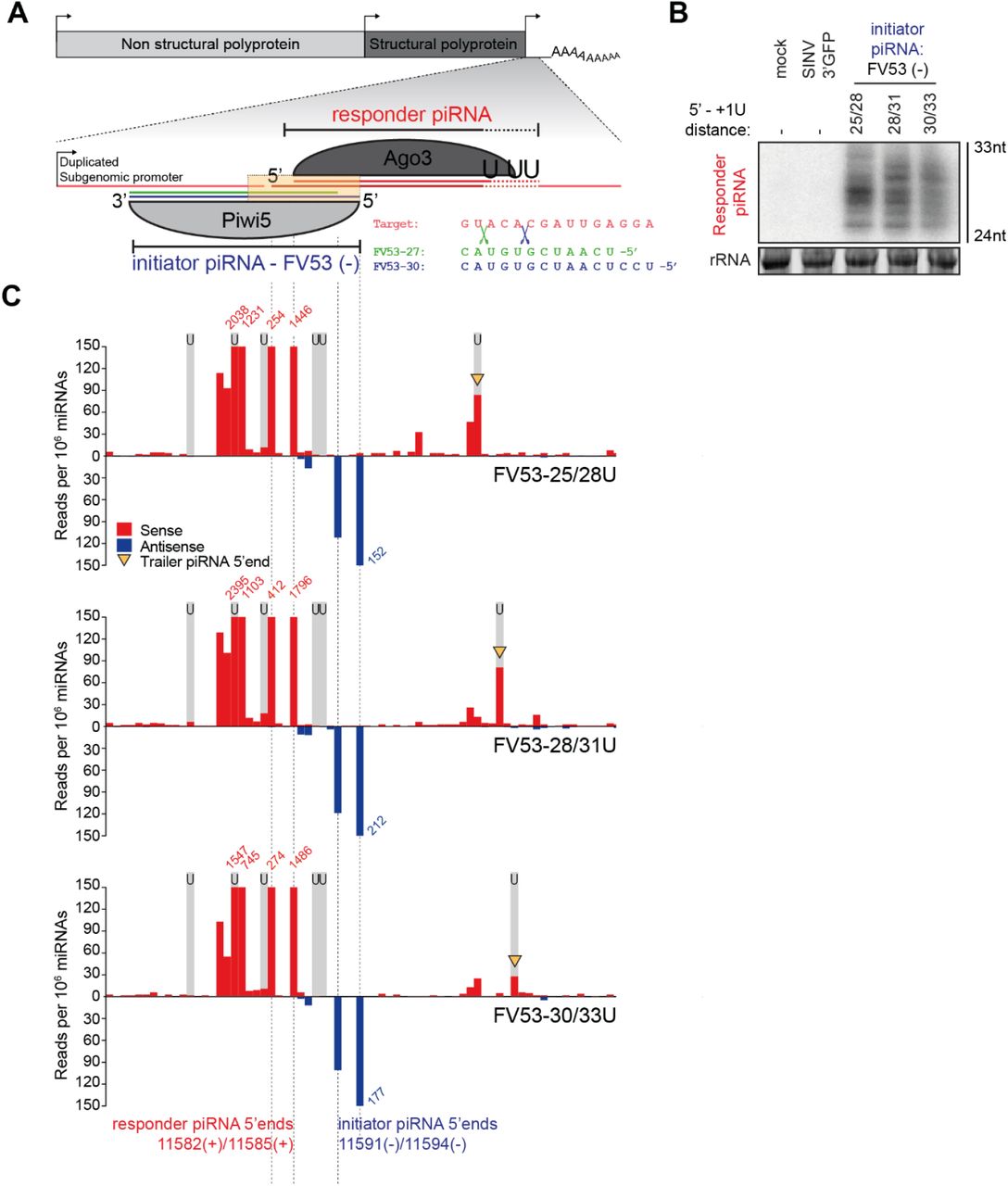
To study sequence determinants for vpiRNA 3’ end formation, we designed a SINV-based reporter system in which exogenous piRNA target sequences can be easily introduced. We modified SINV such that a duplicated subgenomic promoter drives the expression of a non-coding RNA sequence that harbors a target site for an abundant Piwi5-associated initiator piRNA (Figure 2A, Figure S2A). Initiator piRNA-guided recognition of the target site should trigger slicing by Piwi5 and subsequent processing of the resulting cleavage fragment into an Ago3-associated responder piRNA through the ping-pong amplification cycle. We designed viruses with target sites for two abundant endogenous Piwi5-associated initiator piRNAs: one derived from the Ty3/gypsy LTR retroelement gypsy73 (g73 - Figure 2A), and a second originating from an EVE of flaviviral origin (FV53, described in Supplemental text).

(A) Schematic representation of recombinant Sindbis reporter viruses. The enlarged view depicts the reporter locus expressed under the control of a duplicated subgenomic promoter. This non-coding RNA harbors a target site for a Piwi5-associated initiator piRNA derived from the Ty3-gypsy73 retrotransposon (g73 - indicated in blue). Slicing of this target triggers the production of responder piRNAs (indicated in red) that are loaded into Ago3. The distance between the responder piRNA 5’ end and the position of downstream uridine residues in the various viruses used in later experiments are shown.
(B) Northern blot analyses of responder piRNA production in cells infected with the indicated reporter viruses. Numbers indicate the distance between the 5’ end of the responder piRNA and the first downstream uridine residue. SINV 3’ GFP is a virus without an initiator piRNA target site and serves as a negative control, as do mock-infected cells. The positions marking 24 and 33 nt are inferred from an EtBr-stained small RNA size marker. EtBr-stained rRNA serves as loading control.
(C) Visualization of 5’ ends of sense (red) and antisense (blue) piRNAs (24-33 nt) mapping to the reporter locus of the indicated viruses. Dashed lines indicate responder and initiator piRNA 5’ ends and the positions of grey shading indicate the location of uridine residues on the sense strand of the various viruses. The red and blue numbers show piRNA counts (in reads per 106 miRNAs) that exceed the range of the y-axis; yellow arrowheads indicate the 5’ ends of trailer piRNAs.
(D) Size distribution of responder piRNAs produced from the various g73-viruses, as determined by small RNA seep sequencing. Read counts were normalized to the number of miRNAs in each library.
Our previous results indicate that both transposon- and SINV-derived piRNAs have a strong bias towards a uridine residue directly downstream their 3’ ends (Figure 1B, D). To study the importance of +1U for viral responder piRNA production in Aedes mosquitoes, we introduced uridine residues at specified distances from the putative Piwi5 slice site in the viral reporter system (Figure 2A; viruses were named g73-25U, 28U, and 30U according to the responder piRNA 5’ end-to-U distance). Responder piRNAs were readily detected by high resolution northern blotting in Aag2 cells infected with all reporter viruses (Figure 2B), but not in cells infected with control virus expressing GFP from the duplicated subgenomic promoter (SINV 3’GFP). These results indicate that endogenous piRNAs may instruct the cleavage of exogenous viral RNA and the subsequent production of responder piRNAs during acute infection.
Responder piRNA size did not fully reflect the distance between the Piwi5 cleavage site and the downstream uridine residue; while no clear differences in responder piRNA size are observed between g73-25U and g73-28U viruses, increasing the 5’ end-to-U distance to 30 nt (g73-30U) results in a more diffuse distribution of responder piRNA 3’ ends (Figure 2B). These data suggest that downstream uridines may not be absolutely required for determining the 3’ ends of the reporter-derived responder piRNAs or that additional exonucleolytic trimming of pre-piRNA 3’ ends mask a putative endonucleolytic cleavage event directly upstream of the uridine residues.
To discriminate between these two possibilities, we generated small RNA deep sequencing libraries from Aag2 cells infected with the various reporter viruses. We hypothesized that even if a Zuc mediated cleavage event would be masked by exonucleolytic trimming, the presence of downstream trailer piRNAs with 5’ ends coinciding with the U residues would indicate Zuc-mediated endonucleolytic cleavage was responsible for generating the responder piRNA 3’ end. Mapping of piRNA 5’ ends to the genomes of g73-targeted viruses reveals that virtually no antisense piRNAs other than the initiator piRNAs map to the reporter sequence (Figure 2C). These initiator piRNAs trigger the production of highly abundant sense responder piRNAs with characteristic 10 nt overlap of piRNA 5’ ends, indicative of production by ping-pong amplification (Figure 2C). Responder piRNA size distribution for g73-targeted viruses generally recapitulates the results from the northern blot analysis, with reduced 3’ end sharpness for the g73-30U virus (Figure 2D).
We noted that responder piRNAs <26 nt in size are inefficiently produced from any virus (including those with a responder 5’ end-to-U distance of 25 nt: g73-25U and FV53-25/28U). As responder piRNAs are expected to associate with Ago3, we hypothesize that Ago3 may cover the first 25 nucleotides of pre-piRNAs, rendering them inaccessible to endo- or exonucleolytic processing. Indeed, small RNA data from Ago3 immunoprecipitation indicate a clear preference of Ago3 to bind piRNAs in the size range of 26-30 nt (Figure S3A). In line with this, responder piRNAs produced from all reporter viruses are predominantly 26-30 nt in size (Figure 2D).
Intriguingly, in viruses with a responder piRNA 5’ end-U distance ≥28 nt, we uncover the production of a putative trailer piRNA (indicated with yellow arrowheads in Figure 2C) downstream of the responder piRNA. Even in viruses that produce responder piRNAs with diffuse 3’ ends (e.g. g73-30U), the 5’ end of the following trailer piRNA is sharply defined at the downstream uridine. These data suggest that downstream uridines may instruct endonucleolytic cleavage, thus coupling responder (pre-)piRNA 3’ end formation to trailer piRNA 5’ end formation. Accordingly, no trailer piRNA production is observed in cells infected with the g73-25U virus, in which we propose the downstream uridine is inaccessible for endonucleolytic cleavage. The diffuse responder piRNA size profile of g73-30U likely results from trimming of piRNA 3’ ends.
Responder piRNAs are produced through ping-pong mediated slicing
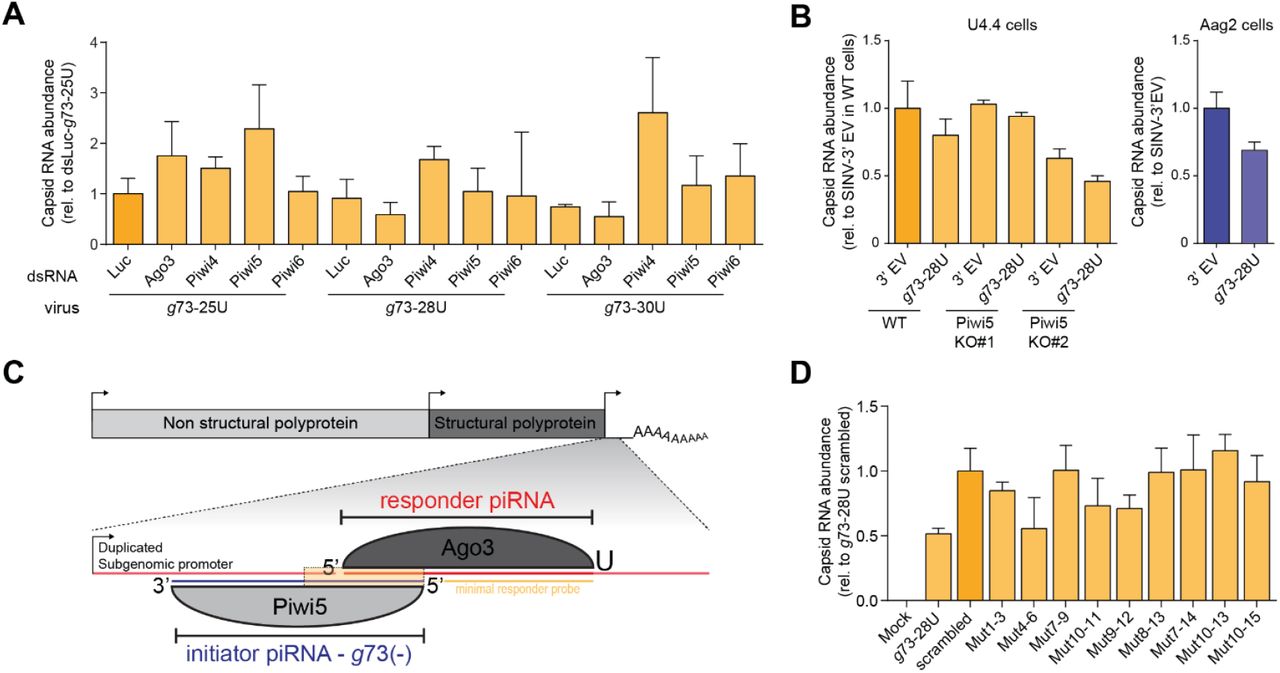
We previously identified Ago3 and Piwi5 as the core components of the ping-pong amplification loop in Ae. aegypti (24,34). We thus wanted to analyze which PIWI proteins are responsible for the generation of the responder piRNAs from our reporter virus. As expected, responder piRNA production was reduced upon knockdown of genes encoding the ping-pong partners Ago3 and Piwi5 and, to a lesser extent Piwi6 (Figure 3A, S4A). Surprisingly, responder piRNA production was increased upon Piwi4 knockdown, which may be explained by increased levels of the g73-derived initiator piRNA in Piwi4 knockdown (Figure 3A). The Piwi5-dependency of g73 responder piRNA production was further validated in Piwi5 knockout Aedes albopictus U4.4 cells (Figure 3B, S4B).

(A) Northern blot analysis of viral responder and g73-initiator piRNA levels in Aag2 cells in which indicated PIWI proteins are depleted by dsRNA transfection. EtBr stained-rRNA serves as loading control.
(B) Northern blot analysis of responder piRNA production in wildtype (WT) and Piwi5 knockout (KO) Ae. albopictus U4.4 cells and Ae. aegypti Aag2 cells infected with the g73-28U virus or a virus that does not express the reporter locus from the second subgenomic promoter (SINV-3’ EV). EtBr-stained rRNA serves as a loading control.
(C) Magnification of the first 15 nucleotides of the g73 piRNA-target site, showing the mutations in the target site sequence of the various viruses. Red bold font indicates mutated residues that are mismatched with the g73 initiator piRNA and the numbers denote positions relative to the g73 initiator piRNA 5’ end.
(D) Northern blot analysis of responder piRNA production in Aag2 cells infected with indicated (mutant) viruses. Responder piRNAs were detected using the ‘minimal responder’ probe that hybridizes to the last 18 nt at the 3’ end of the responder piRNA (indicated in yellow in (C)), which are identical for all viruses. The dashed box denotes an area for which the contrast was adjusted to enhance weak responder piRNA signals (enhanced signal – middle panel). EtBr stained rRNA serves as loading control (bottom panel).
We next investigated targeting requirements for responder piRNA production. To this end, we introduced target site mutations into the seed region (nt 2-8) and around the putative slice site (nt 10-11, Figure 3C, S4C). Responder piRNA production was strongly depleted in viruses in which mutations were introduced in the seed sequence (Mut 1-3, Mut 4-6 and Mut 7-9), compared to a virus bearing the intact target site (g73-28U, Figure 3D). Similarly, introducing mismatches around the slice site resulted in strongly reduced responder piRNA production. As viral RNA levels are virtually unchanged between all viruses, reduced responder piRNA production cannot result from differences in the amount of available substrate (Figure S4D). Weak responder piRNA production was observed in two seed mutants (Mut 1-3 and Mut 4-6) as well as one slice site mutant (Mut10-11) (Figure 3D). These data suggests that low level piRNA slicing may occur even in the absence of full complementarity in the seed region or the slice site, in line with findings in Drosophila (9) and mouse (39).
Zuc-mediated endonucleolytic cleavage defines piRNA 3’ends
Our previous results suggest the involvement of a Zucchini (Zuc)-like endonuclease in the generation of responder piRNAs 3’ ends and trailer piRNA 5’ ends. Hence, we used our viral responder piRNA reporter to identify the mosquito ortholog of Zuc. As the catalytic activity of Zuc lies in its phospholipase D (PLDc_2)-domain, we first identified all Ae. aegypti PLDc_2-domain containing proteins. The neighbor joining tree shows that AAEL011385 has the highest degree of similarity to the various Zucchini orthologs (Figure 4A). The protein encoded by this gene contains a fully conserved catalytic H(X)K(X4)D (HKD)-motif (Figure S5A), suggesting that its endonucleolytic activity is conserved. Moreover, akin to Zuc orthologs in various other species (5,6), AAEL011385 localizes to the mitochondria in Aag2 cells (Figure 4B).

(A) Neighbor joining tree based on PLDc_2 domains from the PFAM database, Ae. aegypti PLDc_2 domain containing proteins and Zuc orthologs from Drosophila (Dm Zuc), silkworm (SmZuc) and mouse (MmMitoPLD).
(B) Confocal microscopy images of Aag2 cells expressing C-terminally 3xflag tagged Zuc. Mitochondria were stained using Mitoview green. On the right, enlargements of the areas indicated by dashed boxes in the image on the left are shown, with the nuclei oriented at the bottom.
(C) Northern blot analysis of viral responder piRNA production in Aag2 cells upon dsRNA-mediated knockdown of Ae. aegypti PLDc_2 domain containing proteins and the PARN ortholog AAEL001426. Numbers indicate the VectorBase gene identifiers (without the AAEL0 prefix). The 24 and 33 nt size markers are inferred from an EtBr stained small RNA marker and rRNA stained. EtBr served as a loading control (same for (F)).
(D) Viral responder piRNA (11585(+)) size distribution as determined in small RNA deep-sequencing libraries from dsLuc and dsZuc treated cells. Read counts were normalized to the number of miRNAs in each library. The inset shows the average responder piRNA read size in Luc- and Zuc knockdown libraries. Bars and whiskers represent mean and SD, respectively.
(E) Read count of the responder piRNA in the reporter locus (left) and to the SINV genomic RNA (right), which is common for the three reporter viruses, in dsLuc and dsZuc treated Aag2 cells. Bars and whiskers show the mean +/- SD (left graph) and mean +/- SEM (right graph).
(F) A sharpness score was attributed to all viral piRNAs upstream of the artificial reporter cassette (see materials and methods). The maximum score (log2(14) = 3.81) would be reached if 100% of reads that map to an individual piRNA had the same length. For each of the different reporter viruses the sharpness score for the top most 275 piRNAs were analyzed upon control and Zuc knockdown. The piRNAs were ranked according to the score in the control condition and the means within 11 bins of 25 piRNAs were determined (orange shade). The difference of piRNA score upon Zuc knockdown is plotted as mean ± SEM. A two-sided student’s t-test was applied to each bin, to assess whether its mean was significantly different from zero. The null hypothesis was rejected at p<0.05. * P < 0.05 and ** P < 0.01
(G) Same as (F), but for transposon piRNAs.
(H) Western blot of indicated PIWI proteins in Zuc-3×flag immunoprecipitations (IP) in Aag2 cells.
To our surprise, we found that AAEL011385 contains a sizeable insertion directly downstream of the catalytic HKD-motif (Figure S5A). Similarly, Drosophila Zuc also contains a (much smaller) insertion in the same location relative to mouse mitoPLD, suggesting it is a variable region beyond Aedine mosquitoes. This non-conserved region is part of a helix that sticks out of the Zuc core structure (36). Homology detection revealed that the large insertion is conserved in the Culicidaefamily, but not in Drosophila, suggesting it is a mosquito-specific insertion. Moreover, during cloning of the AAEL011385 gene, we found that the size of this insertion is increased in Aag2 cells (32 additional amino acids; Figure S5A).
We next aimed to functionally validate AAEL011385 as the ortholog of DmZuc. Indeed, knockdown of AAEL011385 in Aag2 cells resulted in a broader size distribution of viral responder piRNAs (Figure 4C, Figure S5B), which is consistent with reduced sharp endonucleolytic 3’ end generation. Knockdown of the other PLDc_2 domain containing proteins (AAEL003651 and −22490) did not affect responder piRNA size (Figure 4C, Figure S5B), despite very efficient knockdown (88-94% and 96-98% for AAEL003651 and −22490, respectively, compared to the modest, 58-71% knockdown of AAEL011385/Zuc; Figure S5C). Because of the reduction in responder piRNA 3’ end sharpness upon AAEL011385 knockdown and its mitochondrial localization, we formally identify AAEL011385 as the functional Zuc ortholog in Ae. aegypti.
Small RNA deep-sequencing of dsZuc-treated Aag2 cells recapitulated the viral responder piRNA size distribution seen by northern blotting, with a general increase in piRNA length upon knockdown of Zuc (Figure 4D). We propose that upon Zuc knockdown, the viral RNA is cleaved downstream of the uridine residue, either by a hitherto unknown endonuclease or by piRNA-PIWI ribonucleoprotein complexes, as in Drosophila (12). Subsequently, the responder pre-piRNA may be trimmed, giving rise to mature piRNAs with more diffuse 3’ ends in a Zuc-independent manner.
To study the effects of Zuc knockdown on the 3’ end sharpness of viral piRNAs outside of the reporter locus in more detail, we assigned a sharpness score to every individual piRNA, defined by a shared 5’ end (see materials and methods for details). As expected, Zuc knockdown significantly reduced sharpness scores, especially of those piRNAs that in control conditions had the sharpest 3’ ends and are therefore likely to be the most prominent Zuc substrates (Figure 4E). The same effect was observed for piRNAs that mapped to transposon sequences (Figure 4F). Moreover, Zuc knockdown resulted in an overall increase in size of piRNAs produced from sense as well as antisense substrates of various origins, including retrotransposon subfamilies, mRNAs and viral RNAs (Figure S5F), suggesting that Zuc is involved in the formation of 3’ end of piRNAs from all substrates.
We next assessed the global effects of Zuc depletion on vpiRNA levels. While the abundance of the g73-triggered artificial responder piRNA was largely unaffected upon Zuc knockdown (Figures 4G-left), Zuc depletion reduced the overall vpiRNA production from the SINV genomic and subgenomic RNA, which is common to the g73-25U, −28U and −30U viruses (Figure 4G-right).
At the heart of the ping-pong amplification machinery in Ae. aegypti are two PIWI proteins: Ago3, which predominantly binds sense piRNAs, and Piwi5, which mainly associates with antisense piRNAs (24,34). In Drosophila, a strong interaction between Zuc and Aub (the ortholog of Piwi5), as well as weak associations between Zuc and Ago3/Piwi have been observed (40–42). We thus evaluated the interaction between Zuc and the somatic Aedes PIWI proteins. While we readily detect Piwi5 in Zuc immunoprecipitates, we did not observe interaction of Zuc with Ago3 (Figure 4H), nor with Piwi4 and −6 (Figure S5E). The interaction between Zuc and Piwi5 may underlie the comparatively stronger +1U bias observed for piRNAs derived from antisense transcripts.
A subset of responder piRNAs undergoes Nibbler-mediated trimming
In Drosophila, piRNA 3’ ends are generated by the concerted activities of two enzymes: the endonuclease Zuc (7–9) and the 3’ – 5’ exonuclease Nbr (12–14). Aside from its role in piRNA 3’ end formation, Nbr is required for the trimming of microRNAs (miRNAs) (43,44). We set out to identify the functional Ae. aegypti Nbr ortholog by predicting all DEDDy-type 3’ - 5’ exonuclease domain-containing proteins, which were used in a phylogenetic analysis along with Drosophila Nbr (DmNbr). This analysis identified AAEL005527 as a one to one ortholog of DmNbr (Figure 5A). To verify that AAEL005527 is indeed the functional orthologue of Drosophila Nbr, we assessed the effect of its depletion on trimming of two miRNA with heterogeneous 3’ ends in Ae. aegypti: miR-34-5p and miR-184 (45,46). It has previously been shown that miR-34-5p undergoes extensive Nbr-mediated trimming in Drosophila (43,44). Indeed, knockdown of AAEL005527 significantly reduced trimming of both miR-34-5p and miR-184 (Figure 5B), confirming that AAEL005527 is indeed the functional ortholog of Drosophila Nbr.

(A) Neighbor joining tree based on the DEDDy 3’ – 5’ exonuclease domains identified in Ae. aegypti, along with the DEDDy consensus sequence (CDD) and the exonuclease domain of DmNbr.
(B) Northern blot analyses of viral responder piRNAs in Aag2 cells in which Zuc and AaeNbr knockdown was established. miR-34-5p and miR-184 are two miRNAs with heterogenous 3’ ends in Ae. aegypti. mir-34-5p has been shown to undergo Nbr-mediated trimming in Drosophila (43,44).
To evaluate the role of exonucleolytic trimming on responder vpiRNA 3’ end formation in Ae. aegypti, we combined Nbr knockdown with infection using the g73-targeted reporter viruses. Knockdown was efficient and did not have major effects on viral RNA levels (Figure S6A-B). Interestingly, for all viruses tested, Nbr knockdown results in a specific loss of shorter (<27 nt) responder piRNA isoforms, but had no effect on larger isoforms (Figure 5B, Figure S6C). A similar reduction of shorter piRNA isoforms upon Nbr knockdown has previously been observed in Drosophila (13,14). These findings suggest that mosquito Nbr may trim pre-piRNAs generated through a Zuc-mediated endonucleolytic cut, but that only a minor fraction of such pre-piRNAs undergo trimming. Nbr-mediated trimming may be a general hallmark of aging piRNAs, as has recently been proposed for miRNAs (47).
The 3’ - 5’ exonucleases PNLDC1 and PARN-1 are responsible for trimming of piRNA 3’ ends in B. mori and C. elegans, respectively (48,49). While PNLDC1 is not conserved in Ae. aegypti, a clear mosquito ortholog of PARN-1 can be identified: AAEL001426. Knockdown of this gene however, had no effect on responder piRNA 3’ end formation in our viral reporter system (Figure 4C, Figure S5B).
Targeting by an endogenous piRNA triggers trailer piRNA production
While Ae. aegypti displays strong signatures of phased piRNA production (7), it is currently unknown whether de novo produced RNA from cytoplasmic viruses is processed similarly through piRNA phasing. This is especially interesting as the genomes of Ae. aegypti and Ae. albopictus mosquitoes contain large amounts of endogenous viral elements (EVEs). These non-retroviral sequence elements are enriched in piRNA clusters and, accordingly, give rise to abundant piRNAs (26), which may guide the slicing of cognate RNA from acutely infecting viruses. It has been recently shown that piRNAs derived from EVEs are indeed able to target and inhibit newly infecting viruses (31–33), yet, it remains unknown whether piRNA phasing may serve as a mechanism to expand the vpiRNA pool after an initial cleavage by an endogenous, possibly EVE-derived, piRNA.
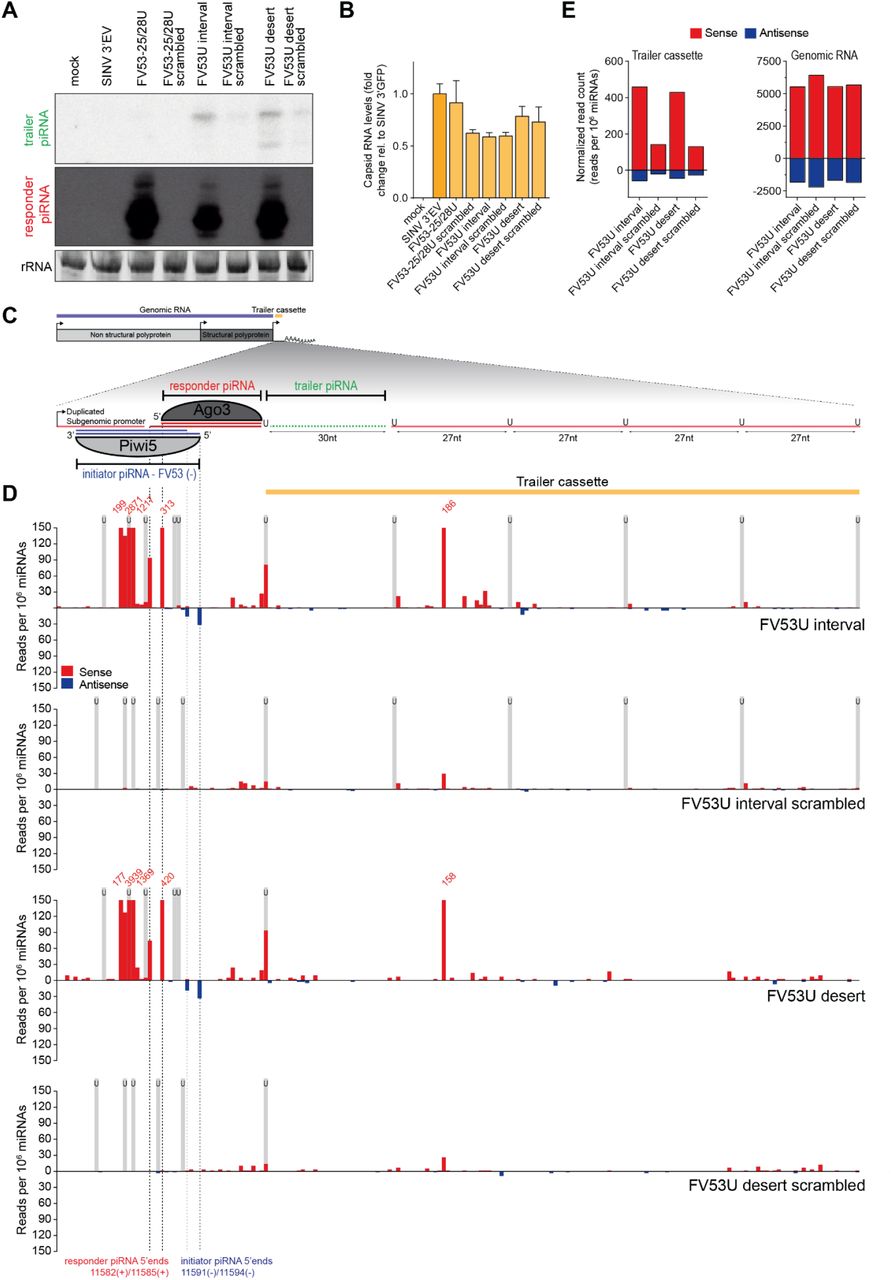
Hence, we employed our viral reporter system to explore whether targeting by a genomically encoded piRNA may trigger phased piRNA biogenesis from an acutely infecting virus. To this end, we introduced an additional non-coding RNA sequence downstream of the g73- and FV53-derived initiator piRNA target sites, which we termed the trailer cassette. To direct sequential Zuc-mediated endonucleolytic cleavage, this cassette contains uridine residues at regularly spaced intervals in an RNA sequence that is otherwise devoid of uridines (U interval viruses, schematically shown in Figure 6C and Figure S7C). As a control, these uridines were replaced by adenosine residues to create a trailer cassette completely devoid of uridines (U desert viruses). In Aag2 cells infected with the g73 interval virus, we detected the first trailer piRNA using northern blotting (Figure 6A). Interestingly, we also observed the production of the first trailer piRNA in cells infected with the g73 desert virus (Figure 6A), suggesting a downstream uridine residue is not required for trailer piRNA production in Ae. aegypti. Trailer piRNA production from viruses targeted by the FV53 EVE-derived piRNA was also observed regardless of the presence of uridine residues in the trailer cassette (Figure S7A). Importantly, while viral RNA levels were similar (Figure 6B), no trailer piRNA production was observed in cells infected with viruses containing a scrambled target site or viruses lacking a trailer cassette (g73-28/FV25-28), indicating that, as expected, trailer piRNA production depends on initial targeting by the endogenous piRNA.
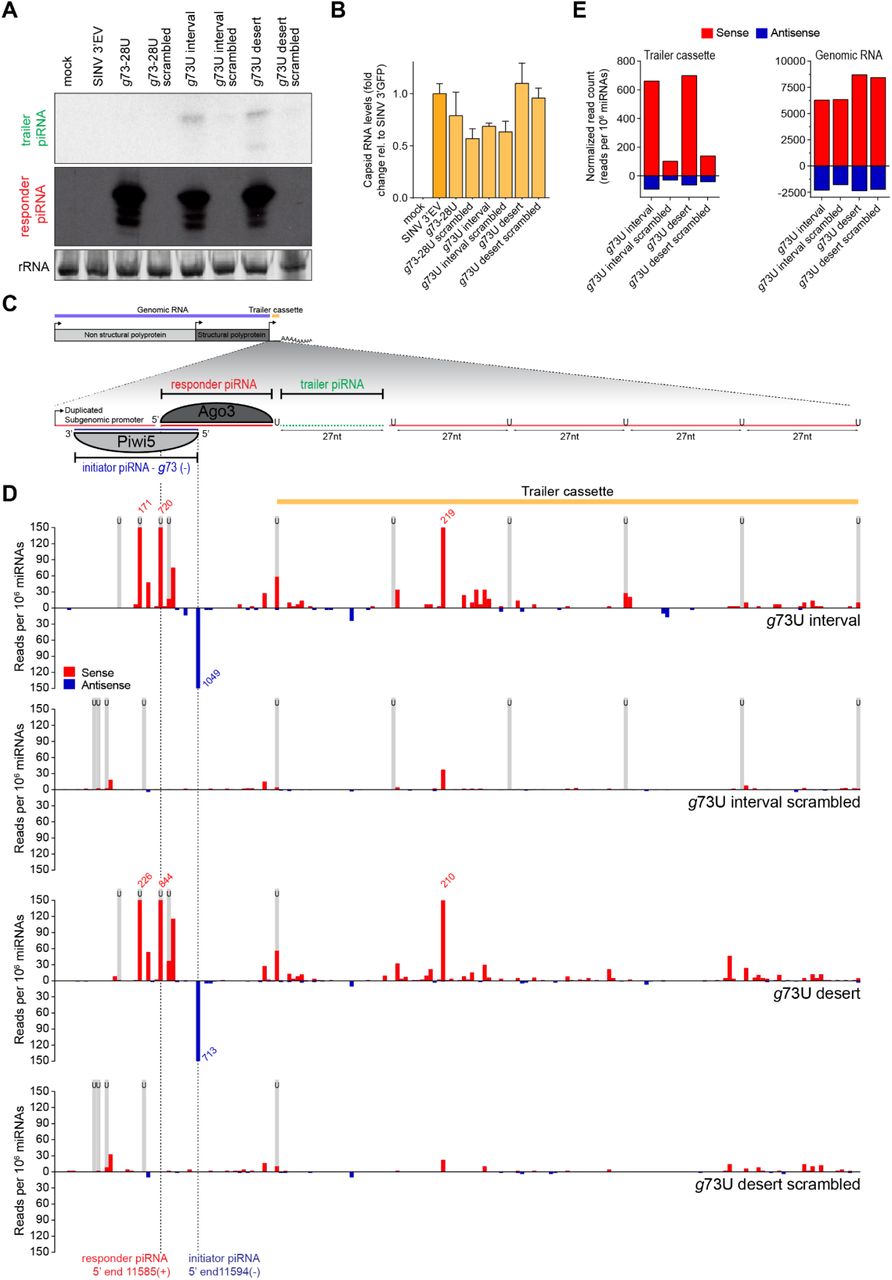
(A) Northern blot analysis of the production of the first trailer piRNA from indicated viruses. As a control, the target site was scrambled to abolish targeting by the g73-derived piRNA. The remainder of the responder piRNA site as well as the trailer cassette are identical to the respective non-scrambled U interval and U desert viruses. As additional controls, a virus bearing an intact target site, but no trailer cassette (g73-28U), and a virus that contains no insert (SINV 3’ EV) were used. The general structure of the g73 U interval virus is shown schematically in (C). rRNA stained with EtBr serves as a loading control.
(B) RT-qPCR analyses of viral capsid RNA production in Aag2 cells infected with indicated viruses. All data are shown as a fold change relative to cells infected with a control virus lacking any insert (SINV 3’ EV). Bars and whiskers show the mean and SD, respectively.
(C) Schematic overview of the g73 U interval reporter virus. The inset shows a magnification of the non-coding reporter RNA expressed under control of the second subgenomic promoter. This reporter RNA contains a target site for a Piwi5-bound g73-derived initiator piRNA, which guides the production of an Ago3-associated responder piRNA. The downstream sequence makes up the trailer cassette and either contains regularly spaced uridine residues (g73U interval) or is completely devoid of uridine residues (g73U desert). Indicated in green is the first trailer piRNA, which was detected in (A).
(D) Normalized counts (in reads per 106 miRNAs) of piRNA 5’ ends mapping to the initiator piRNA target site and trailer cassette of indicated viruses. 5’ ends of initiator and responder piRNA are indicated by dashed lines. Numbers indicate counts that exceed the range of the y-axis and black boxes indicate the position of uridine residues. The position of uridine residues is indicated in grey.
(E) Total number of normalized sense (red) and antisense (blue) piRNA-sized (24-33 nt) reads mapping to the trailer cassette (left) and genomic RNA (right) of the indicated viruses. The areas of virus denoted as the trailer cassette and genomic RNA are shown in yellow and blue in (C).
To assess phased piRNA biogenesis beyond the first trailer piRNA, we deep sequenced small RNAs produced in Aag2 cells infected with interval and desert viruses, as well as their respective scrambled control viruses. Mapping piRNAs to the trailer cassette reveals production of additional piRNAs in cells infected with the g73- and FV53-derived piRNA targeted viruses (Figure 6D, Figure S7D). Significantly fewer piRNAs were produced from the trailer cassette in viruses containing a scrambled target site. In contrast, vpiRNA production from the SINV genome upstream of the artificial reporter and trailer cassettes is unaltered (Figure 6E, Figure S7E), indicating that there are no differences in sensitivity of these viruses for processing by the vpiRNA biogenesis machinery. As both the pattern and level of piRNA production is highly similar between U interval and U desert viruses (Figure 6D-E, Figure S7D-E), the presence of uridine residues to guide Zuc-mediated endonucleolytic cleavage appears to be dispensable for phased production of piRNAs from viral RNA, at least in the context of this reporter virus.
Conclusion
Altogether, our results indicate that during acute infection with a cytoplasmic RNA virus, genomically encoded piRNAs can initiate piRNA production from viral genomic RNA via the ping-pong amplification loop (Figure S8). The endonucleolytic and exonucleolytic activities of Zuc and Nbr are involved in maturation of the 3’ ends of these newly produced piRNAs. Moreover, Zuc is also required for 3’ end formation of endogenous and viral piRNAs, whereas Nbr trims miRNAs, like in Drosophila. Importantly, cleavage of viral RNA by an endogenous piRNA triggers the diversification of piRNA sequence pool by piRNA phasing. These findings suggest that few cleavage events by individual genome-encoded piRNAs are sufficient to launch a piRNA response of bona fide vpiRNAs.
MATERIALS AND METHODS
Cell culture, transfection and infection of Aag2 and U4.4 cells
Ae. aegypti Aag2 and Ae. albopictus U4.4 cells were grown in Leibovitz’s L-15 medium (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), 50 U/ml Penicillin, 50 μg/mL Streptomycin (Invitrogen), 1x Non-essential Amino Acids (Invitrogen) and 2% Tryptose phosphate broth solution (Sigma) at 25°C. Cell lines were maintained by splitting twice weekly according to confluency.
For knockdown experiments, ~1×106 cells were seeded in 6-wells plates and allowed to attach for 16-24 hrs. Subsequently, cells were transfected with gene-specific dsRNA and retransfected 48 hrs later to ensure sustained knockdown. Three hrs after the second transfection, cells were infected with indicated viruses at multiplicity of infection (MOI) of 0.1 and RNA was harvested 72 hrs post infection. In experiments where no knockdown was performed, cells were infected 16-24 hrs after seeding at MOI = 0.1, followed by RNA extraction at 72 hrs post infection
Generation of reporter viruses
Target sites for initiator piRNAs and reporter responder piRNA were introduced into the recombinant Sindbis virus backbone to be expressed from a second subgenomic promoter. The previously described SINV-GFP (50,51) was digested using XbaI to remove the GFP-gene. The reporter locus was subsequently introduced downstream of the duplicated subgenomic promoter by ligation of annealed oligo’s (see below), making use of the overhangs generated by the XbaI enzyme (initiator piRNA target site in red, downstream uridine position is indicated in bold font).

To introduce U-interval and U-desert reporter sequences, the g73-28U and FV53-25/28U viruses were digested using NotI followed by ligation of annealed oligo’s (see below).

Mutagenesis of target site mutant viruses
Target site mutations were introduced into the g73-28U virus backbone by mutagenesis PCR, using the primers shown below. PCR-products were DpnI-treated and In-fusion (Takara Biotech) was using to circularize the plasmid for transformation. After verification of the sequence by Sanger sequencing, viruses were grown as described previously (25).

dsRNA production for knockdown experiments
Gene-specific PCR products bearing T7 promoter sequences at both ends were in vitro transcribed using T7 polymerase. Either the T7 promoter sequence was introduced directly with the gene-specific PCR, or a universal GC-rich tag was introduced in the first PCR, to which the T7 promoter sequence was added in a second PCR. Oligos use to create the T7-promoter tagged PCR products are shown below (T7 promoter sequence in bold font, the universal GC-rich tag in underlined font):

Generation of Piwi5 knockout U4.4 cells
Description and characterization of Piwi5 knockout U4.4 cells is described elsewhere (manuscript in prep).
Phylogenetic analyses
The PFAM PLDc_2 domain (PF13091) consensus sequence was as input for iterative searches against the Ae. aegypti proteome using JackHMMR (https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer) to identify all Ae. aegypti PLDc_2 domain containing proteins. To generate a neighbor-joining tree, the PLDc_2 domain sequences from the identified genes, as well as those from Zuc orthologs in Drosophila, silkworm and mouse were aligned using M-Coffee (http://tcoffee.crg.cat/apps/tcoffee/do:mcoffee). One of the identified genes, AAEL022490, contains two PLDc_2 domains, both of which were used as separate inputs in the subsequent analysis. The same approach using the DEDDy type 5’-3’ exonuclease (cd06141) CDD-consensus sequence was used for the identification Ae. aegypti Nbr. For the alignment of full Zuc proteins, the entire sequence of Zuc as annotated on VectorBase (www.vectorbase.org), the Zuc sequence as determined by us from Aag2 cells and the sequences of Zuc orthologs in Drosophila, silkworm, mouse were aligned using M-Coffee with default settings and visualized using Jalview 2.11.0. The analyses of conservation of physicochemical properties at each position was performed in Jalview 2.11.0., and is based on the Analysis of Multiply Aligned Sequences (AMAS) method (52).
RNA isolation
Cells were lysed in 1 mL RNA-Solv reagent (Omega Bio-tek), followed by RNA extraction through phase separation and isopropanol precipitation. RNA integrity was evaluated on EtBr stained agarose gels, and RNA concentration was determined using the Nanodrop ND-1000.
Small RNA northern blotting
For small RNA northern blotting, 4-12 μg of RNA was size separated by denaturing urea polyacrylamide (15%) gel electrophoresis and transferred to nylon membranes using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (53). 32P labelled DNA probes (sequences shown below) were used to detect small RNAs.

To generate the line graphs shown in Supplementary Figure S5B and S5C, northern blot signals were quantified at the center of each lane. Afterwards, peaks were manually aligned to correct for minor variations in size separation between samples. The average of five neighboring pixels was plotted to smoothen the line graph.
Generation of small RNA deep sequencing libraries
Small RNA deep sequencing libraries were generated using the NEBNext Small RNA Library Prep Set for Illumina (E7560, New England Biolabs), using 1μg RNA as input. As piRNAs generally have 2’-O-methylated 3’ ends, we performed the 3’ adapter ligation for 18 hrs at 16°C to enhance the ligation efficiency of small RNAs bearing such modifications. The rest of the library preparation was performed in accordance with the manufacturers’ instructions. Libraries were sequenced on an Illumina Hiseq4000 machine by the GenomEast Platform (Strasbourg, France).
Small RNA sequencing bioinformatic analyses
RTA 2.7.3 and bcl2fastq were used for image analysis and base calling, and initial quality control was performed using FastQC. All subsequent manipulations were performed in Galaxy (54). First, the FASTX Clip adapter software was used to clip 3’ adapters from the small RNA sequence reads. Subsequently, reads were mapped onto the corresponding recombinant Sindbis virus genomes, transposable element sequences extracted from TEFAM (http://tefam.biochem.vt.edu; downloaded on 10-12-2019), Ae. aegypti transcripts (AaegL5.2 gene set, downloaded from VectorBase) and the Phasi Charoen like virus (PCLV) genome as sequenced from Aag2 cells (Genbank accession numbers KU936055, KU936056 and KU936057) (55). Reads were mapped using Bowtie allowing 1 mismatch, except for the mapping to interval and desert viruses and for 3’ end sharpness analyses, in which cases no mismatches were allowed. Small RNA libraries were normalized to the number of reads mapping to published pre-miRNA sequences deposited in miRBase v.21 without allowing mismatches.
Size profiles were obtained by counting piRNA lengths after mapping with exception of the size profile of small RNAs obtained after PIWI IP (Figure S2E), for which raw reads prior to mapping were analyzed from our previously published data (34). For genome distribution plots, the number of piRNA 5’ ends was determined for each position of the sequence to which small RNAs have been mapped.
For the analyses of piRNA production from the area of the non-coding reporter RNA downstream of the initiator/responder site versus the remainder of the viral genome shown in Figure 6E and Figure S7E, the SINV genome is defined as the area encoding the non-structural and structural polyproteins up to the start of the duplicated subgenomic promoter (nt 1-11385), and the trailer cassette as nt 11610-11753.
Sequence data have been deposited in the NCBI sequence read archive under SRA accession XXXXX.
To generate heat maps of the length profile of individual piRNAs, published small RNA sequencing data was re-analyzed (24). SAM files were converted into interval files and piRNA-sized reads (25-30 nt) were extracted and separated according to the strand they mapped to. For each individual mapped piRNA species, defined by a shared 5’ end, the frequency of reads of different length was determined. Only piRNAs that were supported by at least 20 reads were considered for further analyses. The frequency of read lengths per individual piRNA was imported into Multiple experiment viewer (v 4.9.0; tm4) and k-means clustering based on Pearson correlation was performed using the indicated number of clusters and a maximum number of iterations of 500. The ‘Construct hierarchical tree’ option was enabled.
To analyze nucleotide biases within and downstream of each piRNAs, only piRNA species that were supported by at least 20 reads and had a dominant piRNA length (at least 75% of all mapped reads have the same length) were considered. Combining the Get flanks (v1.0.0) and Get genomic DNA (v3.0.3) tools, the genomic sequence 5 nt downstream of that dominant piRNA length was extracted. Subsequently, each piRNA species was collapsed to one unique sequence and its corresponding downstream sequence. The sequence logo was generated from FastA files that contained either these unique piRNA or downstream sequences. The piRNA logos were cropped in Adobe illustrator to show nucleotide biases of piRNA positions 1 to 12 and +1 to +5.
To analyze the effect of Zuc knockdown on piRNA 3’ ends, small RNAs mapping to the SINV genome up to the subgenomic promotor (nt 1-11385) were considered. For each of the six datasets (3x dsLuc and 3x dsZuc) available for the g73-25U virus, piRNA species were extracted by selecting only those piRNA start sites that were supported by at least fifty 25-30 nt reads in the combined six datasets. For these piRNA positions, all reads in the size range of 25-38 nt were then extracted from the original mapped data. As for the heat map analysis, the frequency of piRNA lengths was counted for each individual piRNA species. To filter out noise due to low read count, only the 275 most abundant piRNAs were considered, which corresponded to approximately 150 reads in the combined six datasets. From this frequency distribution, a sharpness score was calculated for each individual piRNA species based on the maximum entropy (all reads have the same length) minus the observed Shannon entropy:  (56). The mean sharpness score for each piRNA was determined for the three control and Zuc knockdown datasets and the 275 piRNAs were then ranked based on mean sharpness score in the control knockdown from high score (sharp 3’ end) to lower scores (more diffuse 3’ end). Within this ranking an average of the mean sharpness scores was then calculated for 11 bins of 25 individual piRNA positions each, both for the control and the Zuc knockdown. Finally, the difference in sharpness score (ΔSsharp) between control and Zuc knockdown was determined for each bin. The entire analysis was repeated for the six datasets of the g73-28 and g73-30 viruses and the data was combined by calculating the mean + SEM of the ΔSsharp per bin obtained for each type of reporter virus. To determine statistical significance, the ΔSsharp was tested against the null hypothesis that there is no effect of Zuc knockdown on 3’ end sharpness scores (ΔSsharp = 0). A two-sided student’s t-test was performed per bin and the hypothesis was rejected at p<0.05. The sharpness score analysis for transposon mapping piRNAs was performed with the 4400 most expressed piRNAs (400 piRNAs per bin).
(56). The mean sharpness score for each piRNA was determined for the three control and Zuc knockdown datasets and the 275 piRNAs were then ranked based on mean sharpness score in the control knockdown from high score (sharp 3’ end) to lower scores (more diffuse 3’ end). Within this ranking an average of the mean sharpness scores was then calculated for 11 bins of 25 individual piRNA positions each, both for the control and the Zuc knockdown. Finally, the difference in sharpness score (ΔSsharp) between control and Zuc knockdown was determined for each bin. The entire analysis was repeated for the six datasets of the g73-28 and g73-30 viruses and the data was combined by calculating the mean + SEM of the ΔSsharp per bin obtained for each type of reporter virus. To determine statistical significance, the ΔSsharp was tested against the null hypothesis that there is no effect of Zuc knockdown on 3’ end sharpness scores (ΔSsharp = 0). A two-sided student’s t-test was performed per bin and the hypothesis was rejected at p<0.05. The sharpness score analysis for transposon mapping piRNAs was performed with the 4400 most expressed piRNAs (400 piRNAs per bin).
RT-qPCR
For RT-qPCR analyses, DNaseI (Ambion)-treated RNA was reverse transcribed using Taqman reverse transcriptase (Life Technologies) and PCR amplified in the presence of SYBR green, using the GoTaq qPCR system (Promega) according to the manufacturers’ recommendations. Expression levels of target genes were normalized to the expression of the housekeeping gene lysosomal aspartic protease (LAP) for Ae. aegypti samples or Ribosomal Protein L5 (RpL5) for Ae. albopictus samples and fold changes were calculated using the 2ΔΔCT method (57). The following primers were used:

Generation of Zuc expression plasmids
The sequence encoding the Zuc protein was amplified from Aag2 cDNA using CloneAmp HiFi PCR Premix (Takara) and cloned into the pAWG vector (The Drosophila Gateway Vector Collection, Carnegie Science) using In-fusion (Takara) technology.
The following primers were used to amplify the vector and insert:

Subsequently, pAW3F-Zuc was generated by inverse PCR of pAWG-Zuc, which introduced the 3×flag tag to replace the eGFP-tag, followed by In-fusion cloning (Takara Biotech). The primers used for this inverse PCR are shown below (underlined sequence makes up the 3×flag tag, double underlined sequence is the 15 nt overlap required for In-fusion cloning):
Immunofluorescence
Approximately 5×105 Aag2 cells were transfected with 1 μg of the pUb-Zuc-3xflag expression vector using 1 μL X-tremeGENE HP DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. 48 hrs later, cells were fixed on coverslips using 4% paraformaldehyde for 10 minutes at room temperature. Fixed cells were permeabilized in PBS-Triton (0.25%) for 10 minutes and blocked in 10% normal goat serum/0.3M Glycine in PBS-Tween (0.1%). Antibody staining was performed using mouse anti-flag (1:200, Sigma, F1804, RRID: AB_262044) for 1 hr at room temperature, followed by goat anti-mouse IgG Alexa fluor 568 (1:200, Invitrogen, A-11004, RRID: AB_2534072) for 1 hr at room temperature. Following antibody staining, mitochondria were stained using 200 μM Mitoview Green (Biotium), according to the manufacturer’s instruction. Lastly, Hoechst reagent was used to stain the nuclei and the coverslips were mounted onto microscope slides for imaging using the Zeiss LSM900 confocal microscope. In between all steps during the staining procedure, cells were washed three times with PBS.
Co-immunoprecipitation and western blot
For immunoprecipitation of Zuc, ~4.5 × 106 Aag2 cells were seeded and after ~16 hrs, 15 μg of the pAW3F-Zuc was transfected using X-tremeGENE HP DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. Cells were lysed 48 hrs after transfection using 300 μL lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% Igepal CA-630 [Sigma], 10% Glycerol, 1x cOmplete Protease Inhibitor [Roche], 1 mM PMSF). After incubation for 1 hr at 4°C with end-over-end rotation, lysates were centrifuged for 30 min at 15000 × g, 4°C. The supernatant was snap-frozen in liquid nitrogen and stored at −80°C for later use.
For immunoprecipitation, 15 μL M2-Flag bead slurry (Sigma) was equilibrated in lysis buffer, and, along with 450 μL dilution buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 1x cOmplete Protease Inhibitor [Roche], 1 mM PMSF), added to the thawed lysate. After incubation for 2 hrs at 4°C with end-over-end rotation, beads were washed thrice using 500 μL dilution buffer, before harvesting immunoprecipitates by boiling for 10 min in 20 μL 2× sample buffer (120 mM Tris/Cl pH 6.8, 20% glycerol, 4% SDS, 0.04% bromophenol blue, 10% β-mercaptoethanol). After boiling, samples were diluted by adding 20 μL lysis buffer.
Samples were resolved on 10% polyacrylamide gels and blotted to nitrocellulose membranes. The following antibodies, generated in our laboratory, were used for western blotting: rabbit-anti-Ago3, -Piwi4, -Piwi5 and -Piwi6 (all at 1:500) (34,58). Additionally, mouse anti-flag (1:1000, Sigma, F1804, RRID: AB_262044) was used. Secondary antibodies were goat-anti-rabbit-IRdye800 [Li-cor; 926-32211, RRID: AB_621843] and goat-anti-mouse-IRdye680 [926-68070, RRID: AB_10956588].
SUPPLEMENTARY MATERIAL

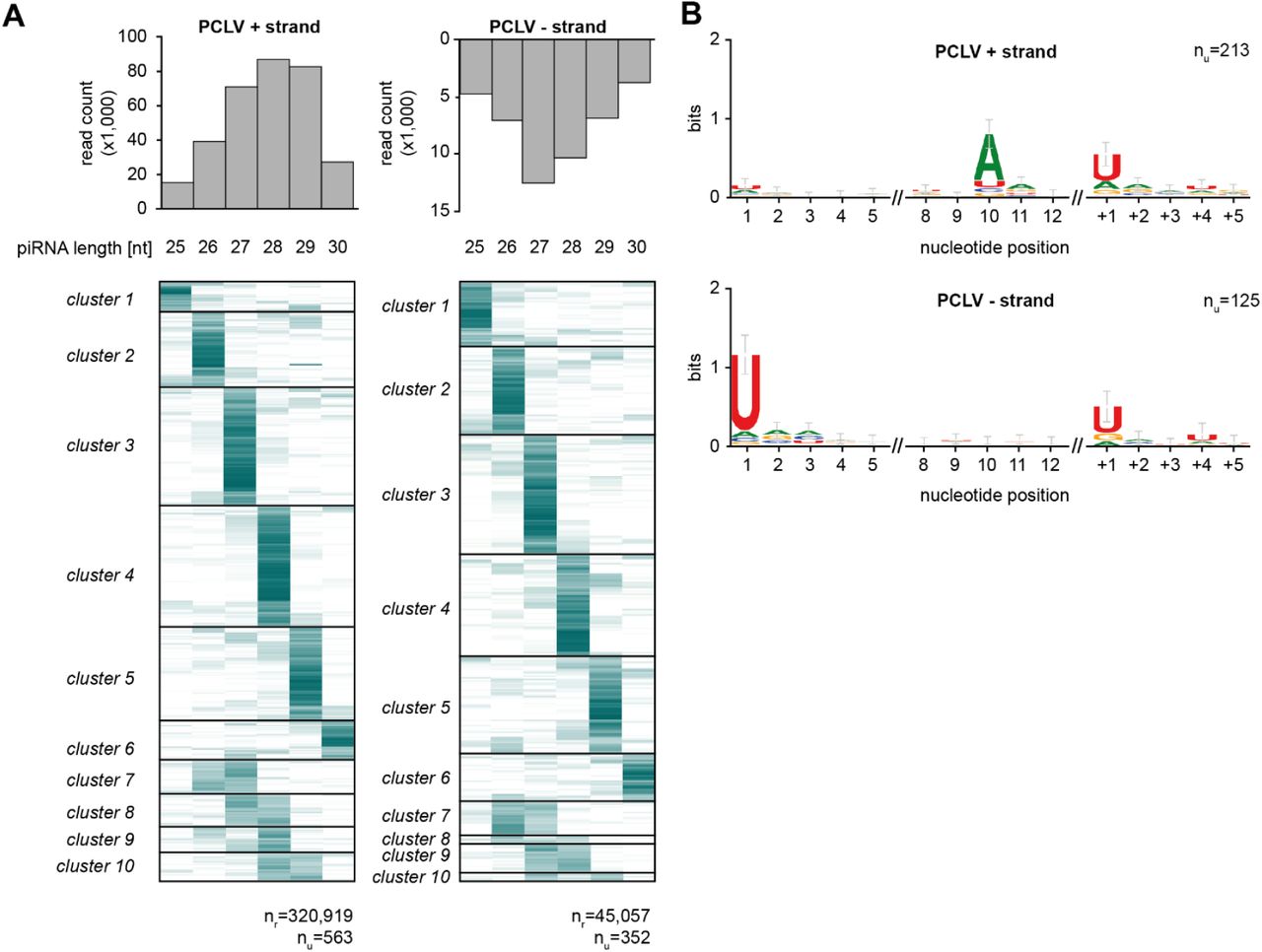
(A) Heat map showing the relative size distribution of individual piRNAs (defined by a shared 5’ end) mapping to the genome of Phasi Charoen-like virus (PCLV). Shades of blue indicate the percentage of reads contributing to the indicated read length, white represents absence of reads of a specific size. The number of unique piRNAs (nu) and the number of reads (nr) that underlie the heat map are indicated. A minimum of 20 reads/unique piRNA was required to be included in the analysis.
(B) Nucleotide biases for the indicated nucleotide positions of PCLV-derived piRNAs and a downstream region. Only piRNAs from (A) that had a dominant length (determined by at least 75% of reads) were considered in this analysis and all reads were collapsed to unique sequences. The downstream region (+1 until +5) shows the bias at the genomic region downstream of the dominant piRNA sequences.
Supplementary text and Figure S2
An EVE-derived piRNA has the potential to trigger responder piRNA production
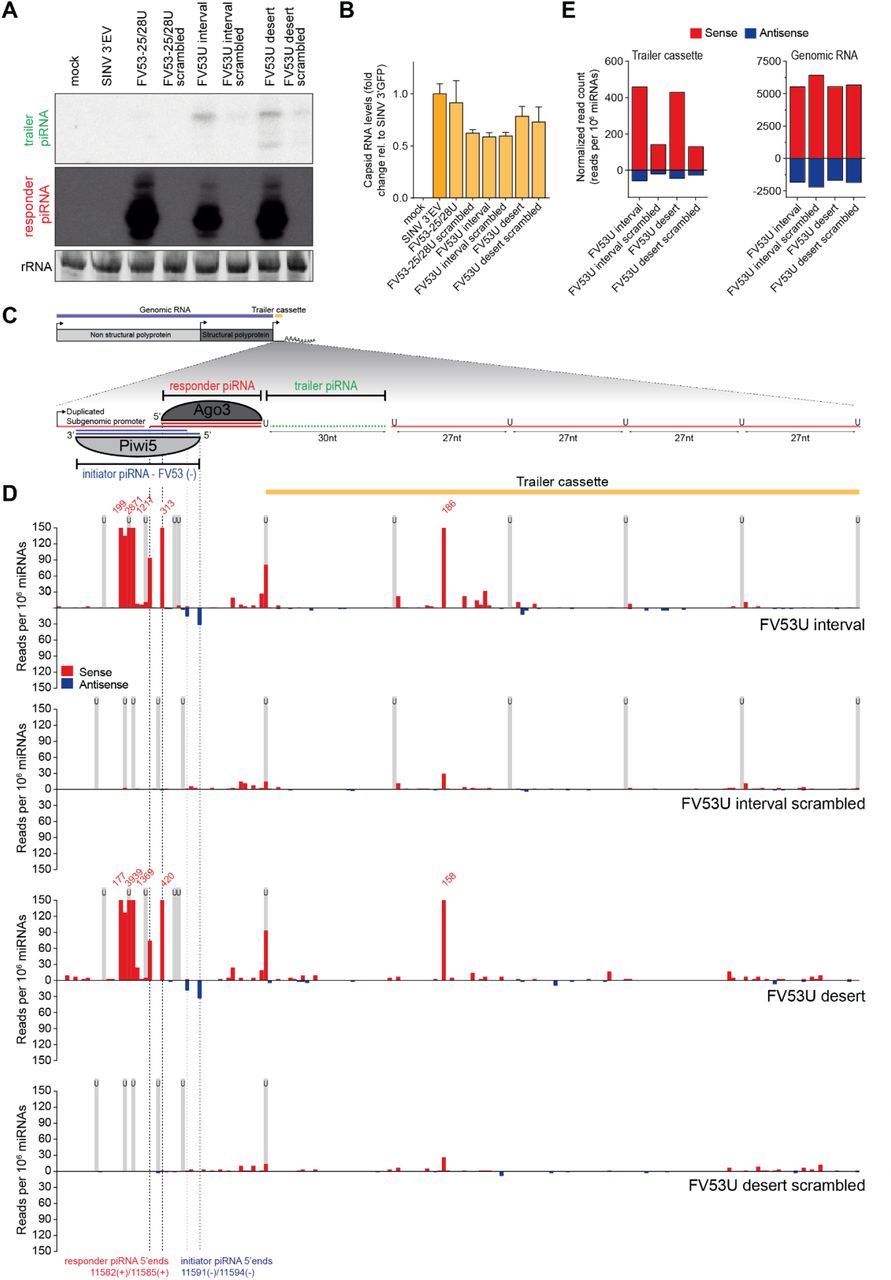
Apart from the g73 piRNA triggered reporter virus described in the main text, we generated a second set of viruses bearing target sites for an abundant Piwi5-associated piRNA derived from an EVE of flaviviral origin (FV53 (26), Figure S2A). As the target site for the FV53-derived initiator piRNAs can be recognized by two piRNA isoforms that align at their 3’ end and differ 3 nt in size, target cleavage may result in the production of two Ago3-bound responder piRNAs isoforms that have an offset of 3 nt (Inset in Figure S2A).
Similar to the g73 target site bearing viruses (Figure 2B), responder piRNA 3’ end sharpness is moderately reduced as a function of increased distance between the initiator piRNA cleavage site and the first downstream uridine residue (Figure S2B), suggesting a role for exonucleolytic trimming in responder piRNA maturation.
For all viruses tested here, we observe abundant responder piRNAs as well as the production of a first trailer piRNA (indicated with yellow arrowheads in Figure S2C). As the trailer piRNA 5’ ends perfectly overlap with the uridine residue, we propose that this residue guides cleavage of the viral RNA, simultaneously generating the responder pre-piRNA 3’ end and the trailer piRNA 5’ end.
Aside from these responder and trailer piRNAs, additional abundant sense piRNAs are produced from the sequence upstream of the initiator piRNA slice site (Figure S2C), suggesting that other, partially complementary, initiator piRNAs may target the sequence and instruct production of these additional sense piRNAs.
(A) Schematic depiction of the SINV-based viral reporter system in which responder piRNA production is triggered by an initiator piRNA derived from an endogenous viral element (FV53). As the Piwi5-associated FV53 initiator piRNA is expressed as two isoforms that align at their 3’ end (blue and green lines), endonucleolytic cleavage may generate two Ago3-bound responder piRNAs (red lines) that differ 3 nt in size. The inset shows a part of the viral target RNA (red), the 5’ ends of the two FV53-piRNA isoforms (27-mer: green and 30-mer: blue) and the scissors represent their respective slice sites (green and blue).
(B) Northern blot analysis of responder piRNAs produced in Aag2 cells infected with indicated FV53-targeted reporter viruses. 24 and 33 nt size markers are inferred from EtBr staining of a small RNA marker, and EtBr stained rRNA serves as loading control.
(C) Visualization of 5’ ends of sense (red) and antisense (blue) piRNAs (24-33 nt) mapping to the non-coding reporter RNA sequence. Predicted initiator and responder piRNA 5’ ends are indicated by dashed lines and positions of uridine residues on the sense strand are denoted by light grey shading. Red and blue numbers indicate read counts (per 106 miRNAs) at positions where they exceed the y-axis range and yellow arrowheads indicate 5’ ends of putative trailer piRNAs.

Size distribution of piRNA-sized (24-33 nt) small RNA reads in Ago3-(A), Piwi4-(B), Piwi5-(C), and Piwi6-IP (D) sequencing libraries. Piwi4-IP is dominated by two very abundantly expressed piRNAs (tapiR1/2-30 and 29 nt in size, respectively), that are involved in the degradation of maternally provided transcripts during embryonic development in mosquitoes (58). Counts are raw, unmapped reads from our previously published data (34,58).

(A) RT-qPCR analyses of subgenomic capsid RNA abundance in the samples used in Figure 3A. RNA levels are shown as a fold change relative to dsLuc treated cells infected with g73-25U. Bars and whiskers show the mean and standard deviation (SD), respectively (same for B and D).
(B) RT-qPCR analyses of capsid RNA abundance in the indicated U4.4 (yellow) and Aag2 (blue) samples used in Figure 3B. Values shown are fold changes relative to WT cells infected with the SINV 3’ EV control virus.
(C) Schematic representation of the g73-28U virus that was used to study the effect of target site mutations on responder piRNA production. Yellow shading indicates the area that is shown in Figure 3C.
(D) RT-qPCR analyses of capsid RNA levels in samples used in Figure 3D. All data are normalized to cells infected with the g73-28U scrambled virus.


(A) Multiple sequence alignment of Zucchini orthologs from D. melanogaster (DmZuc), B. mori (BmZuc) and M. musculus (MmMitoPLD), and AAEL011385 as annotated in VectorBase (VB) and as sequenced from Aag2 cells. Residues shaded in blue are shared between ≥3 of the proteins, and the brown-yellow bars indicate conservation of physicochemical properties at each position.
(B) Quantification of the responder piRNA signal from the northern blot in Figure 4B, showing the responder piRNA size shift upon Zuc knockdown, analyzed for the three g73-derived piRNA targeted viruses separately. The X-axis represents the position on the northern blot from top to bottom, the Y-axis shows responder piRNA signal intensity.
(C-D) RT-qPCR analyses of the knockdown efficiency of indicated genes (C) and viral capsid RNA levels (D). Gene expression levels are shown as fold change relative to dsLuc treated cells infected with the g73-25U virus. Bars and whiskers depict mean and SD, respectively.
(E) Western blot analyses of Piwi4 and Piwi6 in the same Zuc-3xflag-IP material that was used in Figure 4H.
(F) Average size of piRNAs (24-33 nt) derived from the indicated substrates in small RNA deep sequencing libraries of dsLuc and dsZuc treated Aag2 cells. The length profile of two piRNAs (tapiR1/2) involved in degradation of maternal transcripts during embryogenesis (58), is not affected by Zuc knockdown, suggesting that they are generated through an Zuc-independent, non-canonical mechanism. As virtually no antisense reads map to the tapiR1/2 locus, these data are not shown. Bars and whiskers represent mean and SD, respectively.

(A-B) RT-qPCR analyses of the knockdown efficiencies of the indicated genes (A) and viral capsid RNA levels (B). Expression values represent the fold change relative to dsLuc treated cells infected with the g73-25U virus. Bars and whiskers represent the mean +/- standard deviation.
(C) Quantification of the northern blot signal in Figure 5B, showing the effect of Nbr knockdown on responder piRNA size during infection with the indicated viruses. The Y-axis represents responder piRNA signal intensity, the X-axis represents the position on the northern blot from top to bottom.
(A) Northern blot analyses showing the production of the first putative trailer piRNA (marked in green in (C)) from the indicated viruses). As a control, the target site was scrambled to abolish targeting by the FV53-derived initiator piRNA. The remainder of the responder piRNA site, as well as the trailer cassette was identical to the U interval and U desert viruses. Additional controls include a virus that lacks the trailer cassette, but contains an FV53 piRNA-target site (FV25-28) and a control virus without any insert (SINV 3’ EV). A schematic overview of the FV53 interval virus is shown in (C). rRNA stained by EtBr serves as loading control.
(B) Capsid RNA levels in Aag2 cells infected with indicated viruses as determined by RT-qPCR. All capsid RNA levels are shown as a fold change relative to the virus lacking an initiator piRNA target site (SINV 3’ EV). Bars and whiskers denote the mean and SD, respectively.
(C) Schematic overview of the FV53 U interval virus. In this virus, a duplicated subgenomic promoter drives the expression of a non-coding reporter RNA (shown in the magnification), which contains a target site for two isoforms of a Piwi5-bound FV53-derived initiator piRNA (shown in blue). Slicing of the reporter RNA in the ping-pong amplification loop thus may give rise to two differently sized Ago3-associated responder piRNAs (shown in red). Downstream of this responder piRNA, the trailer cassette is located which either contains uridine residues at regularly spaced intervals (FV53 U interval) or is completely devoid of uridines (FV53 U desert). The first trailer piRNA which was detected in (A) is shown in green.
(D) Visualization of normalized counts of 5’ ends of sense (red) and antisense (blue) piRNA-sized reads (24-33 nt) mapping to the non-coding reporter RNA. The 5’ ends of the two FV53-derived initiator piRNA isoforms, as well as the 5’ ends of putative responder piRNAs are indicated by dashed lines. Numbers indicate normalized read counts where they exceed the range of the y-axis and grey shading indicates the position of uridine residues.
(E) Quantification of the number of sense (red) and antisense (blue) piRNAs (in reads per 106 miRNAs) mapping to the trailer cassette (left) and genomic RNA (right) of the indicated recombinant Sindbis viruses. The areas of the viruses denoted as trailer cassette and genomic RNA are indicated in (C) in yellow and blue, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Genomically encoded endogenous viral elements (EVE) give rise to a pool of Piwi5-associated initiator piRNAs that have the potential to target newly infecting viruses. Upon acute infection with a virus containing a cognate sequence, EVE-derived piRNAs trigger the production of Ago3-bound responder piRNAs from the viral RNA, which are generated and maturated by the combined activities of Zuc and Nbr. Additionally, targeting of the viral RNA by the ping-pong machinery prompts the processing of the downstream RNA into additional trailer piRNAs, thus expanding the piRNA sequence pool that is able to target viral RNA.
ACKNOWLEDGEMENTS
We thank the members of the laboratory for critical discussion of this manuscript. Furthermore, we thank Bas Pennings for his help with cloning experiments. This work was financially supported by a Consolidator Grant from the European Research Council under the European Union’s Seventh Framework Programme (grant number ERC CoG 615680) and a VICI grant from the Netherlands Organization for Scientific Research (grant number 016.VICI.170.090).
REFERENCES




