

Abstract
Nitric oxide is (NO) known to be produced by most Eukaryotic organisms although the NO production mechanism is only known for animals (Cánovas et al., 2016). Mammals have a set of different nitric oxide synthases (NOSs) with basically similar mechanism for producing NO under different circumstances (Velayutham and Zweier, 2013; Yu et al., 2014). NO-production is characteristically induced as part of innate immunity reactions to bacterial Microbial Associated Molecular Patterns (MAMPs). We knew from our previous work that the plant pathogen Fusarium graminearum quickly upregulates genes characteristic for an innate immune response so we set out to test if it also produces NO as part of this response and find the responsible genes if it does.
We found that F. graminearum produces NO in response to bacterial MAMPs and that the NO production system must be similar in fungi as in mammals where NO is activated by a homo-dimerization of nitric oxide synthetase proteins (NOSs) containing an N-terminal cytochrome P450 domain (CYP) and a C-terminal NADPH dependent cytochrome P450 reductase (NCP) domain. The electrons are transferred from the C-terminal NCP domains to the CYP domain in the paired protein to produce NO. We could not find a candidate NOS in the fungus. Thus, we tested the hypothesis that an NCP-protein similar to the NCP part of a NOS is brought together with a CYP-protein to produce NO in another way than the dimerization of a classic NOS. We found that in F. graminearum NO is produced by an FgNCP and an FgCYP located to the endoplasmic reticulum membrane where both proteins are predicted to be N-terminally attached by a transmembrane or embedded hydrophobic alpha helix. Deletion of any of these proteins lowered pathogenicity to wheat and radically reduced NO-production. Knockout of the FgNCP also completely blocked deoxynivalenol synthesis needed for pathogenicity indicating that the FgNCP also delivers electrons to another CYP (TRI4) located at the ER. The FgCYP we found to be involved in NO-productions is the same or similar to proteins involved in Eukaryote sterol synthesis (CYP51) reducing lanosterol on the way to the final main sterols different for different Eukaryotes. Lanosterol enriched membranes are known to be inhibited in endocytosis and we found that the deletion of the FgCYP producing NO also completely stopped endocytosis. We further tested consequences of these indications of more than one function and suggest the CYP-protein is most likely an FgCYPNO,ERG involved in both NO and ergosterol synthesis and the NCP is involved in NO, trichothecene and ergosterol synthesis, an FgNCPNO,TRI,ERG. The two proteins shown here to be responsible for NO production in F. graminearum are both highly conserved in Eukaryotes from amoeba to human and homologues are likely candidates for the production of NO in many other eukaryotes including mammals. The multiple functions of these proteins can be part of the explanation for the links between chronic inflammation, sterols and blood pressure in human.
Introduction
Organisms rely on NO as signaling and effector molecule for numerous physiological and cellular processes. NO is a diffusible, short lived, reactive free radical that acts as a secondary messenger (Velayutham and Zweier, 2013; Yu et al., 2014). Upon microbial infection, the induced production of NO is a hallmark of the basal defense response also known as innate immunity. Plants and animals produce NO upon the detection of Microbial-Associated Molecular Pattern (MAMP) (Zeidler, 2004; Ulett and Potter, 2011; Velayutham and Zweier, 2013). MAMPs are conserved molecular patterns shed from microbes which are recognized and responded to by host organisms (Reviewed by Newman et al., 2013).
The production of NO is catalyzed by the enzyme Nitric Oxide Synthase (NOS) which converts L-arginine to citrulline and NO through the intermediate N-hydroxy-L-arginine (NOHA) as reviewed by (Stuehr, 1999). The archetypal NOS consist of an N-terminal oxygenase part containing the arginine binding site, a heme and a tetrabiopterin domain; a central calmodulin (CaM) binding site; and the C-terminal reductase part contain FAD, FMN and an NADPH binding site. The enzyme is active when it is in its homodimer form and is regulated by Ca2+ and CaM (Stuehr, 1999; Gorren and Mayer, 2007).
NOSs are thus proteins containing two main parts, a Cytochrome P450 (CYP) heme binding part and a NADP Dependent P450 (NDP) heme-reductase part. To become active they dimerize aided by CaM and Ca2+ and the Heme-reductase part (NDP) transports electrons to the hemes in the heme binding past (CYPs) in the paired protein (Gorren and Mayer, 2007). NO synthesis is initiated with the heme binding and activating of O2 and the reductase domain transfers NADPH-derived electrons to the oxygenase domain. NOSs are subjected to NO-self-regulation since NO binds to the iron in the heme and blocks NO synthesis at high NO concentrations (Abu-Soud et al., 2001; Gorren and Mayer, 2007). NOSs are similar to cytochrome P450s due to the heme group and reductase domain which are present in both enzymes (Gorren and Mayer, 2007).
In animals, NO is generated by the very well characterized mammalian NOSs that has 3 different isoforms. Two of them are constitutively expressed and located in the brain (neuronal NOS; nNOS) and within the endothelial cells (eNOS) regulating vasodilation of artheries. During microbial infection, the inducible NOS (iNOS) which is located within macrophages produce NO and mediate the elimination of bacteria (Bogdan et al., 2000). In humans, the iNOS is part of a structural complex that contains the cationic amino acid transporter (CAT1), arginosuccinate synthetase (ASS), arginosuccinate lyase (ASL), the NOS and the heat shock protein 90 (HSP90). ASL and ASS are enzymes responsible for the production of the substrate arginine that is fed directly to the NOSs. Disruption of the structural integrity of the complex was found to be detrimental towards NO production (Erez et al., 2011).
Although evidences of NO have been reported in fungi and plants, in most cases, their NOS orthologs have still not been found by using sequence homology (Fröhlich and Durner, 2011; Rőszer, 2012; Samalova et al., 2013). In Arabidopsis, a NOS-associated enzyme (AtNOA1) was discovered and found to contribute to the induction of a NO burst in response to the MAMP lipopolysaccharides (LPS) (Guo, 2003; Zeidler, 2004; Guo and Crawford, 2005; Hong et al., 2008; Yu et al., 2014), The AtNOA1 sequence did not bare similarities to the archetypal NOS but instead was very similar to a snail protein that exhibited NOS-like activity (Guo, 2003) and was later found to be a cGTPase and not to have anything to do directly with NO production (Moreau et al., 2008).
In fungi, the genes encoding enzymes responsible for the catalysis of NO have not been identified yet. The Aspergillus species have been reported to contain genes with sequence homology to the mammalian NOS, but the enzyme activity was not tested (Gorren and Mayer, 2007). Studies have shown that yeast NOS-like proteins can be detected in filamentous fungi using antibodies, but the sequence of the detected proteins have not been reported yet, (Kanadia et al., 1998; Domitrovic et al., 2003) nor is there any similarities between the NOS-sequences to the yeast genome sequence (Rőszer, 2012). Nevertheless, NO has been shown to affect various stages of the fungal lifecycle ranging from germination to sporulation (Samalova et al., 2013; Wang and Higgins, 2005; Gong et al., 2007; Prats et al., 2008; Rőszer, 2012).
Nitrite has also been shown to be a substrate for NO-formation in animals and plants, illustrating redundancies for the production of this important radical (Rockel et al., 2002; Jansson et al., 2008; Lozano-Juste and León, 2010). Although the nitrate reduction pathway was shown to be involved in NO production in plants, the same was not observed when nitrate reductase or nitrite reductase or the nitrate and nitrite reductase were deleted in the fungus Magnaporthe oryzae. The deletion mutants still produced NO and showed no signs of altered phenotype (Samalova et al., 2013).
We had selected plant pathogenic fungus Fusarium graminearum strain PH1 (teleomorph Gibberella zeae) to test if fungi possess innate immunity reactions towards bacteria since this fungus has become a model fungus for molecular biology. The fungus is a severe pathogen of many grasses including wheat and causes Fusarium head blight (FHB), crown rot (FCR) and root rot (FRR) (Wang et al., 2015). At the same time, it is a soil borne fungus active in the plant rhizosphere (Wang et al., 2015) where it has to co-exist with a large number of bacteria, neutral, beneficial and detrimental to the fungus (Ipcho et al., 2016). We found that F. graminearum reacted with quick transcriptomic responses (1-4h) to addition of purified bacterial MAMPs by upregulating genes typical for Eukaryotic innate immunity responses (Ipcho et al., 2016). We decided to use our previous study as a starting point to investigate if NO formation is part of the fungal innate immunity response, and if it is, identify the proteins responsible for synthesizing nitric oxide.
Hypotheses
NO is produced in response to purified bacterial MAMPs as part of an innate immunity response (Ipcho et al., 2016).
NO is produced as for normal NOSs from arginine or from NO2 through the Nitrate reduction system.
If 2 is right NO might be produced by a non-conventional NOS where the heme protein and the heme-reductase belong to separate proteins that together produces NO.
We here report a study where we first investigate the similarities between NO production in F. graminearum stimulated by addition of bacterial MAMPs and find that it reacts similar to NOSs in mammals and nitrite additions also stimulates NO production confirming hypothesis 1 and 2. Investigating co-regulation in our large transcriptome dataset from F. graminearum challenged with bacterial MAMPs we found a tentative heme-reductase homologous with the heme-reductase part of rat mNOS and knocked this out and found that it is a F. graminearum-NADPH-Cytochrome P450 heme reductase involved in NO production (FgNCP). We further characterized this protein and found it localizes to endoplasmic reticulum ER and is indeed necessary for NO production. We used a web tool to find all F. graminearum predicted heme-proteins (NCBI, Batch CD-Search) and combined this with our transcriptomics data from bacterial MAMPS challenged F. graminearum to find heme-proteins co-regulated with FgNCP under conditions when NO is formed after bacterial MAMPs challenge. Finally, we used a new webtool, DeepLoc, to find which of the co-regulated genes encodes for a protein predicted to be located to the ER membrane. We found one putative F. graminearum Cytochrome P450 reductase (FgCYP) and knocked this out and localized the protein and could show that it is a FgCYP involved in NO production at the ER. This confirms hypothesis 3. Further analysis suggest FgNCP has 3 separate functions, NO, ergosterol and mycotoxin deoxynivalenol (DON) synthesis while the FgCYP is involved both in NO and ergosterol synthesis. Since both proteins are highly conserved in Eukaryotes including other fungi, mammals, plants and protozoa this could indicate that it is an ancient system for NO-production in response to bacterial challenges that is mixed up with Eukaryote sterol production. Some of the possible consequences of such a mix is discussed and a conceptual model presented in the discussion.
Results and discussion
NO production in response to bacterial MAMPs
NO is known to be elicited in response to bacterial MAMPS in plants and animals. We thus exposed the fungus to various MAMPs. The level of accumulated NO by the fluorescent dye was measured after 10 hours of elicitation. Similar to in mammals full length flagellin FFLG but not the short FLG22 used to elicit NO responses in plants (Zipfel and Felix, 2005) was found to be the most consistent elicitor of NO in the fungus (Fig. 1A) and was used as a reference in the subsequent investigations although the commercial flagellin preparation contained small non-declared amounts of sucrose (Ipcho et al., 2016).

(A) Accumulated increased NO production measured as increase of fluorescence of the NO probe DAF-FM DA during 10h post stimulation by adding different bacterial MAMPs. Water was used as negative control to test if the disturbance by addition triggers NO production. FLG22=the short flagellin peptide plant flagellin receptors reacts to. FFLG=full length flagellin, LOS=lipo-oligosaccharides, PGN=peptidoglycan, EF18= Elongation factor 18. Three replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average of bars with non-overlapping error bars ≪0.05. (B) Accumulated relative increased NO production measured as increase of fluorescence of the NO probe DAF-FM DA during 10h post stimulation by adding full length flagellin (FFLG) as positive control together with different inhibitors. FFLG=Full length flagellin, ZST=ZSTK474 PI3K inhibitor, wortmannin=PI3K inhibitor, bpV(Hopic)=PTEN inhibitor, W7=calmodulin inhibitor, EGTA=Calcium chelator, LNNA=L-NG-nitro-L-arginine NOS inhibitor. Three replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average of bars with non-overlapping error bars ≪0.05. (C) Relative increase in fluorescence of the intracellular calcium probe Fuo4 subtracted the fluorescence in the negative control with only water. Error bars show standard error of means (N=3).
Test if NO production is sensitive to standard NOS inhibitors
In the mammalian system, iNOS has been shown to be regulated by signaling components such as phosphatidylinositol-3-kinases (PI3K), Phosphatase and tensin homolog (PTEN), Calcium influx and Calmodulin (Günzl and Schabbauer, 2008; Ma et al., 2008). It is also well documented that NOSs use arginine as a substrate to generate NO (Mori and Gotoh, 2004). To query if the NO producing enzyme in F. graminearum was governed by similar characteristics as in the mammalian system, a series of signaling inhibitors were applied to elicited fungal cultures. PI3K was inhibited with ZSTK474 and wortmannin, and PTEN was inhibited with BpV(HOpic). Inhibition of both signaling component showed decreased level of NO production. Extra-cellular calcium and the intracellular calmodulin were inhibited with EGTA and W7 respectively. Chelation of calcium ions showed a moderately decreased NO response (Fig. 1B) whereas inhibition of calmodulin completely inhibited the production of NO. The arginine analogue L-NNA, which cannot be metabolized by an iNOS was added to elicited cultures as a competitive inhibitor. The results showed that the accumulation of NO signal was decreased compared to the control indicating that the NOS-system response to bacterial MAMPs in F. graminearum uses arginine as substrate as also do mammalian NOSes in contrast to what seems to be the case for NO production during spore germination in M. oryzae (Samalova et al., 2013).
Test if elicitation with bacterial MAMPs increases intracellular calcium levels
Calmodulin is known to interact with and activate NOS (Lee and Stull, 1998; Li et al., 2003; Ma et al., 2008; Nagpal and Panda, 2015) when Ca2+ levels increase intracellularly. To test if calcium signaling is involved in the MAMPs detection intracellular Ca2+ levels following bacterial MAMPs challenges was investigated and intracellular calcium increases above water controls using the intracellular fluorescence calcium indicator Fluo4 AM that can be loaded into cells. Our results show increasing green fluorescence indicating increasing intracellular Ca2+ concentration triggered by the bacterial MAMPs treatments (Fig. 1C).
Test of standard substrates for NO production by NOSes
The ability of F. graminearum to utilize exogenous chemicals as substrates was tested. Arginine, the established substrate in mammalian systems was added into bacterial MAMPs challenged cultures. Interestingly, the NO production rate increased very fast as a result of arginine addition and then completely stopped (Fig. 2A). To assess the possibility of NO production from nitrite (Fig. 2B) this were exogenously injected into MAMPs challenged fungal cultures. Interestingly, in both experiments with exogenous arginine and nitrite, the amount of NO produced approximately doubled at 4h as compared to 2h. However, contrary to the arginine experiment, NO accumulation continued post spiking. The effects of exogenous nitrite and arginine added in different sequential combinations to test if the effects were additive. The results showed that the addition of nitrite promoted a higher production of NO when arginine was added. This was followed by the typical arrest of NO production post arginine addition. When arginine was added exogenously first, a NO burst was observed followed by complete arrest of NO production. Addition of nitrites following the arrest did not promote further NO production (Fig. 2C and D). In conclusion our results indicate that both arginine and nitrite can be substrates for oxidative or reductive formation of NO (Samalova et al., 2013) but it is only the arginine additions that results in fast cumulative increases in the DAF-FM fluorescence. This indicates that it is mainly the arginine that is responsible for high transient concentrations of NO that can be used for fast signaling. The very fast increases seemed to self-inhibit NO formation from arginine as would be expected if it is formed by a heme protein similar to iNOS in mammals (Abu-Soud et al., 2001) giving further support that arginine is the main substrate for quick NO formation in response to bacterial MAMPs.

(A) Effect on NO production in relation to control of adding exogenous arginine or (B) nitrate on NO production. Both were added to a final concentration of 5mM. Rate of NO production increased very quickly after addition and in the case of arginine the fast increase stopped accumulation. This inhibition s less the nitrite additions. (C) Sequential spiking nitrite and arginine to flagellin induced fungal cultures. First spiking by nitrite followed by a second nitrite spiking or an arginine spiking. (D) First spiking arginine followed by arginine spiking or nitrite spiking. Arginine spiking limits total NO accumulation but this does not happen with nitrite spiking. Error bars are standard error of mean (N=3).
Finding NOS candidate genes in F. graminearum
Our experiments indicated that the NO generating system in F. graminearum must be very similar to the mammalian system (Gorren and Mayer, 2007). Yet there are no classic NOS candidate genes in fungi (Cánovas et al., 2016). The NOS-domain heme containing domain PF02898.15 is what is normally used as a signature for finding nitric oxide synthetase enzymes (Crane, 1998). Since no normal NOS containing a PF02898.15 domain is present in any F. graminearum protein there must be a different organization of domains. We know from the literature that NOSes need to dimerize to produce NO and this dimerization is Ca2+ dependent. It also means that the electron transport goes from the heme-reductase in one protein to the heme in the other (Gorren and Mayer, 2007). Thus, there could be another way to bring together a NOS-like heme domain containing protein and NOS like heme reductase containing all the other domains of a NOS. The heme domain in a NOS has similarities to a CYtochrome P450 heme (CYP) and there are many of that kind of proteins in F. graminearum. We thus turned our interest to find a possible NADPH-Cytochrome P450 reductase (NCP) containing all the other domains necessary for a NOS-NCP involved in NO-production (Gorren and Mayer, 2007). To indicate which pathway an enzyme is involved in we from now indicate that in subscript after the enzyme abbreviation. The enzyme we are thus looking for is a F. graminearum NCP involved in NO production, a FgNCPNO. (from now we write gene and protein abbreviations in bold where they are introduced in the text) We used the amino acid sequence of the C terminal non-heme part of the Mouse iNOS (NP_035057.1) as a bait and made a BLAST search against the . graminearum proteome. We only found 2 candidate FgNCPNO (FGSG_ FGSG_08413 and FGSG_09786) with high similarity, containing all necessary domains (Gorren and Mayer, 2007) (Supplementary File SF1). Both genes contain regions that indicate they are NADPH dependent Cytochrome P450 heme reductases (NCPs) that could be involved in transporting electrons from NADPH to a Heme protein that produces the NO from reaction between arginine and O2. They could be the heme-reductases but then they need to transport the electron to an unidentified heme containing protein.
To prioritize which gene to study in detail we decided to use our previous transcriptome data (Supplementary Data1) to test if any of the 2 candidate genes are co-regulated with a putative F. graminearum Nitric Oxide Dioxygenase (FgNOD) needed to protect a NO producing cell from the NO itself produces (Gardner, 2012). We calculated the correlation coefficients (Pearson) for transcription of the putative FgNCPNO candidates and the two putative FgNODs for all F. graminearum gene in 113 transcriptomes from data from experiments where F. graminearum has been exposed to different bacterial MAMPs (see methods) and sorted the genes in order of highest to lowest correlation (Supplementary File SF2). We found that only FGSG_09786 was highly correlated with the two putative NODs, FgNOD1 and FgNOD2 (FGSG_00765 and FGSG_04458). Both are similar to NO dioxygenases in Candida albicans that are experimentally verified (Missall et al., 2004) and also to a NOD annotated for Fusarium sporothrichiodes (Supplementary File SF2) FgNOD1 is almost identical to the F. sporothricioides protein (100% cover E-value 0 and identity 98%) and FgNOD2 is similar to FgNOD1 (90% cover, E value 2e-117 and identity 45.61%). These correlations indicate that only FGSG_09786 is a putative FgNCPNO (Fig. 3). The other putative FgNCPNO (FGSG_04458) was not highly expressed and is thus not encoding a likely FgNCPNO since it even showed a slight negative correlation with the two putative FgNODs (Fig. 3). FgNOD1 is also more closely co-regulated with FgNCPNO indicating FgNOD1 might be the main NOD responsible for limiting NO stress assuming FgNCPNO takes part in NO generating activities.
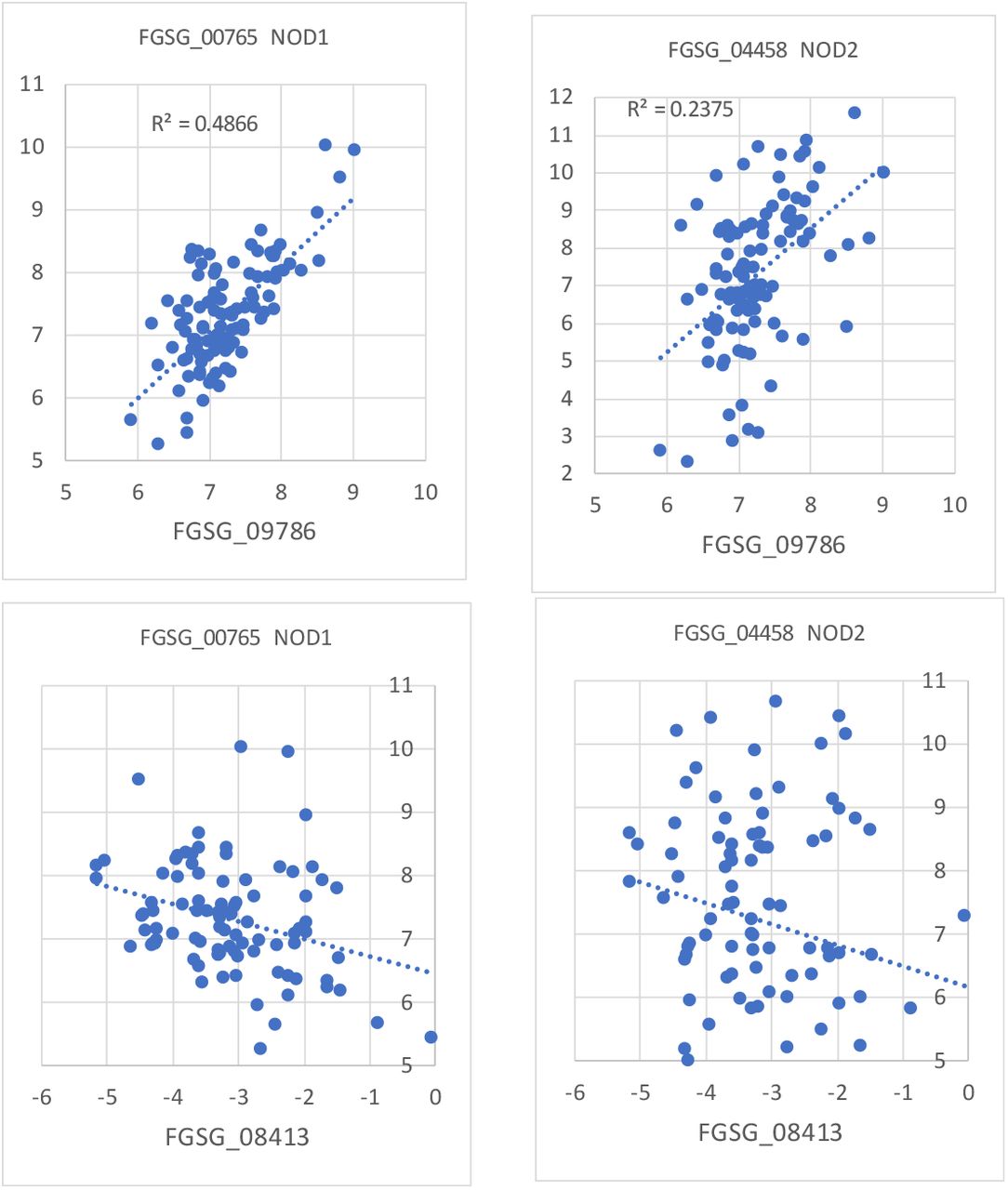
Correlations of expression between the two putative FgNCPNO and the two FgNODs in the transcriptome dataset for fungal interactions with bacterial MAMPs. For both axes Log2 values for the relative expression are used and scales for both axes are set the same to aid visual comparisons.
Deletions of putative FgNCPNO,FgNOD1 and FgNOD2 lowered NO production rate in response to bacterial MAMPs
NO production of mutants
Deletion and GFP complementation strains for FgNCPNO, FgNOD1 and FgNOD2 were constructed (see methods) and tested for NO production (Fig. S1). We found that the ΔFgNCPNO had radically lower rate of accumulation of the DAF-FM fluorescent NO reporter stain than the WT indicating a reduced NO production rate (Fig. 4A). When focusing on the difference between controls and induced responses using bacterial free washings from washed bacteria (see methods) the NO response to the MAMPs was markedly diminished (Fig. 4B). Of the two NOD deletions only ΔFgNOD1 showed a tendency to accumulate NO-signals faster than WT and could therefore be the main counterpart to the NO generating system in F. graminearum.

(A) Relative increase rate in fluorescence units of the NO probe DAF-FM per minute in water controls hyphae treated with treated with bacterial MAMPs (sterile bacterial washings, see methods). (B) Same data but showing the increased rate compared to controls. FGSG_09786=ΔFgNCPNO, FGSG_00765= ΔFgNOD1 and FGSG_04458=ΔFgNOD2. Four replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average of bars with non-overlapping error bars ≪0.05. The experiment was repeated twice.
Subcellular localization of FgNCPNO, FgNOD1 and FgNOD2 and NO production
FgNCPNO-GFP localizes to ER like structures in spores and hyphae (Fig. S2 I) that could also be shown to co-localize with an ER-tracker (Fig. 5A) and an mCherry labelled ER-marker protein (Fig. 5B) while FgNOD1-GFP localizes to cytoplasm in germinating conidia while FgNOD2-GFP localizes to cytoplasm and dots (Fig S2 II and III). Since the FgNCPNO-GFP localizes to ER we tested if NO production also takes place preferentially in the ER to further check the likelihood that it is involved in NO production. To test this, we incubated an mCherry labelled ER reporter strain with a NO-sensitive probe for as short time as possible. Fluorescein diacetate (FDA) is a non-fluorescent hydrophobic viability probe that loads through membranes easily and in the cytosol the two acetate groups are cleaved off by always present esterases leaving relatively hydrophilic fluorescein that is strongly fluorescent accumulating in the cytosol if the membrane is intact. The membranes are however slightly permeable to fluorescein; thus, the external medium will be colored within minutes as are basically also all intracellular compartments. DAF-FM is a non-fluorescent fluorescein derivative that only becomes strongly fluorescent after reacting with NO. DAF-FM is loaded in the diacetate form DAF-FM DA in the same way as FDA. Thus, we wanted to load the fungus with DAF-FM DA as fast as possible and see where the first green signal appears. Thus, we wanted to load the fungus as fast as possible and see where the first green signal appears. It turned out that we did not need to use MAMPs to elicit NO as the background NO formation rate was nearly too fast for us to get good images without green signals leaking out from the fungus. Good signals appeared already minutes after addition of the NO probe. As can be seen NO is produced in ER but also in non-ER dots (Fig. 5C).

(A) Cellular localization of FgNCPNO (FGSG_09786) in hyphae of F. graminearum in relation to ER-tracker and (B) mCherry labelled ER Marker protein (see methods). (C) Localization of NO signal (DAF-FM) in relation to ER Marker protein (ER).
We can thus conclude that the heme reductase FgNCPNO is involved in NO-production and the NO production takes place in the ER where the heme reductase is located but also in dots that might be peroxisomes where NO3 reduction is known to take place. FgNOD1 is also the FgNOD that could be the main responsible for limiting NO in nucleus and cytoplasm and FgNOD2 might be active in the dots (maybe peroxisomes) (Fig S2 II and III). Our transcriptome data also supported that FgNOD1 is the main NOD co-regulated with FgNCPNO (Fig. 3) since it is most co-regulated.
Phenotypic effects of deletion of NO generating and NO protecting proteins
Phenotype analysis showed that deletion of the FgNCPNO had no large effect on growth rate (Fig. 6A and Fig. S3A) but conidia formation was inhibited (Fig. 6B) and there was no formation of sexual spores (Fig. S3B). Pathogenicity was also reduced (Fig. 6C and Fig. S4) and this is probably mainly caused by the abolishment of DON (Fig. 6D) production since DON is needed for full pathogenicity (Proctor et al., 1995). NO signaling is known to be involved in conidial production (Ding et al., 2020) so the lowered conidia production could be a result of this lack of NO (Fig. 6B). There was otherwise little or no effect of the FgNCPNO mutation on stress phenotypes indicating cell wall and membrane changes except the ΔFgNCPNO was a bit more sensitive to Na+ and K+ ionic stresses (data not shown). The NOD deletions had very little effect growth phenotypes and on infection phenotypes which could indicate that protection from NO is not very important for pathogenicity or normal growth (Fig. 6, Fig. S6 and S7).

Inhibition effect of deletions and complementations (C) compared with intact PH1 genes. Y=(PH1/Mutant or complement). FGSG_09786=ΔFgNCPNO, FGSG_00765= ΔFgNOD1 and FGSG_04458=ΔFgNOD2. (A) Colony diameter on CM agar. (B) Inhibition of conidiation. (C) Disease inhibition. (D) Inhibition of mycotoxin formation DON. Three replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average for bars with non-overlapping error bars ≪0.05.
Finding a putative heme protein that works together with FgNCPNO to produce NO
Since we now have found the NO-producing heme-reductase (FgNCPNO) and know where it is located, we turned our interest towards the heme protein that should be the one actually making the NO using electrons delivered from the FgNCPNO. The expected heme binding protein should have the following characteristics except being a predicted heme binding. 1. It should be co-regulated with the FgNCPNO at least in transcriptome data when fungus is exposed to bacterial MAMPs. 2. It should be predicted to be located at the ER membrane. We found that we could not trust PSORTII for localization prediction since it wrongly localizes the ER-localized FgNCPNO. (Table S5).
There was one heme protein on top of our list for genes co-regulated with FgNCPNO when exposed to bacterial MAMPs, FGSG_07925 (Supplementary File SF5). We knocked out and complemented it with FGSG_07925::GFP but this protein did not localize to the ER (Fig. S6) or showed any notable phenotype except a decrease in conidiation (Fig. S7). We later found that the localization prediction server DeepLoc 1.0 (Almagro Armenteros et al., 2017) correctly localized FgNCPNO to ER as we had found experimentally. Using DeepLoc and our transcriptomic MAMPs data we found that only FGSG_01000 was predicted to make an ER localized heme containing protein that is also co expressed with FgNCPNO and the expression is in a close to 1/1 ratio of max expression with the max expression of FgNCPNO (Table S8 and Supplementary File SF5) making it a better candidate for a F. graminearum CYtochrome P450 involved in NO production, a FgCYPNO
Deletion of putative FgCYPNO: Localization, NO production rate and effect on phenotype
We decided to knock out FgCYPNO (FGSG_01000) and test for NO-production. We successfully knocked it out (Fig S9) and could show a significantly decreased NO production for ΔFgCYPNO to the same level as for the ΔFgNCPNO (Fig. 7A) using our newly developed technique employing confocal microscopy to get cleaner data and avoid fluorescent specks not related to the NO-production (See methods). As predicted the FgCYPNO localizes to ER-like structures (Fig.7 B). We can conclude that the putative FgCYPNO is ER located and responsible for NO production.

(A) NO production measured as LOG2 (NO signal/biomass signal) using confocal microscopy of large areas with many hyphae in WT=PH1, FGSG_01000= ΔFgCYPNO FGSG_09786= ΔFgNCPNO deletions and WT for NO formation rate. Three replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average of bars with non-overlapping error bars ≪0.05. (B) FgCYPNO-GFP (FGSG_01000-GFP) localizes to ER like structures. Size bar 10μm.
Phenotype analysis of the ΔFgCYPNO showed some effect on growth (Fig. 8A) but more effect on stress tolerance (Fig. 8B and Fig. S10A) and on pathogenicity (Fig. S10 B and C) and decrease in DON production (Fig. 8C). However, this decrease in DON production was notable but it was not completely absent as it was for the ΔFgNCPNO. The stress phenotypes indicate also that FgCYPNO plays a role in cell wall and plasma membrane integrity.

(A) Phenotypic effects of stress treatments by addition of stressors to CM-medium. SDS=0.01%, KCL=1M, H2O2=5 mM, Congo red CR=1 mg/ml, CFW=200μl/ml, NaCl=1M. Three replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average of bars with non-overlapping error bars ≪0.05. (B) Effect of FGSG_01000= ΔFgCYPNO on DON mycotoxin production. Three replicates were used and error bars shows 95% confidence intervals for the mean values. Thus, the P value for null hypothesis for the same average of bars with non-overlapping error bars ≪0.05.
Phylogenetic analysis of the two proteins involved in NO production
A phylogenetic analysis showed a high degree of conservation of both the NCPNO and the CYPNO in a wide range of Eukaryotes (Fig. 9). Most of the putative CYPNO-proteins were to our surprise annotated as CYP51 (ERG11 yeast) involved in sterol synthesis while the NCPNO-proteins were annotated as NADP-dependent heme (P450) reductases as expected (Supplementary File SF5 and 6). The domain organization for all these proteins were almost identical to the domain organization for mouse iNOS but separated on two proteins with predicted N-terminal hydrophobic attachment to the ER-membrane (Fig. 10 and Supplementary File SF5 and 6). The FgCYPNO contain a CaM-binding domain that could indicate its activity is positively regulated by calcium as also our result indicates (Fig. 2 and 3). FgNCPNO seemed to be involved in DON production necessary for full pathogenicity this production might also be Ca regulated as has also be shown (Kim et al., 2018). We find that the FgNCPNO often also contain a signal for a putative CaM-binding region using the CaMELS webtool (Abbasi et al., 2017) that could explain why toxin production (Kim et al., 2018) and pathogenicity is dependent on Ca.

Phylogenetic analysis of the two proteins (FGSG_09786 and FGSG_01000) involved in NO production. (A) All are annotated as NADPH-cytochrome-P450 oxidoreductases (NCPs) in the database and some are known to be involved in sterol synthesis. (B) All are CYPs in the database and, except FgTRI4 involved in DON synthesis, are all annotated as lanosterol 14-alpha-demethylases (ERG11 in yeast) involved in converting lanosterol to the next step in sterol synthesis. The FGSG_04092 and FGSG_11024 only becomes activated late during infection where Tri4 is highly activated in planta according to transcriptome data (not shown). For full analysis and protein sequences see Supplementary file SF6 and SF7.

Domain structure of mouse iNOS (bottom) compared to the best hits for genes homologues to the CYP-part of the iNOS left of the hydrophobic helix (TM-marked) region and the heme-reductase part (NCP) to the right according to HMMER. The NCP Erg11 involved in ergosterol synthesis is the protein most like the FgCYPNO (FGSG_01000) and a P450 mouse reductase (NCP) is similar to the FgNCPNO involved in NO production. All these pairs except the iNOses have N-terminal TM regions predicted to be associated and inserted in the ER membrane by DeepLoc. All CYP like proteins are predicted to be strongly Cam binding and a few of the NCPs to the right are also predicted to be strongly CaM-binding. The rest of the NCPs are predicted to have cam-binding sites but are predicted to themselves be Cam-binding to a lesser degree (under the confident threshold level 1 (Abbasi et al., 2017) but might still have Cam binding possibility. For full analysis and protein sequences see Supplementary file SF5.
Can FgCYPNO also be involved in ergosterol biosynthesis?
The FgCYPNO is well conserved in fungi but mainly implicated being a cytochrome P450 (CYP51) having a role in ergosterol synthesis converting lanosterol to the next step in the sterols synthesis on the way to ergosterol in fungi and cholesterol in animals and other sterols in other eukaryotes (Lepesheva and Waterman, 2007). The final products, ergosterol or cholesterol, are necessary for endocytosis. Lack of the final product ergosterol in the membrane and instead accumulation of lanosterol should therefore inhibit endocytosis (Pichler and Riezman, 2004) as has been shown previously (Kim et al., 2017). Thus, if the FgCYPNO is also involved in ergosterol synthesis, endocytosis should be stopped in the ΔFgCYPNO strain (Suchodolski et al., 2019). In the ΔFgCYPNO strain endocytosis was shut off as FM4-64 was not taken up and quickly internalized (Fig. 11A) indicating the deleted heme protein (FgCYPNO) is likely involved in sterol production and actually a FgCYPNO,ERG.

(A) Staining endocytosis uptake using the endocytosis probe FM4-64 for 1h. Wt =PH1 takes up the stain by endocytosis and the stain accumulates at the ER around the nucleus. ΔFgCYPNO,ERG (FGSG_01000) is completely blocked for endocytosis and the stain stays at the plasma membrane in a normally looking hyphae and FGSG ΔFgNCPNO,TRI,ERG (FGSG_09786) is not severely blocked in endocytosis. DIC image to show that FGSG_01000 appears normal in intracellular structures. Size bars 10μm. (B-E) Co-regulation and expression during the course of infection of the two NO-producing genes compared with the main DON-producing genes in a large transcriptome dataset with transcription during plant infection from a previous study (Zhang et al., 2019). Expression axes are log2 expression and each dot represents data for the genes from one transcriptome. (B) Correlation between DON producing FgCYPTRI (FGSG_03535, Tri4) and FgNCPNO,TRI,ERG (FGSG_09786)(left). Expression of FgCYPTRI during the course of infection 0-144 hours post infection (HPI) (middle). Correlation between DON producing FgCYPTRI (FGSG_03535, Tri4) and FgCYPNO,TRI,ERG (FGSG_09786)(right). (C) Expression of FgCYPNO,ERG (FgNOD1 and FgNOD2 0-144 HPI showing NO upregulation of the genes linked to NO-production. (D-E) Regulation of FgNCPNO,TRI,ERG (FGSG_09786) compared to FgNCPERG (FGSG_08413) and FgCYPTRI (FGSG_03535) and FgCYPNO,ERG (FGSG_01000) during the course of infection. (D) Expression of FgNCPNO,TRI,ERG increases during the course of infection (Left) as do FgCYPTRI right) while FgNCPERG slightly decreases (left) and FgCYPNO,ERG does not change (right). (E) Negative correlation between FgNCPNO,TRI and FgNCPERG. (F) Test if FgCYPNO,ERG (FGSG_01000) and FgNCPERG (FGSG_08413) are compensatory upregulated to maybe aid ergosterol synthesis in the ΔFgNCPNO,TRI,ERG (FGSG_09786). Y axis represent the ratio of expression of the respective genes in the FgNCPNO,TRI,ERG (FGSG_09786) mutant compared to the WT (PH1). Both genes potentially involved in ergosterol synthesis are upregulated and that could explain why ergosterol dependent endocytosis were normal in the ΔFgNCPNO,TRI,ERG. Three replicates were used and error bars shows 95% confidence intervals for the mean values.
Regulation of the genes involved in ergosterol, NO and DON production in published in planta data during the course of infection
We have previously downloaded publicly available transcriptomic in planta data from plant infection plant infection experiments (Zhang et al., 2019). Especially the first dataset from a time course of infection is interesting since deletion of FgNCPNO completely stopped DON production (Fig. 6D). From the literature it is known that FGSG_03535 situated in the co-regulated trichothecene cluster (the TRI cluster) is a P450 heme protein named TRI4 (Tokai et al., 2007). It localizes to the ER (Kistler and Broz, 2015) that consequently here should be called a FgCYPTRI since it is a CYP necessary for making a reduction step in the DON synthesis. If FgNCPNO is ALSO a reductase delivering electrons to FgCYPTRI it should be co-regulated with FgCYPTRI in the in-planta data and also be upregulated during the course of infection (Hours Post Infection, HPI). In the transcriptome dataset from in planta the FgCYPTRI is strongly co-regulated with the FgNCPNO during infection and FgCYPTRI is upregulated after 24HPI. The NO producing FgCYPNO is however not co-regulated with FgNCPNO during infection (Fig. 11B) indicating that FgNCPNO should be named FgNCPNO,TRI since it seems involved in reducing heme proteins needed for both NO formation as well as DON synthesis.
Since there are only two NCPs with the right structure (FGSG_09786 and FGSG_08413) (Fig. 3) and we could not find any or a very small effect on endocytosis when deleting FgNCPNO,TRI (Fig. 11A) the other heme reductase could be involved in ergosterol production also delivering electrons to FgCYPNO,ERG and consequently both heme reductases could complement each other in ergosterol synthesis. Therefore, the remaining heme reductase with the relevant domain organization could have a function as a reductase not involved in DON or NO production but only in ergosterol production, a FgNCPERG, and the FgNCPNO,TRI would then be a reductase involved in ergosterol production as well, a FgNCPNO,TRI,ERG. If this is the case, and since there is no increased NO formed during the first 144HPI indicated by the absence of upregulation of the FgCYPNO,ERG, FgNOD1 and FgNOD2 (Fig. 11C), there should be an inverse relationship between FgNCPNO,TRI,ERG and FgNCPERG expression during the course of infection since the FgNCPNO,TRI,ERG get increasingly occupied with the FgCYPTRI for producing DON. This is indeed the case (Fig. 11D-E) pointing to that both these proteins are involved in ergosterol synthesis feeding FgCYPNO,ERG with electrons.
A way to directly test this conclusion is to investigate FgCYPNO,ERG and FgNCPERG expression in WT (PH1) and compare with expression of the same genes in the ΔFgNCPNO,TRI,ERG strain that does not make DON but is capable of endocytosis (Fig. 11A) that needs ergosterol to work normally. We made this comparison using RT-qPCR and can show that FgCYPNO,ERG is strongly and significantly upregulated and FgNCPERG is slightly but not significantly upregulated in the ΔFgNCPNO,TRI,ERG compared to the WT(PH1) indicating that these genes are compensatory regulated in the mutant (Fig. 11F) explaining why the ΔFgNCPNO,TRI,ERG still show endocytosis (Fig. 11A).
Conclusion
Similarities and differences to NOS in mammals
There are obvious similarities between the ER located NO generating and lanosterol reducing system (Fig. 9 and 10) and mammalian NOS. For NOS to be activated a calcium dependent dimerization is needed (Gorren and Mayer, 2007). NO production in F. graminearum is stimulated by MAMPs that also leads to increases in concentration of cytosolic Ca2+. Since the ER located proteins involved in NO production in F. graminearum are predicted CaM interacting (Fig. 10) and Ca2+ increase intracellularly induced by bacterial MAMPs (Fig. 1C) as do NO production (Fig. 1A-B,2) CaM most likely bring these proteins together on the ER membrane activating NO production (Fig. 12A). The ER-located CYPNO,ERG and NCPNO, ERG,TRI reducing system seems to be very conserved from Protista to Mammals (Fig. 9-10). Electron transfer proteins where electrons are transferred between domains in the same proteins seem to be made efficient by quantum tunneling of electrons between precisely spaced domains (Stuchebrukhov, 2010; Fereiro et al., 2018). This need for tunneling should impose serious constrains on evolution of essential proteins and can be an explanation for the very high conservation between the involved proteins and why there evolves double and triple functions for the involved proteins that then can act in different combinations with other proteins.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Model for the arrangement of domains in the ER localized CYPNO and NCPNO implied in this study compared with the localization of domains in a NOS-dimer (Gorren and Mayer, 2007). (B) Conceptual model for the arrangements and re-arrangements of the CYPs and NCPs at the ER membrane needed for NO, ergosterol and DON production. The main proteins of this study FgCYPNO,ERG and FgNCPNO,TRI,ERG seem to have 2 respective 3 functions in F. graminearum. NO binds to FgCYPNO,ERG heme and self inhibits NO and ergosterol formation at Lanosterol known to reduce endocytosis as well as FgCYPTRI since DON formation seem to be shut off by NO-donors (Ding et al., 2020).
Plant pathogenicity and NO generation
It has recently been shown that NO is formed in F. graminearum as response to-plant molecules pre plant contacts in the rhizosphere. A plant sensing receptor involved in triggering the NO downstream responses resulting in NO-formation and further downstream responses have been identified (Ding et al., 2020). Our results indicate that fungal transcripts for the FgCYPNO,ERG needed for NO production appears to decrease when in planta which would meet the challenge of not producing NO inside a plant since that can activate plant defenses (Samalova et al., 2013; Ding et al., 2020). Instead, in planta the electron transfer from the FgNCPNO,TRI,ERG now takes part in DON production partly using the NO generating enzymes (see above). DON is an inhibitor of translation (Diamond et al., 2013) and the innate immune responses in Eukaryotes are dependent on quick upregulation of defense protein (Rauscher and Ignatova, 2015). A shift from NO to DON production in planta to be able to attenuate eventually turned on plant defenses could be an evolutionary adaptation of F. graminearum. In the native north American grasses F. graminearum seems to have evolved to colonize as an endophyte it does not trigger disease with high DON production (Lofgren et al., 2018). A production of large amounts of NO in planta would trigger the SA defenses and recruit PR proteins as would exposure of ergosterol. Limiting plant access to ergosterol that is a known fungal MAMP that plants reacts to should also avoid trigging plant defenses (Granado et al., 1995; Klemptner et al., 2014). The changed roles of the proteins in planta could thus be an evolutionary adaptation to an endophytic lifestyle where plant defenses stressing the fungus and DON responses balances the interaction with the plant allowing the fungus to grow inside the plant without plant disease development. The higher levels of DON production produced in wheat and other Eurasian grasses might be caused by the fungus being mistaken for a pathogen by these grasses making it a de facto pathogen (Lofgren et al., 2018) as a response to the stresses imposed by the pathogen defense system in wheat.
Endocytosis and NO
Heme interaction with O2 in the oxidative formation of NO from arginine is inhibited at high NO concentrations since NO binds heme better than O2 and blocks the NO production (Abu-Soud et al., 2001), thus iNOSes are self-inhibited. Since the heme in the FgCYPNO,ERG is also used for reducing lanosterol in the ergosterols synthesis NO should also halt ergosterol synthesis (Park et al., 2017) at lanosterol inhibiting endocytosis (Martinez et al., 1996; Pichler and Riezman, 2004; Kim et al., 2017). Interestingly, a complete stop in endocytosis was previously observed in Rhizoctonia solani hyphae confronted with a bacterial biological control strain of Burkholderia vietnamiensis (Cuong, 2010; Cuong et al., 2011; Nicolaisen et al., 2018) while nitric oxide was produced (Cuong, 2010). It was those observations that inspired this study and the study of fungal innate immunity (Ipcho et al., 2016) in F. graminearum with considerable growth in the plant rhizosphere (Wang et al., 2015) and an effective transformation system for knockout studies.
A shutting off of endocytosis due to high NO interacting with heme triggered by bacterial challenges is likely an ancient system protecting primitive eukaryotes from pathogenic bacteria utilizing the endocytosis mechanism to gain entry the interior of the cells (Veiga and Cossart, 2005).
LDL is in mammals taken up by endocytosis (Herbert and Erridge, 2018) and is dependent on CYP51 function (Tamboli et al., 2008). Endocytosis is known to be downregulated by NO (Martinez et al., 1996). If the heme containing CYP51 (homologue to FgCYPNO,ERG) takes part in cholesterol synthesis is inhibited by the NO it may produce due to inflammation this could possibly also explain inflammation induced decreased LDL uptake and resulting health problems linked to chronic inflammation (Herbert and Erridge, 2018).
Conclusion and overall conceptual model for what happens at the ER
From our results it appears that the evolutionary ancient but very conserved sterol synthesis system in Eukaryotic cells is also involved in NO production. In F. graminearum the same system seems to have a role in producing the mycotoxin deoxynivalenol that inhibits translation in the plant host cell. In vitro the fungus makes ergosterol, if severely challenged with bacteria NO forms likely stopping ergosterol synthesis at lanosterol shutting off endocytosis preparing the fungus for bacterial defenses. In planta if attacked by plant defenses the system seem to shift to production of DON that inhibits plant translation and attenuates plant defenses (Fig. 12.B). If the cholesterol synthesis system in mammals also generates NO when receiving inflammation signals this can potentially cause the known effects on LDL-metabolism and blood pressure linked to chronic inflammation (Herbert and Erridge, 2018).
Materials and Methods
Maintenance and growth of Fusarium graminearum for plate-reader assays
The wild type strain of Gibberella zeae ((Schwein) Petch) (anamorph Fusarium graminearum (Schwabe) PH1 (NRRL31084) was obtained from the Agricultural Research Service Culture Collection, National Center for Agricultural Utilization Research, Peoria, IL, USA. The wild type and mutant strains were stored as spores in 10% glycerol at −70°C. Cultures were maintained on minimal Defined Fusarium Medium (DFM) at 21oC, in the dark and shaking at 150 rpm if necessary. To prepare mycelium for DNA extraction the strains were cultured in YPG liquid medium (Yoder and Christianson, 1998). Spore production was induced by fungal growth in liquid RA medium (succinic acid, 50 g L-1; sodium nitrate, 12.1 g L-1; glucose, 1 g L-1 and 20 mL L-1 50X Vogels salts without nitrogen or carbon source) for 3 days. The spore suspension was filtered through Miracloth; washed and centrifuged twice at 5000 × g for 15 min.
NO elicitation from F. graminearum using purified MAMPs in a plater reader
Production of NO was measured using a 96 well plate assay with a variety of potential elicitors including MAMPs. In each well, 2000 spores were cultured overnight in 100 μl of DFM (diluted 1:50) at 21°C in an incubator without light. The growth medium was removed from the overnight culture and sterile Milli-Q water was added for an overnight equilibration. The water was removed from the equilibrated culture and replaced by 100μl elicitor, supplemented with 50ng 4-amino-5-methylamino-2,7-difluorofluorescein diacetate (DAF-FM DA) (D23844; Invitrogen, CA, USA) to detect NO production. The elicitors used were 0.01% w/v glucose; 0.01% w/v sucrose; 0.01% w/v glutamate; 0.01% w/v arginine; 0.01% w/v potassium nitrate; 0.01% w/v potassium nitrite; sterile milliQ water; sterile Copenhagen tap water; 100 ng mL-1 full length flagellin (FFLG) (containing 0.01% sucrose; tlrl-pstfla; Invivogen, CA, USA); 50 μg mL-1 lipooligosaccharides (LOS; LPS without the O-antigen), (Silipo et al., 2005, 2012); 50 μg mL-1 peptidoglycan (PGN) (Erbs et al., 2008); 100 nM flagellin 22 (Peptron Inc., South Korea); 100 nM elongation factor 18 (Peptron Inc., South Korea). We chose to use the 22 amino acid peptide from flagellin (FLG22) and the 18 amino acid from elongation factor Tu (EF-Tu; EF18), because they are known to be the epitopes that elicit innate immunity in plants (Newman et al., 2013). Fluorescence per well was measured a 4×4 matrix (1400 gain) per well using a FLUOstar OPTIMA plate reader (BMG Labtech, Ortenberg, Germany) with emission filters at 485nm and excitation filter at 520nm. Measurements were taken every 30 minutes for the duration of the experiment. The basal fluorescence reading (T0) at the start of the experiment was used as the control, unless stated otherwise. The accumulation of NO as histograms is reported with readings at 10hr (T10) post elicitation or as graphs of accumulated fluorescence from produced NO. FFLG containing 0.01% sucrose was found to be the most consistent inducer of NO and was used as a control in all the subsequent experiments. It was difficult to get a between experiment reproducible mycelial density at the bottom of the microtiter wells. To account for this biological variation between various experiments and to ease comparison, the readings from other treatments were scaled to the FFLG reading.
NOS inhibition assays
The assays were performed as described above but prior to elicitation, the culture were exposed to the inhibitors for 2 hours [10mM L-NNA, NG-nitro-L-Arginine;L-NG-Nitroarginine (80220, Cayman Chemicals, MI, USA), 100μM W7, N-(6-Aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride (A3281, Sigma, MO, USA); 500nM ZSTK474 (S1072, Selleckchem, TX, USA); 100nM Wortmannin (W1628, Sigma, MO, USA); 25nM BpV(HOpic) (203701, Calbiochem-Millipore, MA, USA)] and elicited with flagellin (containing 0.01% sucrose). Calcium ions in the flagellin solution were chelated with EGTA (E3889, Sigma, MO, USA) prior to elicitation. The level of NO accumulated after 10 hours post elicitation was compared between the treated cultures and untreated culture.
NOS spike assays
The fungus was cultured and elicited with flagellin (FFLG) as described above. Post elicitation, the chemical of choice [Arginine (A8094); Potassium Nitrate (P6030); Potassium Nitrite (60417) from Sigma] was added to a final concentration of 5mM per well. The increase or decrease in the production of NO was monitored. For sequential spiking assays with arginine and potassium nitrite, the first substrate was added at 4 hours and the second substrate 2 hours later, (i.e. at 6 hours).
Procedure to find a putative FgNCPNO
This procedure is described in detail (Supplementary File SF1) and is here outlined as follows. A mouse iNOS was looked up at NCBI. Using HMMER the domains of the protein was identified and the NCP-part of the protein was used for a protein blast (NCBI) against the F. graminearum PH1 predicted proteins. Two good hits (FGSG_09786 and FGSG_08413) were found for putative NCPNOs. If any of these produce NO the NO producing one is likely co-regulated with a nitric oxide dioxygenase (NOD) to protect against too much NO formed. Two putative NODs were found in F. graminearum PH1 (Supplementary File SF2). To get an indication which of the two putative NCPNOs are most likely a NCPNO a transcriptome dataset for F. graminearum with full transcriptomes obtained for many different conditions of bacterial MAMPs exposure was used to investigated for co-regulation with the identified putative NODs. This dataset was previously partly published (Ipcho et al., 2016) and is now available in full (Supplementary Data1 https://figshare.com/s/4a02adb12cb48dba6d7a (reserved DOI: 10.6084/m9.figshare.12361823)). FGSG_09786 but not FGSG_08413 was co-regulated with both NODs (Fig.3 and Supplementary File SF3) and is the most likely the FgNCPNO taking part in NO production.
Procedure to find a putative FgCYPNO
When the NCP responsible for NO production (FGSG_09786) had been knocked out, found to be responsible for NO production (a NCPNO) and to be localized to the ER it was time to look for an heme-containing FgCYPNO that can work together with NCPNO at the ER membrane. The genome of F. graminearum PH1 contain numerous genes annotated as coding for heme binding proteins. To get a more complete list we searched the whole genome for heme domain containing proteins using the Batch Conserved Domain search tool to identify all occurrence of putative domains (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi). The result of this search (Supplementary Data2 https://figshare.com/s/a853e4ff951355eb0228 (reserved DOI: 10.6084/m9.figshare.12386501)) was then searched for proteins predicted to be heme binding and these proteins were listed. All heme proteins were then sorted after their co-regulation with the FgNCPNO (FGSG_09786) in the transcriptome data set for F. graminearum exposed to bacterial MAMPs (Supplementary File SF4). FGSG_07925 was found to be most correlated with FgNCPNO but was not predicted to localize to ER or have any phenotype that could be expected for a true FgCYPNO (Fig. S6 and S7). The ER localized FgNCPNO was not correctly predicted by PSORTII (Table S5) but correctly predicted to localize to the ER membrane by DeepLoc. Thus, we used DeepLoc to predict the protein localization of the most correlated proteins predicted to be heme-binding (Supplementary File SF4) and found that FGSG_01000 is the most likely FgCYPNO. This procedure is described in more detail in the supplementary file (Supplementary File SF5).
Knockout and complementation of FgNCPNO (FGSG_09786) and FgCYPNO (FGSG_01000)
To replace the FgNCPNO (FGSG_09786) gene, a 932-bp fragment upstream of FGSG_09786 and a 1176-bp fragment downstream of FGSG_09786 were amplified with specific primers (Table S11). FGSG_09786 replacement constructs were finally generated by a split-marker approach (Yu et al., 2004). Subsequently, the resulting constructs were transformed into protoplasts of the wild-type strain PH-1. Hygromycin-resistant transformants were screened by PCR with two pairs of primers (Table S11). Deletion of FGSG_09786 was further verified by Southern blot with the Digoxigenin High Prime DNA Labeling and Detection Starter Kit I (Roche, Mannheim, Germany). Using a similar strategy, FgCYPNO (FGSG_01000), two FgNCPNO (FGSG_00765, FGSG_04458) and FGSG_07925 deletion mutants were constructed. All primers used in this study are listed in Table S11. At least two positive transformants for every gene were used for phenotypic analyses. To generate pFGSG_09786-GFP fusion vector, the primers FGSG_09786CF and FGSG_09786CR (Table s11) were used to amplify the full-length sequence of the FGSG_09786 gene and its native promoter from the WT genomic DNA. It was then cloned into a pKNTG vector using One Step Cloning Kit (Vazyme Biotech Co., Ltd, China). The same method was used to construct pFGSG_00765-GFP and pFGSG_04458-GFP and pFGSG_07925-GFP vectors using their respective primers.
Preparation of OMVs as MAMPs source for NO production
Outer membrane vesicles carrying MAMPs are shed by Gram negative bacteria (Tashiro et al., 2010; Cecil et al., 2019). OMVs where harvested in the following way: An inoculum of Escherichia coli DH5α was inoculated to 10 ml 1/10 DFM medium (Ipcho et al., 2016) in a 50ml in a sterilized centrifuge tube and incubated over night at 28°C. The bacterial cell harvested by centrifugation at 4200g for 5 minutes. Supernatant was removed and the pellet washed by resuspended in 10 ml sterile MilliQ water and centrifugation 2 times. Finally, the pellet was resuspended in 1 ml Milli Q water and incubated at 28°C for 1 hour to allow OMV to form. After Incubation the suspension was harvested for OMVs by sterile filtration using a sterile 0.2 μm filter. The resulting liquid containing OMVs were used for MAMPs treatments.
NO measurements using mycelial plugs
Milli Q water agar (Ipcho et al., 2016)was poured into a sterile Omnitray (Nunc) which is in principle a microtiter plate with one 8×12 cm big rectangular well (Hennessy et al., 2017). Fungal cultures of strains to be tested was grown on 1/10 DFM medium in 9 cm diameter petri dishes containing 10 ml DFM agar medium (Ipcho et al., 2016). When fungal mycelium was tested for NO production 5 mm diameter plugs were cut away from the edge of the mycelium to avoid getting DFM nutrients interfering with the test (Olsson, 1994). A DAF-FM DA (ThermoFisher, D23844) staining solution was prepared as 2μl/ml of a stock solution (2μg/ul in DMSO) was added to the bacterial MAMPs suspension of OMVs or to Milli Q water giving a final concentration of 4 μg/ml DAF-FM DA. To perform the assay, 10 ul droplets of the DAF-FM DA staining solution with or without bacterial MAMPs was added on top of the water agar in the Omnitray. Agar plugs were immediately put mycelium down on top of the droplets, one agar plug per droplet. In each Omnitray there were place for 4 rows of 4 replicate plugs. F. graminearum PH1 was always used as positive and negative control taking up 2 rows so it could be compared with mutant or complement strains with or without bacterial MAMPs in the remaining two rows. The amount of fluorescence from DAF-FM as a result of its reaction with NO was recorded for the whole plate automatically (GFP-filter) at roughly 0.5 h intervals. Recordings was made through the bottom of the plate and a lid was kept on to avoid drying of the plate. The plate reader was set up to produce one big image for the whole plate that could be exported as a TEXT image and imported into the freeware ImageJ to produce a TIFF Z-stack that can be measured for the fluorescence development over time in any defined area (Hennessy et al., 2017). The fluorescence increase from the hyphae of each agar plug could then be analyzed and compared. For the first hours the increase in fluorescence was linear indicating a linear accumulation of fluorescent DAF-FM product due to a constant rate of NO production. The slope of the curves was used as relative measures of rates of NO formation to compare MAMPs treatments with controls. The slopes for the fluorescence MAMPs/Water increase over time was used to compare the relative NO response of the strains to the MAMP treatments.
Subcellular localization of NO production and measurements of NO production over time using confocal microscopy using the NO probe DAF-FM DA
NO was produced by F. graminearum PH1 mycelium also without MAMPs addition only a bit slower. To localize the intracellular sites of production NO DAF-FM DA was added directly to a microscope slide. Preliminary experiments showed that the color development was very fast. This was good since fluorescein derivates like the resulting DAF-FM will relatively quickly leak out or move to the vacuole as is common for fluorescein. We therefore tried to minimize the time to get as good localization as possible while still getting a good signal. Thus we, picked fresh mycelium from a colony grown in a CM agar medium and transferred that to a microscope slide an added 10 ul water containing 2 ul DAF-FM DA solution to a final concentration of 4 μg/ml and immediately observed for NO production localization under a Nikon A1 confocal microscope for subcellular localization in NO production. Since we knew that both FgNCPNO responsible for NO production is localized to the ER (Fig. 5B) we used a F. graminearum strain carrying an mCherry tagged ER-marker protein (ER-mCherry-HDEL) (Adnan et al., 2020) to confirm that NO is formed at the ER where the NO-forming proteins are localized (Fig. 5C).
Looking at the microscope images it was obvious that there were specks of autofluorescence that could interfere with the DAF-FM signal. To avoid these problems and get an alternative method for measuring NO formation from both FgNCPNO and FgCYPNO we chose to use the confocal microscope as a fluorimeter. Slides were prepared as above. Method for analysis: A time series of images of the mycelium was recorded using a 10X objective for the GFP signal and for the DIC signal. First Import the ND2 time series Z-stack into ImageJ as separate Tiff images. Separate GFP images and DIC images in separate folders and rename images so the number order index comes first in the name and import these as separate 16 bit TIFF bw into the freeware ImageJ (NIH, https://imagej.nih.gov/ij) to make two TIFF Z stacks. Define a measure area (Region of Interest, ROI) in the DIC image that does not contain air bubbles and relatively much hyphae. Import the ROI to the fluorescence image and check that in this ROI there is no autofluorescence specs (dots that do not fade with time). Do the same for an area without hyphae as close to the previous area as possible to get a background control area. Measure and record the Z-profile for the fluorescence in the measure area and the control area. Use the edge filter to transform the DIC image to an image where the average light intensity is proportional to the biomass (Olsson, 1995) and measure and record this for the first image in the sequence (there is no change over time for the DIC image). Analyzing the signal over time we found that the fluorescence signal followed a logistic decay curve probably as a combined function of accumulation of fluorescence signal and fading due to the strong laser illumination. To maximize the sensitivityof the analysis, we calculated the area under the fitted decay curve corrected for the biomass and used that as a relative measure for the NO formation for the PH1, FgNCPNO and FgCYPNO (Fig. 7A).
Colony growth, conidiation, sexual reproduction, pathogenicity and DON production phenotypes
Infection assays on flowering wheat heads and wheat coleoptiles of cultivar XiaoYan 22 were conducted as described previously (Ding et al., 2009; Zhang et al., 2012). For assaying DON production, fungal cultures were grown for 7 days at 25°C in liquid trichothecene biosynthesis (LTB) medium (Jiang et al., 2016). Subsequently, a competitive enzyme-linked immunosorbent assay (ELISA)-based DON detection plate kit (Beacon Analytical Systems, Saco, ME, USA) was used to measure DON production. Growth rates on were measured on complete medium (CM) plates and conidiation assayed in liquid carboxymethyl cellulose (CMC) medium as described previously (Zheng et al., 2012). The formation of perithecia was tested on carrot agar medium (Wang et al., 2011).
Endocytosis assay using FM4-64
Agar blocks (5 mm in diameter) were transferred from CM solid medium to 50ml centrifuge tubes containing CM liquid medium. After 24h shaking, 10 μ;l of the fungal hyphae suspension was mixed with FM4-64 to a final concentration of 4 μg/ml and endocytosis of the stain was observed using a Nikon A1 confocal microscope using settings for TRITC (Fig. 11A). Endocytosis was estimated 1h after stain addition when most of the FM4-64 stain have been endocytosed and distributed to intracellular organelles if endocytosis is not blocked.
Comparing FgCYPNO and FgNCPNO like proteins in eukaryotes, describing the domain structure and predicting CaM binding
These comparisons are described in detail (Supplementary File SF6 and SF7) and is here outlined as follows. First NCBI was blast searched for all predicted proteins in Eukaryotes that are similar to FgNCPNO. From this result a set of organisms were selected that had good hits for the whole length of the protein. Organisms were chosen closely related to F. graminearum and progressively evolutionary further away. The best hits for organisms for the FgCYPNO were then chosen and phylogenetic trees for both proteins were generated (Fig. 9). DeepLoc was used to check that all similar proteins localize to ER and CaMELS (CalModulin intEraction Learning System) https://camels.pythonanywhere.com/) was used to find likely CaM binding sites while HMMER was used to predict domain structure for all these proteins (Supplementary File SF6 and SF7). All proteins were found to have similar domains and domain order and very similar domain sizes and even distances between domains (Fig. 10).
Statistics
MS Excel was used to calculate all statistics presented in the figures. We have often chosen to use 95% confidence interval bars instead of performing normal t-tests to compare with controls since it simplifies for the reader and is more stringent. Thus, bars with non-overlapping 95% confidence intervals are significantly different (P-value for null hypothesis ≪ 0.05).
Conflict of interest statement
The authors declare that they have no conflict of interest.
Acknowledgements
Thanks to Professor Peter Stougaard, Department of Environmental Science - Environmental Microbiology and Circular Resource Flow, for initial discussions at the start of the work. The Minnesota Supercomputing Institute is kindly acknowledged for computing resources and support for producing the additional transcriptomic that we now publish but was not published with our previous publication (Ipcho et al., 2016) We also thank The Velux Foundation (Denmark) that financed that initial Copenhagen part of the research.
Footnotes
↵* Co-first authors
Authors contributions divided per authors
WZ Deletion and complementation work on the heme reductase the NODs and the first Heme protein we deleted and phenotype analysis. Manuscript writing and correction
HL Deletion and complementation of Heme protein and phenotype analysis including of NO measurements for Confocal microscopy. Manuscript writing and correction
SI MAMPS stimulation and NO measurements including inhibitor analysis, methods development for carrying out these analyses and experiments and performed the transcriptomic experiments, Manuscript writing and correction.
WF Phenotype analysis including growth, conidiation and pathogenicity. Manuscript correction.
RH NO measurements of effect of HemeR and NOD deletions. Manuscript correction.
BOH Preparation of transcriptomic data for co-regulation analysis. Manuscript correction.
GD Acquisition of financial support for final research. Manuscript correction.
ZW Acquisition of financial support for final research. Manuscript correction.
MAN Initial idea, discussions, and preparation of MAMPs for testing. Acquisition of financial support for initial research. Manuscript correction.
SO Initial idea. Acquisition of financial support for initial research. Hypotheses generation from data and from literature as well as from bioinformatics, co-regulation analysis, phylogenetic and structure analysis, overall responsible for driving the work forward and methods development. Suggesting confirmatory analyses and experiments for additional functions. Main responsible for manuscript writing and coordination of the manuscript writing and corrections.
References




