

Abstract
Protein quality control is an essential cellular function and it is mainly executed by a large array of proteases and molecular chaperones. One of the bacterial HtrA protein family members, the homo-oligomeric DegP-protease, plays a crucial role in the Escherichia coli (E. coli) protein quality control machinery by removing unfolded proteins or preventing them from aggregation and chaperoning them until they are properly folded within the periplasm. DegP contains two regulatory PDZ domains, which play key roles in substrate recognition as well as in the transformation of DegP to proteolytic cage-like structures. Here, we analyse the interaction and dynamics of the PDZ-domains of DegP underlying this transformation in solution by high-resolution NMR spectroscopy. We identify an interdomain molecular lock guiding the interactions between both PDZ domains, regulated by fine-tuned protein dynamics and potentially conserved in proteins harboring tandem PDZ domains.
Introduction
An effective protein quality control (PQC) system consisting of molecular chaperones as well as proteases is essential for each living organism 1,2. Within the periplasmic space of Gram-negative bacteria, this crucial task is carried out mainly by the following three proteins: Skp, SurA, and DegP 3. These proteins play a central role in the quality control of outer membrane proteins (Omp) and protect bacteria from the accumulation and aggregation of misfolded proteins within the periplasm. Skp and SurA are responsible for controlling protein folding through their function as holdase proteins 4,5. In contrast, DegP has been shown to have a tightly-regulated dual activity under different stress conditions in vivo 3: Primarily, it functions as a serine protease to rescue cells during heat stress but it also possesses chaperoning activity at reduced temperatures 6. DegP was shown to be essential for E. coli survival as a degP gene knockout is lethal at elevated temperatures (42°C) 7. However, it was possible to show that DegP protease activity is not mandatory since overexpression of protease-deficient forms of DegP is already sufficient for E. coli to recover 8 under stress conditions, suppressing the lethal phenotypes 9.
On a structural level, DegP consists of a protease domain connected to two adjunct PDZ domains named PDZ1 and PDZ2 after the first proteins PSD-95, Dlg1, and ZO-1 in which these domains were observed 10. In its protease-inactive state DegP forms a homo-hexameric complex 11, which upon activation is able to re-arrange into dodecameric or 24-meric cage-like forms, suggested to encapsulate substrate proteins before proteolytic cleavage 12–14. The common building block of the different cage-like structures is a DegP trimer, thus DegP is assumed to undergo large structural transitions from its hexameric resting state via a trimeric state towards the higher oligomeric states 15,16. This structural transition itself is supposed to be mediated by modulations of the PDZ1–PDZ2 interaction 17. Nevertheless, the underlying mechanism and eventual structural adaptions remain only partially understood as previous structural studies could only reveal the respective endpoints of the different oligomeric assemblies. The current model suggests that peptides containing a hydrophobic carboxy-terminal tail initially bind to the PDZ1-domain, thereby triggering subsequent structural re-arrangements, which in turn activate DegP 18. Contrary, the PDZ2-domain inhibits protease activity as long as DegP disassembles into trimers and subsequently transforms towards the cage-like structures 17.
To address the underlying structural adaptions upon-temperature activation of DegP, we investigated full-length DegP as well as its PDZ1-PDZ2 domains at the atomic level using high-resolution NMR spectroscopy. Based on local chemical shift perturbations we identified an intermolecular methionine–tyrosine interaction controlling the transition from hexameric to trimeric DegP. Detailed analysis of the inherent protein dynamics revealed a finely tuned temperature-dependent allosteric network within PDZ1 controlling the interaction with PDZ2. Remarkably, the crucial tyrosine-methionine residues are highly conserved within the HtrA proteins indicating a fundamental activation mechanism modulated via the intermolecular lock between their PDZ domains.
Materials and Methods
Cloning of different DegP domain constructs
The pDS4 plasmid contains wild-type DegP (E. coli K12 strain) lacking its amino-terminally signaling sequence within a pET28b-vector backbone yielding DegP with an amino-terminal His6-Tag (purchased from GenScript). The individual PDZ domains, the PDZ1–PDZ2 construct as well as the SUMO-tag were amplified by PCR from pDS4 and pET28_SUMO_Hsc70 (a kind gift of B. Bukau (Heidelberg))19 plasmids, respectively. The different DegP-PDZ constructs and the SUMO-tag were fused by standard PCR-methods and cloned into a pET28a (−) vector via the NcoI and XhoI restriction sites by standard cloning techniques yielding PDZ constructs fused to an amino-terminal His6-SUMO-tag. Plamids and primers used in this study can be found in Supplementary Table 1.
Protein expression and Purification of PDZ1-PDZ2 domains
pDS10 (PDZ1), pDS11 (PDZ2), and pDS12 (PDZ1–PDZ2) plasmids were chemically transformed into E. coli BL21 Star™ (λ DE3) cells. The transformed cells were grown at 37°C in 1 l LB medium containing 50 μg/ml kanamycin until an OD600 ≈ 0.5 was reached. Expression was induced by addition of 0.4 mM IPTG and cells were left to grow overnight at 25°C. Cells were harvested by centrifugation at 8,000*g for 20 min at 4°C and subsequently resuspended in 25 ml of lysis buffer (25 mM Hepes, 0.5 M NaCl, 10 mM Imidazole, pH 7.5). The resuspended cells were incubated on ice with one Complete EDTA-free protease inhibitor (Roche) tablet as well as with 100 U of DNAse (ArcticZymes) for 30 min and subsequently lysed with a Q700 sonicator (Qsonica) (10 s on, 30 s off, 30 % power). Cell debris was removed by centrifugation at 18000*g for 45 min at 4 °C and the supernatant was applied to a Ni2+-Sepharose 6 Fast Flow (GE Healthcare) loaded gravity column, followed by step-wise elution with 20 ml of lysis buffer supplemented with 100 mM and 500 mM imidazole, respectively. Fractions containing the target proteins were dialyzed against PBS and the His6-SUMO-tag was removed by enzymatic cleavage using with human SENP1 protease at 4°C overnight (Addgene #16356) 20. The cleaved proteins were applied again to a Ni2+–column and the flow-through was collected. The proteins were concentrated using Amicon centrifugal filters (5k MWCO or 10k MWCO, Millipore) and purified further by size exclusion chromatography (Superdex 75 Increase, GE Healthcare) in NMR buffer (25 mM potassium-phosphate, 1 mM EDTA 1 mM TCEP, pH 7.0).
For His6-tagged full-length DegP, an additional unfolding/refolding step was introduced. To this end, after cell lysis and centrifugation, the supernatant was loaded to a HisTrap HP 5 ml column (GE Healthcare) equilibrated with lysis buffer supplemented with 6 M Guanidinium chloride. For on-column refolding, the bound protein was washed with a 6–0 M Guanidinium chloride linear gradient (15 CV, 0.5 ml/min) overnight and subsequently eluted with a step elution of 100 mM and 500 mM imidazole. In the next step, DegP was purified by size exclusion chromatography (Superdex 200 Increase, GE Healthcare) in NMR buffer.
Isotope labeling
We obtained [U-15N] and [U-15N,13C] labeled PDZ-domain constructs by growing the expression cells in M9 minimal media 21, supplemented with (15NH4)Cl and D-(13C)-glucose, respectively. For full-length DegP, the cells were in addition grown in D2O-based M9 minimal medium, yielding [U-2H,15N] and upon the usage of D-(13C,2H)-glucose [U-2H,15N,13C] proteins, respectively. For specific isoleucine methyl group labeling, we supplemented D2O-based M9 minimal medium with isoleucine precursor (2-Ketobutyric acid-4-13C,3,3-d2 sodium salt hydrate) 1 hour prior to induction resulting in [U-2H, Ile-δ1-13CH3] labeled DegP. All isotopes were purchased from Merck.
NMR spectroscopy
NMR measurements were performed on Bruker Avance III HD 700 MHz or 800 MHz spectrometers, running Topspin 3.5 and equipped with a cryogenically cooled triple-resonance probe. All experiments were performed in NMR buffer (25 mM potassium-phosphate, 1 mM EDTA, 1 mM TCEP pH 7.0).
For the sequence-specific backbone resonance assignment experiments of DegP and the different PDZ-domain constructs the following experiments were recorded: 2D [15N, 1H]-TROSY-HSQC 22 and for full-length DegP the following TROSY-type 3D experiments 3D trHNCA, 3D trHNCACB, and 3D trHNCO 23 whereas for the different PDZ-domain constructs standard through-bond 3D HNCA, 3D HNCACB, 3D HNCO, 3D CBCA(CO)NH 24 experiments were used. Aliphatic side-chain resonance assignment for the PDZ1-PDZ2-construct was performed based on 2D [13C, 1H]-HSQC spectra with/without constant time (CT) version, as well as 3D (H)CC(CO)NH, H(CC)(CO)NH, and HCCH-TOCSY-experiments 24. Assignment experiments were performed at 50 °C if not indicated otherwise.
For the quantitative analysis of signal intensities, the amplitudes were corrected by differences in the 1H-90° pulse-length, the number of scans, and the dilution factor 25. NMR data were processed with a combination of NMRPipe 26 and mddNMR2.6 27 as well as analysed with CARA 28.
For titration experiments, 2D [15N, 1H]-TROSY-HSQC experiments were acquired with 4 scans and 2048 × 256 complex points in the direct and indirect dimensions, respectively. The chemical shift changes of the amide moiety were calculated as follows:

The chemical shift changes of the methyl groups were calculated as follows:

Secondary chemical shifts were calculated relative to the random coil values by Kjaergaard and Poulsen 29. Further, a weighting function with weights 1–2–1 for residues (i−1)−i−(i+1) was applied to the raw data 5,30.
NMR backbone dynamics
For the analysis of the dynamic properties of the different DegP constructs, the following relaxation experiments were measured: 15N{1H}-NOE 31, T1(15N) 31, T1ρ(15N) 32 and TROSY for rotational correlation times (TRACT) for determining T2α(15N) and T2β(15N) 33. Nonlinear least square fits of relaxation data were done with MATLAB (MathWorks) and the Dynamics Center 2.5 (Bruker Biospin). R2(R1ρ)(15N) values were derived from T1ρ 34 using equation 3:
 with
with  where ω is the spin-lock field strength (2 kHz) and Ω is the offset from the 15N carrier frequency.
where ω is the spin-lock field strength (2 kHz) and Ω is the offset from the 15N carrier frequency.
Error bars for R1(15N), R1ρ (15N), R2α(15N), and R2β(15N) were calculated by a Monte Carlo simulation embedded within Dynamics Center 2.5 (Bruker, Biospin), and for R2(R1ρ) (15N) by error propagation. Error bars for the 15N{1H}-NOE were calculated from the spectral noise. Analysis of the obtained relaxation rates was performed with Tensor2 35 using an anisotropic diffusion tensor on the NMRbox web server 36.
NMR sidechain dynamics
Experiments were performed on a [U-2H, Ile-δ1-13CH3]–PDZ1-PDZ2 sample at a temperature of 37°C in 99.9% D2O based NMR buffer. Side chain methyl order parameters  were determined by cross-correlated relaxation experiments 37,38. Single-(SQ) and triple-quantum (TQ) 1H–13C experiments were collected at a series of delay times. Ratios of the peak intensities were fitted for eight values ranging between 2 and 24 ms using the following equation where T is the relaxation delay time and δ a factor to account for coupling due to relaxation whith external protons:
were determined by cross-correlated relaxation experiments 37,38. Single-(SQ) and triple-quantum (TQ) 1H–13C experiments were collected at a series of delay times. Ratios of the peak intensities were fitted for eight values ranging between 2 and 24 ms using the following equation where T is the relaxation delay time and δ a factor to account for coupling due to relaxation whith external protons:

 values were determined using equation 5, using their fitted η values and the separately determined rotational correlation time (τc) of 7.7 ns for 37°C:
values were determined using equation 5, using their fitted η values and the separately determined rotational correlation time (τc) of 7.7 ns for 37°C:

Where μ0 is the vacuum permittivity constant, γH the gyromagnetic ratio of the proton spin, rHH is the distance between pairs of methyl protons (1.813 Å),  is the generalized order parameter describing the amplitude of motion of the methyl 3-fold axis, Θaxis,HH is the angle between the methyl symmetry axis and a vector between a pair of methyl protons (90°), and P2(x) = ½ (3x2−1). Finally, the product of the methyl order parameter and the overall correlation time constant,
is the generalized order parameter describing the amplitude of motion of the methyl 3-fold axis, Θaxis,HH is the angle between the methyl symmetry axis and a vector between a pair of methyl protons (90°), and P2(x) = ½ (3x2−1). Finally, the product of the methyl order parameter and the overall correlation time constant,  , was determined.
, was determined.
Multiple quantum (MQ) methyl relaxation dispersion experiments 39 were recorded as a series of 2D data sets using constant time relaxation periods (T) of 30 ms (800 MHz; 25°C, 37°C, 43°C, and 50°C) and CPMG (Carr-Purcell-Meiboom-Gill) frequencies ranging from 67 to 1000 Hz. R2,eff, the effective transverse relaxation rate was calculated according to the following equation:
 where I (or I0) are the intensities with and without the presence of a constant-time relaxation interval of duration T, during which a variable number of 13C 180° pulses are applied leading to νCPMG = 1/(2δ), where δ is the time between successive pulses. Dispersion data were fitted numerically to a two-site exchange model using the program ChemEx (available at https://github.com/gbouvignies/chemex/releases).
where I (or I0) are the intensities with and without the presence of a constant-time relaxation interval of duration T, during which a variable number of 13C 180° pulses are applied leading to νCPMG = 1/(2δ), where δ is the time between successive pulses. Dispersion data were fitted numerically to a two-site exchange model using the program ChemEx (available at https://github.com/gbouvignies/chemex/releases).
NMR magnetization exchange
2D methyl magnetization exchange experiments were recorded to quantify the exchange between the locked (l) and open (o) state of the Met280-13Cε1Hε methyl group as outlined by Farrow et al.40. The 13C-EXSY-SOFAST-HMQC experiments 41 were recorded with varying delays between 50 and 1000 ms at 37°C. To ensure equilibration of the initial magnetization a long relaxation delay (1s) was chosen. The signal intensities of the observed three peaks of M280-13Cε1Hε, namely, the auto peak of the locked conformation, Ill, and the auto peak of the open conformation Ioo as well as the cross peak originating from the transition from the locked to the open state, Ilo were determined. Due to signal overlap the second cross-peak, Iol, could not be determined from the spectra, therefore it was assumed that it follows a similar build-up than Ilo. All data points were simultaneously fitted to the following theoretical equations:

Where λ1,2, xl and xo are defined according to the following relationships:
 and the rate of transition from the locked to the open conformations, klo, the rate of transition vice versa, kol,the longitudinal relaxation rate in the locked conformation, R1l, as well as the one of the open conformation, R1o, were fitted. Il(0) and Io(0) denote the initial amounts of longitudinal carbon magnetization associated with the locked and the open conformations at the beginning of the mixing periods.
and the rate of transition from the locked to the open conformations, klo, the rate of transition vice versa, kol,the longitudinal relaxation rate in the locked conformation, R1l, as well as the one of the open conformation, R1o, were fitted. Il(0) and Io(0) denote the initial amounts of longitudinal carbon magnetization associated with the locked and the open conformations at the beginning of the mixing periods.
NMR Diffusion
Translational diffusion coefficients were measured by recording a series of 1D 13C-edited spectra at different temperatures, using a pulse scheme (13C-edited BPP-LED) that is similar to an 15N-edited BPP-LED experiment with 15N and 13C pulses interchanged 42. To compensate for the different J-coupling of the methyl-groups compared to the amide groups, a gradient duration δ of 3.2 ms was used instead of 4.8 ms used in the 15N-filtered version. In addition, a diffusion delay T of 400 ms and a t of 0.1 ms were used. The strength of the encoding/decoding was increased stepwise. The resulting 1H signal was integrated over the methyl 1H frequency range to obtain intensities as a function of encoding/decoding gradient strength. In order to construct the calibration curve given in Fig. 1g the following published translational diffusion coefficients were used: Immunoglobin binding domain of streptococcal protein G, GB1 (MW = 6.2 kDa) 43, lysozyme (MW = 14.1 kDa) and Interleukin-10 (MW = 37.2 kDa) 44, bacterial HsiV (MW = 230 kDa), ½ proteasome from T. acidophilum, α7α7 (MW = 360 kDa), and the α7 single ring variant of the proteasome (MW = 180 KDa) 45.

a, Cartoon representation of the suggested structural changes that DegP undergoes transitioning from its hexameric resting state via its trimeric state towards the higher oligomeric protease-active states. The two PDZ domains are depicted in purple and cyan for the PDZ1- and PDZ2-domains, respectively, whereas the protease domains are shown in grey and brown. A potential substrate is indicated in blue. b, Cartoon representation of the crystal structure of the hexameric assembly of DegP (PDB-ID: 1KY9) with the protease domain depicted in grey and both PDZ-domains in purple. c, 2D [15N,1H]-NMR spectrum of [U-2H,15N,13C]-DegPY444A. The sequence-specific resonance assignment obtained from 3D TROSY-type triple-resonance experiments is indicated. d, Measurement of the molecular diffusion constant with a 13C-methyl-filtered diffusion BPP-LED experiment. The logarithm of the signal intensity is plotted against the squared gradient strength of the applied pulsed-field gradients. [U-2H, Ile-δ1-13CH3]–DegP was measured at different temperatures as indicated. Solid lines are a linear fit to the data. e, Obtained molecular diffusion constants plotted against the temperature. The broken line serves as a guide to the eyes only. f, Calibration curve for estimating the molecular weight based on the determined molecular diffusion constants. Linear correlation of the logarithm of the molecular weight (MW) plotted against the molecular diffusion constants. Published diffusion constants of proteins with known molecular weight are indicated as green dots (see Methods for details). Temperature dependent data for DegP is shown in a blue gradient as indicated in panel d.
Bio-Layer Interferometry (BLI)
BLI experiments were performed on an Octet RED96 system (Fortébio) at 25°C as outlined in detail before 46. Briefly, the respective ligands were biotinylated using the biotinylation kit EZ-Link NHS-PEG4-Biotin (Thermo Fisher Scientific). The biotin label was resolved in H2O and directly added to the protein solution in a final molar ratio of 1:1 in 25 mM MES, pH 6.0, followed by gentle mixing at room temperature for 45 min. Unreacted biotin was removed with Zeba Spin Desalting Columns (7 MWCO, Thermo Fisher Scientific). Biotin-labeled proteins were immobilized on the streptavidin (SA) biosensors (Fortébio) and the biosensors were subsequently blocked with EZ-Link Biocytin (Thermo Fisher Scientific). Analytes were diluted and applied in a dose-dependent manner to the biosensors immobilized with the biotinylated ligand. Bovine serum albumin (BSA) powder (Sigma-Aldrich) and Tween-20 were added to final concentrations of 0.1% and 0.05%, respectively, to avoid non-specific interactions. Parallel experiments were performed for reference sensors with no analyte bound and the signals were subsequently subtracted during data analysis. The association period was set to 30 s and dissociation to 40 s. Data measurements and analysis were performed by using the Data acquisition 10.0 and the Data analysis HT 10.0 (Fortébio) software, respectively.
Data availability
The sequence-specific NMR resonance assignments for the PDZ1-PDZ2, PDZ1, and PDZ2-constructs were deposited in the BMRB under accession codes XXX, YYY, and ZZZ. All other relevant data are available from the corresponding author upon reasonable request.
Results
Solution NMR of full-length DegP
Currently, only static high-resolution structures of DegP by X-crystallography 13 as well as medium-resolution structures by cryo-electron microscopy (cryo-EM) 47 are available, providing an incomplete picture of the functional cycle of DegP. While these structures provide important insight into the organization of the different oligomeric states of DegP, they do not reveal the underlying structural transitions. Therefore, we investigated DegP by high-resolution NMR-spectroscopy to obtain detailed insight into its structural and dynamical properties, which give rise to the different oligomeric states. Due to its large size (~150 kDa as a trimer) and the existence of different oligomeric forms (Fig. 1a), DegP presents a challenging system for solution NMR studies. Here, we present the first [15N,1H]-NMR spectra of uniformly-labeled [U-2H,15N]-DegP (Supplementary Fig. 1a–c). The spectrum obtained at 25°C contained only a very limited number of resonances located exclusively in the central region of the spectrum, indicative of unfolded and loop regions in line with the large size of the protein. In contrast, increasing the temperature to 37°C and subsequently 50°C resulted in well-dispersed spectra containing approximately 120 well-separated resonances (Supplementary Fig. S1b,c). The dispersion of these peaks [10–6 ppm in the 1H-dimension] showed that a folded domain of the full-length DegP was observable under these conditions, indicating its flexible attachment to the rest of the protein. In the next step, we assigned the observed backbone resonance by standard TROSY-type 3D experiments employing the known trimerization mutant DegPY444A [14], which provided further spectral improvement (Fig. 1c and Supplementary Fig. 1d). We were able to sequence-specifically assign large parts of the two carboxy-terminal PDZ domains (150 out of a total of 188 PDZ1–PDZ2 resonances). The obtained secondary chemical shifts indicated that the structure in solution is to a large extent in agreement with the secondary structure elements observed previously within the different crystal structures (Supplementary Fig. 1e–g). The observation of a large set of resonances of the DegP PDZ domains indicates that these domains are not in direct contact with other parts of the protein and tumble independently in solution. Furthermore, the increased spectral quality at elevated temperatures suggested that full-length DegP transitions between different oligomeric states, suggesting an inherent temperature switch for DegP activation.
In order to determine the oligomeric state of full-length DegP in solution, we tried to use the TRACT experiment 33 to probe the DegP size at the elevated temperatures (data not shown). However, due to the lack of signals at lower temperatures, this approach could not be used at temperatures below 37°C. Moreover, the inherent overestimation of the flexible regions in the TRACT experiment prevented in-depth analysis 33. To circumvent these issues, we specifically labeled the methyl groups of the methyl bearing isoleucine residues, which yielded high-quality spectra already at 25°C, and measured pulsed-field gradient diffusion experiments on the methyl groups 42. The obtained translational diffusion coefficients showed a clear temperature dependence (Fig. 1d). With increasing temperature the values increased from 3.4•10−7 cm2s−1 at 25°C to 4.82•10−7 cm2s−1 and 4.95•10−7 cm2s−1 at 43°C and 50°C, respectively (Fig. 1d,e). Comparing these values to known literature values clearly indicates that DegP is in a hexameric state (MW = 300 kDa) at 25°C whereas at 43°C and higher the protein is in a trimeric state (MW = 150 kDa). At 37°C we obtained diffusion coefficients that were slightly elevated compared to 25°C (Fig. 1 d,e) indicating the partial presence of a trimeric species as initial steps of the structural transition, which is estimated to be about 20–25% based on the increase in the diffusion coefficient.
Isolated PDZ1–PDZ2 bi-domains in solution
Based on the initial observation of the highly flexible PDZ-domains within the full-length DegP and their possible contribution to the inherent temperature switch, we subsequently focused on the next step on the isolated PDZ1–PDZ2 domains. High quality resonance assignment data of the PDZ1–PDZ2 domain construct allowed almost complete sequence-specific assignment of the observed resonances in the [15N,1H]-TROSY HSQC spectrum (Supplementary Fig. 2), yielding 87% backbone and 95% aliphatic side-chain assignment. The missing amino acids were part of the interdomain linker (356–366) and located in flexible loops within PDZ1, this exchange broadening is due to the enhanced amide-proton exchange rates at 50°C within these unstructured regions 48. Using the combined 13Cα and 13Cβ chemical shifts of the protein backbone, we identified the secondary structure elements of the PDZ1-PDZ2 domain construct in solution. The observed structural elements, five α-helices and eight β-sheets, are in agreement with the apparent secondary structure elements of the available crystal structure (Supplementary Fig. 2c,d). However, we detected three additional short β-sheet region regions ranging from amino-acids 370–405 in the linker region between the two PDZ-domains, indicating that the domains are not completely decoupled. As we had observed large temperature-dependent effects within the spectra of full-length DegP, we also assigned the PDZ1–PDZ2 construct a 25°C and investigated its secondary structure propensities (Supplementary Fig. 3). The secondary chemical shifts were almost identical to the ones obtained at 50°C, indicating no structural adaptions. Nevertheless, a distinct lack of resonances could be observed for residues within the amino- and carboxy-terminus of the PDZ1–PDZ2 domain construct in the 25°C data set: the resonances for residues M268, G269, L276, A277, M280, R286, G287, S291, N435, I436, G439, D440, and I443–M447 were broadened beyond detection, indicating the presence of a dynamic intermolecular interaction between different PDZ1-PDZ2 molecules on the NMR intermediate timescale, i.e. with kinetic rates ranging from 1000 to 10 s−1 [49].
Probing the interactions between PDZ1 and PDZ2
To follow up on the temperature-dependent line-broadening effect observed for the PDZ1–PDZ2 domains in solution as well as previous observations within a cryo-EM structure, we characterized the PDZ1:PDZ2 interaction in further detail. Previous cryo-EM data indicated that the PDZ1–PDZ2 domains are essential for assembling and interlocking the DegP 12–meric and 24–meric cages and that removal of the PDZ2 domain prevents cage formation altogether leading to primarily trimeric protein 17. Therefore, we prepared individual constructs of the PDZ1 and the PDZ2 domain and confirmed the sequence specific resonance assignments (Supplementary Fig. 4). In order to map possible interaction areas between these two domains at different temperatures, we titrated the PDZ1 and the PDZ2 domains with each other at four temperatures (25–50°C), as these temperatures within the full-length protein represent the hexameric, the ~20% trimeric, as well as the full trimeric state as indicated by the NMR diffusion data (Fig. 1d–f). The observed spectral changes indicated that the extreme amino-terminal and carboxy-terminal regions of PDZ1-PDZ2 domains interact strongly at 25°C and the chemical shift perturbations (CSPs) lessened with increasing temperature, with M280 showing the largest chemical shift perturbations (Fig. 2 and Supplementary Fig. 5). Remarkably, whereas the affected resonances did not have any interfaces on the available hexameric crystal structure (Supplementary Fig. 1f), they were observed within the 24–meric structure (Fig. 2e and Supplementary Fig. 1g), indicating the possibility of a stabilizing sulfur-π aromatic motif 50. Moreover, a distinct set of residues showed larger chemical shift perturbations along with an enhanced intensity loss at 25°C compared to higher temperatures (Fig. 2c,d). Specifically, M280 and Y444 were the residues most affected, implying that these residues might provide the molecular switch locking the hexameric resting state at lower temperatures. To test this hypothesis we constructed the corresponding mutants (PDZ1M280A, PDZ2Y444A) and evaluated if the interaction between the domains was abolished. The titration experiments clearly showed that the interaction was completely abolished if either M280 or Y444 was mutated (Supplementary Fig. 6) highlighting the importance of both amino-acids forming the intermolecular lock.

a, b, 2D [15N,1H]-NMR spectra of [U-15N]-PDZ1 (a, purple) and [U-15N]-PDZ2 (b, black) as well as after addition of two molar equivalents of the respective other domain. Titrations were measured at 50°C. c, d, Detected chemical shift perturbations at four temperatures ranging from 25°C–50°C as indicated of the PDZ1 domain (c) and the PDZ2 domain (d). Severely affected residues experiencing line-broadening are indicated as grey bars in the panels. The dotted lines represent a significance level of 0.05 ppm of the CSPs. e, Perturbated resonances mapped on the crystal structure of DegP 24–mer (PDB-ID: 3CS0). Mainly affected residues, CSPs twice the S.D. on PDZ1 are highlighted in purple (G266, G269, K278, M280, D283, and R286), whereas residues in PDZ2 are highlighted in green (A433, N435, I443, Y444, and L445). The enlargement shows the central M280–Y444 interaction. f, Kinetic analysis by biolayer interferometry (BLI) of the PDZ1-PDZ2 interaction. PDZ1 binding to the biotinylated-PDZ2 domain was probed at 25°C. Analyte concentrations are indicated in the figure. Non-linear least-square fits to the experimental data are indicated by black lines.
To also quantify the interaction between the individual PDZ domains we used Bio-Layer Interferometry (BLI) steady-state analysis at 25°C for obtaining the dissociation constants (KD) of 7.8 ± 0.9 μM for the binding of PDZ1 towards biotinylated-PDZ2 (Fig. 2f and Supplementary Fig. 4c). We did not test the interaction vice versa with biotinylated PDZ1 in detail, because initial tests yielded a dissociation constant weakened by a factor of ~20, indicating that the binding interface on PDZ1 is affected by the close proximity of the Biotin-label and the identified interaction site (data not shown).
The M280–Y444 interaction locks the hexameric state
To further analyze the basis of the M280–Y444 interaction we initially checked the temperature effects on the resonance lines of the side-chains within the PDZ1-PDZ2 construct. For Y444 we observed a Cα resonance line-broadening to a similar extent than observed initially for the amide moiety (Figs. 2b and 3a). This behavior is also reflected by the Y444δ ring resonances, which lose about 60% of their intensity at 25°C compared to 50°C in contrast to the Y444ε resonances, which show stable intensity over the tested temperature range (Fig. 3b). Subsequently, we investigated the effects on the M280 side, where we focused on its methionine-ε-methyl group, which we assigned by employing the mutation variant PDZ1-PDZ2M280A (Fig. 3c). Probing the M280ε resonance at different temperatures revealed the presence of two distinct states, which we termed open (50°C) and locked (25°C) (Fig. 3d). In the transition temperatures of 37°C and 43°C, respectively, we could observe two distinct peaks for the open and locked conformations, which are in the slow exchange regime >s−1 on the NMR timescale. Integrating the peak intensities at the different temperatures revealed that at 37°C the open and locked conformation coexist in a 1:1 ratio, whereas at 43°C and higher the open conformation dominates and at 25°C the locked one (Fig. 3e). In order to quantify the interconversion between the open to the locked state we used NMR exchange spectroscopy (Fig. 3f,g)40,41. The obtained rate for the interconversion between the different states was kex = 0.33 s−1 from the open to the locked state.

a, Focus on the Cα resonance region around Y444 of a 2D [13C,1H]-NMR spectrum of [U-13C,15N]-PDZ1–PDZ2 at 25°C–50°C as indicated. The inset shows the respective δ2[1H]-1D cross–sections through the N349α and Y444α resonances. b, Methionine methyl region of a 2D [13C,1H]-NMR spectrum of [U-13C,15N]-PDZ1–PDZ2 at 25°C–50°C as indicated. Inset shows the signal intensities of the aromatic Y444ε and Y444δ resonances in the dependence of the temperature. c, Overlay of [U-13C,15N]-PDZ1-PDZ2M280A (blue) and PDZ1-PDZ2 (yellow) at 50°C used to assign the M280ε resonance. d, Methionine-ε-methyl region of a 2D [13C,1H]-NMR spectrum of [U-13C,15N]-PDZ1–PDZ2 at 25°C–50°C as indicated. Spectra were manually shifted along the 1H dimension to illustrate the transition between open and locked state. e, Analysis of the open and locked state. Lines are guide to the eyes only. f, Magnetization exchange spectroscopy showing that the open state (o) and locked state (l) of M280 are in a dynamic equilibrium at 37°C as indicated.g, The exchange rate (kex) is determined from the built-up of the exchange peaks. h, Frustration analysis was calculated for the DegP crystal structure (PDB-ID: 1KY9) via the Protein Frustratometer 2 [51]. Minimally frustrated interactions are indicated as blue lines, highly frustrated interactions as red lines.
As for example molecular chaperones were previously shown to recognize locally frustrated regions 30,52 we reasoned that local frustration of the PDZ1-PDZ2 domains 51 could be the driving force for the intermolecular PDZ1-PDZ2 interaction (Fig. 3h). Strikingly, two of the most frustrated regions within the PDZ1-PDZ2 construct reside in helix α1 harboring M280 and sheet β8 with Y444. As frustration arises from conflicts between the constrained connectivity of amino acids resulting in energetically unfavorable local regions, this observation indicates that the M280–Y444 lock reduces this energetically unfavorable state in cellular ground states.
Backbone dynamics of the PDZ1–PDZ2 construct reveal a fine-tuned temperature adaption
In order to understand the molecular basis of the M280 mediated temperature-switch in more detail, we next evaluated the inherent backbone dynamics of the PDZ1–PDZ2 domain construct over a broad range of timescales employing NMR relaxation measurements 53. To investigate the motions initially on the fast-timescale (pico-to nanosecond) the steady-state 15N{1H}-NOE (hetNOE) and the 15N longitudinal (R1) relaxation rates were measured. In general, we observed a planar profile for the folded segments with average hetNOE values of 0.78 and R1-rates of 1.82 s−1, indicating a stable protein fold (Fig. 4a and Supplementary Fig. 7a). As expected, for the loop regions and the termini we observed values indicative of increased flexibility, namely lower hetNOE values and increased R1-rates.

a, Local backbone dynamics on the ps–ns timescale probed by hetNOE measurements at 50°C. The amide moieties of PDZ1-PDZ2 construct are shown as spheres and the hetNOE values are indicated by the green to blue gradient. b, Obtained transversal relaxation (R2(R1ρ)) rates reporting on μs-motions at 50°C are plotted on the amide moieties and are indicated by the yellow to red gradient. c, Temperature-dependence of the average hetNOE, longitudinal relaxation (R1) rate, transversal relaxation (R2(R1ρ)) rate, and resulting τc over the indicated temperature range. Errors are the S.D.. d,e, Temperature dependence of the subnanosecond order parameter S2 (d) and the exchange rate Rex (e), reporting on the flexibility on the μs–ms timescale, calculate using the Lipari-Szabo model-free approach 55,56. f, Temperature dependence of the Rex-rates and zoom in on the region around Met280. Rotation relative to panel d is indicated. The excerpt from the obtained exchange rates in this region is also shown. Residues for which no data could be obtained are indicated with a *. The full data set is given in Supplementary Fig. 7d. g, The cartoon representation of the temperature dependence of the inherent exchange rates (μs–ms) of helix α1 modulating the M280 locking mechanism.
Next, we analysed the contributions of motions in the micro-to millisecond regime. For these slow time-scale motions, we analyzed the 15N transverse relaxation rates. First, we measured the R2-rates derived from the R1ρ rates (R2(R1ρ)), which report on the motions on the lower μs-timescale, because under the used spin-lock radio frequency (RF) field of 2.000 Hz all exchange contributions (Rex) slower than 80μs would be leveled out (Fig. 4b and Supplementary Fig. 7b). In line with the previous analysis of the fast timescale motions, we observed generally a planar profile for the folded segments. In addition, we observed slightly enhanced transversal relaxation rates for the loop regions and for helix α1 as well as the carboxy-terminal sheet β9, containing M280 and Y444, respectively. Furthermore, we measured the transverse relaxation of the slowly relaxing 15N[1H] doublet component (R2β) revealing exchange contributions (Rex) on the higher micro-to millisecond timescale 54, which resulted in similar profiles as observed for R2(R1ρ). In summary, the transversal relaxation rates indicate the existence of local conformational exchange.
In the next step, we assessed the obtained relaxation rates globally in a temperature dependent manner (Fig. 4c). Whereas we observed no differences in the hetNOE values over the tested temperature range, we could observe temperature dependent modulations for R1 and R2(R1ρ), mainly reflecting the change in molecular size as assessed by the R1/R2(R1ρ) derived τc, showing a similar temperature dependent transition as initially observed for full-length DegP by diffusion measurements (Fig. 1d–f).
To obtain more detailed insight into the roles of individual amino-acids involved into the PDZ1-PDZ2 interaction we employed the Lipari-Szabo model-free approach 55. This analysis revealed for the nanosecond timescale order parameters S2, reporting on the fast motions of the N-H vector, indicating distinct temperature dependent differences for the two domains (Fig. 4d). Whereas the core of PDZ2 (e.g. central β-sheet) was showing only slightly temperature modulated S2 values of ~0.8, the core of PDZ1 showed slightly lower values, with a minimum achieved at 37°C with ~0.6 for its central β-sheet (Fig. 4d and Supplementary Fig. 7c).
The exchange rates, Rex, additionally determind by the Lipari Szabo approach 55, report on exchange conformational exchange on the micro– to millisecond timescale. In agreement with the initially determined transversal exchange rates, we observed enhanced Rex-rates for the residues residing in helix α1 (Fig. 4e and Supplementary Fig. 7c). Looking at the exchange rates in a temperature-dependent manner reveals that at 25°C no conformational exchange is observable in helix α1, whereas from 37°C onward micro-to millisecond exchange contributions take place, with the largest exchange rate observed for M280 (Fig. 4e,f). Based on the observed exchange rates a picture emerges, that the extend of motions of M280 are maximized at 37°C and upon temperature dependent dissociation of the PDZ1 and PDZ2 domain this residue remains flexible on the micro-to millisecond timescale (Fog. 4f and Supplementary Fig. 7c,d).
Dynamics of the isoleucine side-chains
To derive the origin of the enhanced local exchange contributions of helix α1, that provide the basis for modulating the PDZ1-PDZ2 interaction we exploited the increased sensitivity of methyl groups providing insight about side-chain dynamics. We chose specific labelling of isoleucine methyl groups as these are well-dispersed among the PDZ-domains providing eleven specific probes. First, we measured [1H,13C]-NMR spectra at different temperatures between 25°C and 50°C (Fig. 5a). Comparing the spectra at 50°C and 25°C revealed that a subset of peaks underwent severe line-broadening (e.g. I304δ1, I310δ1, I318δ1, I371δ1, I408δ1) accompanied by chemical shift changes (Fig. 5b,c). Whereas the latter can be attributed to a slight change in compactness of the protein core, which is also evidenced by the generally decreased global backbone dynamics (Fig. 4d), the lack of signal intensity for a subset of methyl groups indicates the existence of conformational exchange at the lower temperatures. To assess the nature of these motions we first determined the product of the side-chain order parameters and the correlation time of the overall molecular tumbling (S2•τC), providing insight about the extent of the amplitude of motions of the methyl axis in the picosecond regime (Fig. 5d, e). The obtained residue-wise profiles at 37°C indicate a large degree of flexibility for the studied methyl-groups. Whereas groups residing in the core of the PDZ2-domain show the least extend of motions on the ps–ns timescale, methyl groups in the core of the PDZ1 uniformly show a larger degree of flexibility to a similar extent as observed for the backbone fast-timescale motions (Fig. 4a).

a, Isoleucine methyl region of a 2D [13C,1H]-NMR spectrum of [U-13C,15N]-PDZ1–PDZ2 at different temperatures as indicated. b, c, The temperature dependence of the isoleucine signal intensities (b) and chemical shifts (c) for the PDZ1-PDZ2 construct. The broken lines in panel b serve as a guide to the eyes only. d, e, Local isoleucine methyl-group dynamics on the ps–ns timescale probed by methyl triple-quantum (TQ) relaxation experiments measurements 37,38 at 37°C showing the product of the local order parameter and the overall tumbling constant, S2•τC. The isoleucine methyl groups of PDZ1-PDZ2 construct are shown as spheres and the obtained by the green to the blue gradient (d). S2•τC. values plotted against the sequence (e). f, Exemplary 13C methyl CPMG relaxation dispersion profiles at different temperatures as indicated. Non-flat profiles indicate ms-dynamics. g, ΔR2eff values, obtained from the difference of R2eff at the lowest and highest CPMG frequency υCMPG. h, Structural view of the amplitude of the CPMG relaxation dispersion profiles ΔR2eff at 37°C. i, Rate constants of the dynamic process (τex) obtained from a global fit of the CPMG relaxation dispersion data.
As several methyl resonances appeared to be involved in conformational exchange processes we next quantified these by employing multiple quantum Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion measurements 39, sensing motions on the micro–millisecond timescale. Measuring CPMG experiments at temperatures ranging 25–50°C revealed that the residues experiencing μs–ms motions reside mainly in PDZ1 domain, namely methyl-groups I304δ1, I310δ1, I318δ1, as evidenced by non-flat dispersion profiles (Fig. 5f–h). Quantitative analysis of the dispersion data showed that this motion occurred on a 0.7 ms (25°C) and 0.8 ms (37°C) timescale (Fig. 5i). The observation of the increased timescale of the side-chain motions at 37°C compared to 0.7 ms at 43°C and 0.4 ms at 50°C, indicates pronounced modulations of inherent side-chain dynamics, which is directly coupled to the M280 dynamics, following a similar temperature-dependent modulation of its backbone conformational exchange (Fig. 4f,g). Having established the role of the isoleucine methyl groups in driving the conformational switch, we next set-out to confirm this observation within the full-length DegP. Based on the side-chain assignment of the isolated PDZ1-PDZ2-domain construct (Supplementary Fig. 2e) we were able to unambiguously assign all eleven resonances within the DegP full-length construct (Fig. 6a,b). In order to study the transformation from the low temperature hexameric-state to the high-temperature trimeric-state within full-length DegP, we measured [1H,13C]-NMR spectra over the temperature range from 25°C to 50°C (Fig. 6c). At the low temperatures, several residues (e.g. I310δ1, I408δ1) show reduced intensity comparable to the situation within the PDZ1-PDZ2 construct. Using the measured intensities for I310δ1 and I408δ1 from 25°C to 50°C together with transition state theory we extracted thermodynamic values for the transition from the locked to the open state. Fitting the data to a 2-state model yielded an activation enthalpy of ΔH=241.7 ± 88 kJ mol−1 and an entropy of ΔS=0.78 ± 0.14 kJ mol−1 K−1 with a transition midpoint at 36.8 ± 1.3°C. The obtained activation enthalpy is perfectly in line with the obtained values for a comparable methionine–phenylalanine interaction motif of 110 kJ/mol 57, providing further indication of an sulfur-π aromatic motif stabilizing the intermolecular PDZ1–PDZ2 interaction.

a, 2D [13C,1H]-NMR spectrum of [U-2H, Ile-δ1-13CH3]–DegP at 50°C. The indicated resonance assignments were transferred from the assigned PDZ1-PDZ2 construct. b, Chemical shift changes of the isoleucine methyl groups of DegP compared to the PDZ1-PDZ2 (bottom). The isoleucine methyl groups of one DegP monomer are indicated with spheres (top), and the chemical shift changes compared to the PDZ1-PDZ2 construct are indicated by the yellow to red gradient. c, Excerpt of the isoleucine methyl region of a 2D [13C,1H]-NMR spectrum of [U-2H, Ile-δ1-13CH3]–DegP at different temperatures as indicated. d, The temperature dependence of the selected I310δ1 and I408δ1 signal intensities. Solid lines represent a fit to a 2-state model for the extraction of thermodynamic parameters. Inset shows the populations of the open and the locked state at the different temperatures.
Conclusion
Despite being studied extensively and the existence of several high-resolution crystal structures of DegP in different states multiple questions remained about how its structural transitions are achieved. Within the known crystal structures of the hexameric state, the DegP12 and DegP24 structures, an intermolecular PDZ1-PDZ2 interaction always coordinates the arrangements of the DegP trimers to stabilize larger oligomeric assemblies 12. Thereby, the structures indicated a distinct arrangement of the PDZ-domains for the hexameric state compared to the larger oligomeric states (Supplementary Fig. 1f,g). However, our solution NMR results presented here clearly show that in solution DegP is locked in the hexameric state via the same M280–Y444 interaction as observed initially within the larger oligomeric states indicating that this interaction is crucial for modulating the different oligomeric states within DegPs functional cycle. Although the participation of Y444 was already known based on the characterized DegPY444A mutant 14, our data revealed that the M280 is not only participating in this interaction, but effectively driving the temperature dependent transition.
Studying the backbone in combination with the methyl-group side-chain dynamics over the transition temperature range revealed inherent dynamics within the core of the PDZ1 domain (I304, I310) which likely allosterically trigger the conformational transition of M280 between its locked and open state by dynamics on different timescales. Our data clearly indicates that this interaction is stabilized via a sulfur-π interaction of the M280 and Y444 side-chains, a motif lately receiving recognition as an important stabilizer of protein interactions 50,57. Taken together our backbone and side-chain dynamical data draw a detailed picture of how fine-tuning the dynamics of PDZ1 domain governs the temperature dependent oligomerization of DegP (Fig. 7). Whereas the lack of motions at 25°C directly explains the locked conformation at low temperatures, the situation is presumably different at 37°C, where we observed the largest exchange contributions. The large amplitude of motions at this temperature (Figs. 4g and 5i) reveals a fine-tuned dynamical compensation, which can easily adapt to slight changes in temperature to either lock the interaction (at lower temperature) or unlock the interaction and consequently activate also the protease function of DegP (at higher temperature).

In cellular ground-states (e.g. low temperature) the dynamics of M280 are reduced to stabilize the PDZ1-PDZ2 interaction via the sulfur-π interaction between there side-chains. At 37°C the increased motions in helix α1 accommodate for local motions in the whole protein still resulting in a rather 1/1 ratio of open and closed conformations. Under heat shock conditions, M280 exhibits still a large degree of flexibility due to the lack of interaction partner, as it cannot compensate anymore for the overall increased dynamics and the PDZ1-PDZ2 interaction is broken.
Remarkably, the involved residues, M280 and Y444, are highly conserved within different HtrA-proteins carrying two PDZ-domains (Supplementary Fig. S8). Our finding might therefore be indicative of a wide-spread mechanism as it is already known that carboxy-terminal aromatic/hydrophobic residues in a variety of mammalian multi-PDZ domain proteins govern their homo- and heterodimer formation suggesting a possible evolutionary conserved mechanism of stabilizing PDZ-PDZ interactions 58,59.
Author Contribution
B.M.B. conceived the study and designed the experiments together with D.Š.. D.Š. performed all experimental work. D.Š., J.T., and B.M.B. analyzed and discussed the data. D.Š., J.T., and B.M.B. wrote jointly the paper.
Supporting information

a,b,c, 2D [15N,1H]-NMR spectrum of [U-2H,15N]-DegP measured at the indicated temperatures. d, Gel elution profiles of DegP variants. Recorded at 8°C in NMR-buffer on a Superdex S200increase column. The column void volume and the molecular weights of a standard calibration curve are indicated. e, Secondary chemical shifts in a solution of the full-length DegPY444A protein variant compared to the hexameric X-ray structure (PDB-ID: 1KY9). f,g, Comparison of the PDZ domains orientation between different monomers in the crystal structures of the hexamer (f; PDB-ID: 1KY9) and 24–meric cage assembly (g; PDB-ID: 3CS0). Whereas in the hexameric state this interaction is proposed to be mediated mainly via a direct interaction between PDZ1s, PDZ2s are found to be non-constraint at the hexamer edges, however within the 24-mer the interaction between PDZ1 and PDZ2 of different molecules is achieved by a Y444 and M280 interaction indicating stabilization by a sulfur-π-aromatic motif.

a, 2D [15N,1H]-NMR-spectrum of [U-15N,13C]-PDZ1–PDZ2. The sequence-specific resonance assignment obtained from 3D triple-resonance experiments is indicated. b, Cartoon representation of the crystal structure of the PDZ1–PDZ2 domains (PDB-ID: 1KY9) with the assigned resonances highlighted in orange and unassigned residues in grey. c, Secondary backbone 13C chemical shift analysis plotted against the PDZ1–PDZ2 residues. Dark shaded bars present residues experiencing line-broadening in [15N,1H]-NMR-spectra that could be assigned unambiguously via [13C,1H]-NMR-spectra based TOCSY-type experiments. Positive values indicate α-helix whereas negative values β-sheet secondary structure elements. The secondary structure elements derived from the crystal structure (PDB-ID: 1KY9) are highlighted in grey. d, Representative strips for residues G74–L83 from a 3D HNCACB spectrum of the PDZ1-PDZ2-domains. e, Methyl-region of a 2D [13C,1H]-NMR-spectrum of [U-15N,13C]-PDZ1–PDZ2 with the assignment of the methyl side-chain-resonances indicated.

a, 2D [15N,1H]-NMR-spectrum of [U-15N,13C]-PDZ1–PDZ2 at 25°C. The sequence-specific resonance assignment obtained from 3D triple-resonance experiments is indicated. b, Secondary backbone 13C chemical shift analysis plotted against the PDZ1–PDZ2 residues. c, Difference of the secondary chemical shifts at the different temperatures (Δ(Δδ(13Cα)−Δδ(13Cβ)) are shown in dark red. d, Combined chemical shift differences of the amide moieties plotted versus the PDZ1–PDZ2 amino-acid residue number.

a, 2D [15N,1H]-NMR-spectrum of [U-15N,13C]-PDZ1. The sequence-specific resonance assignment obtained from 3D triple-resonance experiments is indicated. b, Representative strips for residues A299–D308 from a 3D HNCACB spectrum of the PDZ1-domain. c, 2D [15N,1H]-NMR-spectrum of [U-15N,13C]-PDZ1. The sequence-specific resonance assignment obtained from 3D triple-resonance experiments is indicated. d, Representative strips for residues A398–D407 from a 3D HNCACB spectrum of the PDZ1-domain. e,f, Secondary backbone 13C chemical shift analysis plotted against the PDZ1 (e) and PDZ2 (f) residues. The secondary structure elements derived from the crystal structure (PDB-ID: 1KY9) are highlighted in grey.
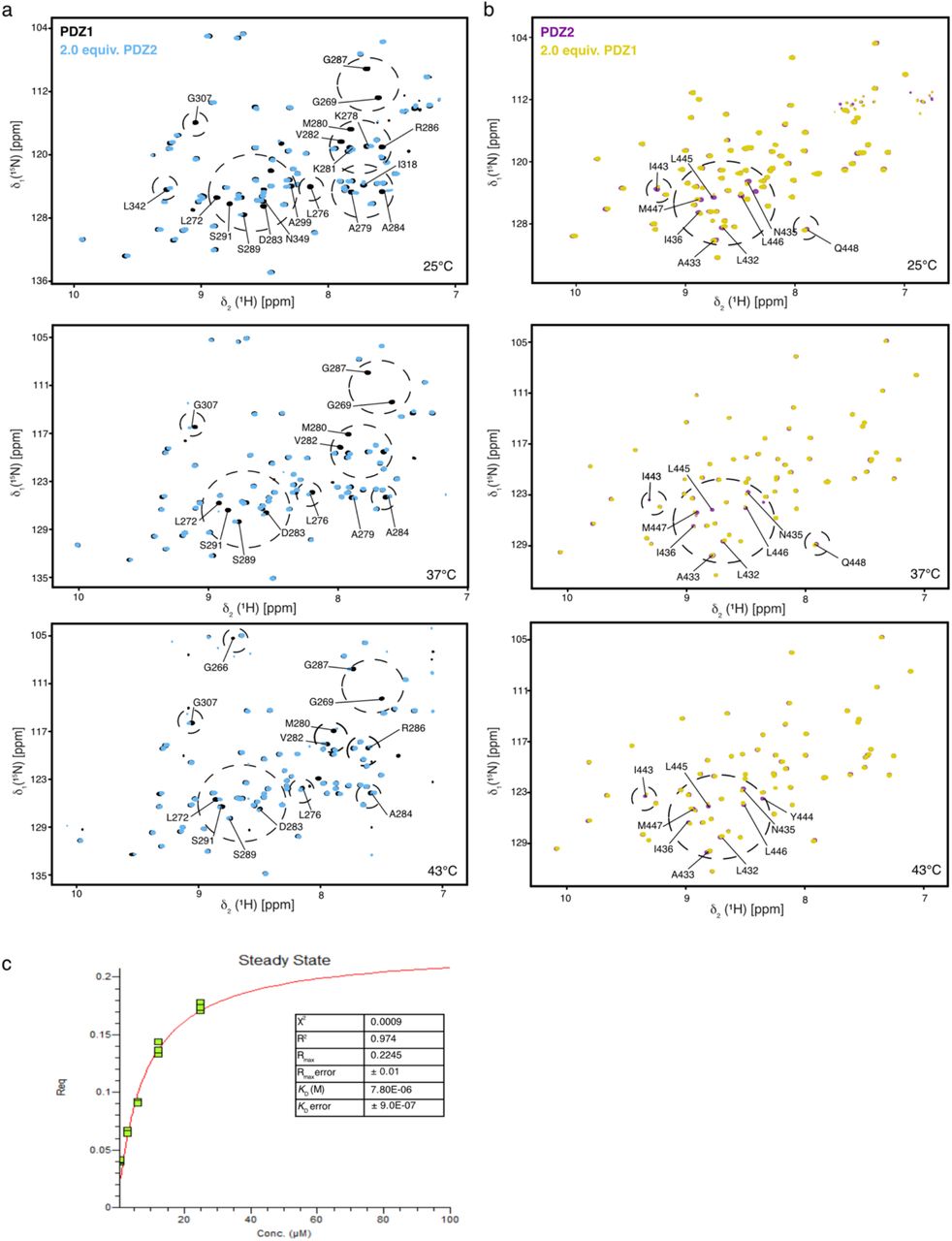
a, b, 2D [15N,1H]-NMR spectra of [U-15N]-PDZ1 (a, purple) and [U-15N]-PDZ2 (b, black) as well as after addition of two molar equivalents of the respective other domain. Titrations were measured at 25°C, 37°C and 43°C, as indicated. c, Steady-state analysis of the BLI-curves shown in Fig. 2f. Curves were measured and analyzed in triplicates (green data points). The red curve is the least-squares fit to the data. The resulting KD and indicators for the fitting quality (R2, χ2) are indicated.

a, b, 2D [15N,1H]-NMR spectra of [U-15N]-PDZ1M280A (a, purple) and [U-15N]-PDZ2Y444A (b, black) as well as after addition of two molar equivalents of the respective other wild-type domain. Titrations were measured at 25°C.
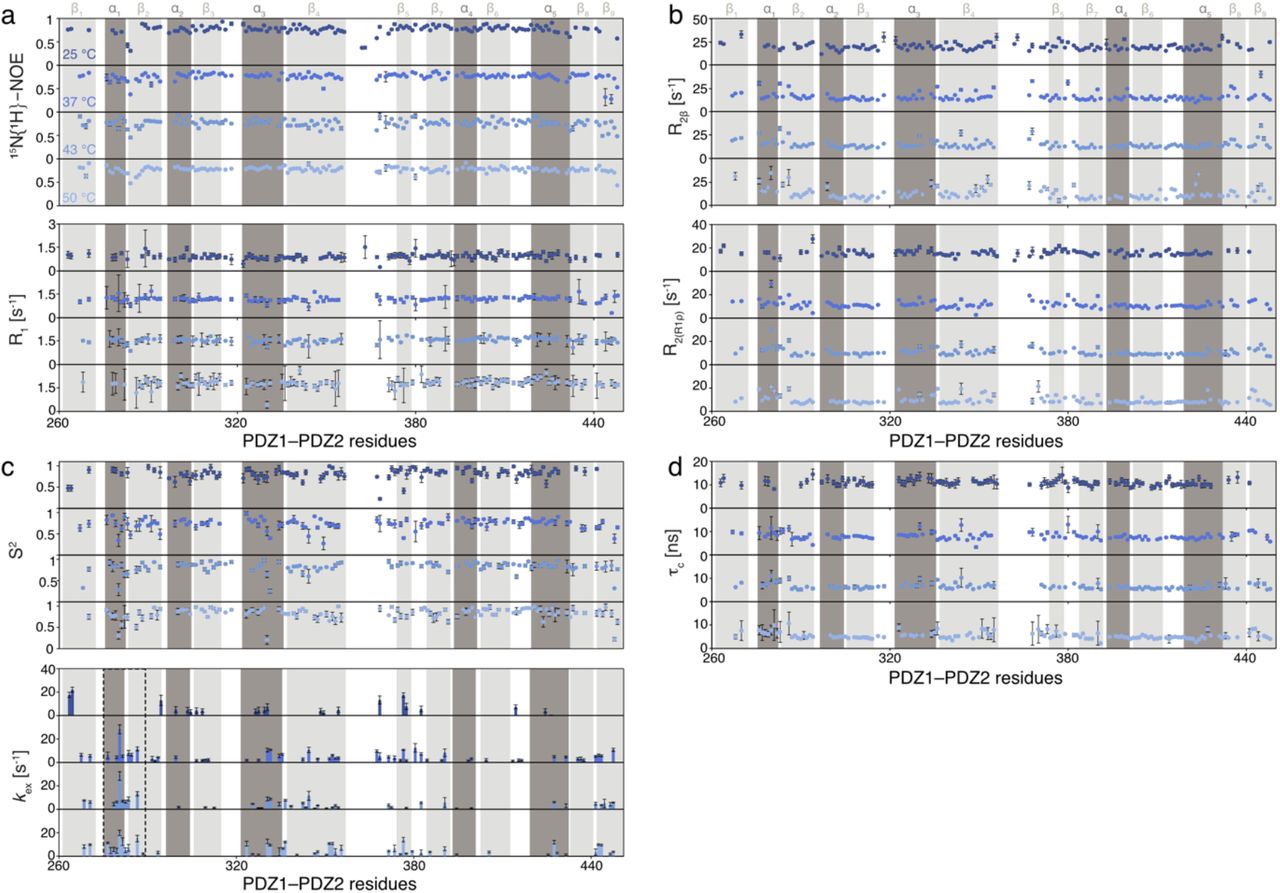
a, Local backbone dynamics on the ps–ns timescale probed by hetNOE and R1 measurements. b, Obtained transversal relaxation rates (R2β and R2(R1ρ)) reporting on μs-ms motions. d, Temperature dependence of the sub-nanosecond order parameter S2 and the exchange rate kex,, reporting on the flexibility on the μs–ms timescale, calculated using the Lipari-Szabo model-free approach. d, Rotational correlation time (τc) determined from the ratio of R1 and R2(R1ρ). All data are plotted against the PDZ1–PDZ2 residues at the indicated temperatures ranging from 25–50°C.

a,b, Cartoon (a) and surface (b) representation of the DegP PDZ1-PDZ2 domains displaying the degree of conservation with the indicated gradient ranging from 0 (variable) to 9 (conserved). Positions of M280, 7/9 conservation score, and Y444, 8/9 conservation score are indicated. Analysis was performed via the ConSurf (Tel Aviv University, Israel) server 1,2.
Plasmids and respective primers used in this study.
Acknowledgements
We thank G. Mas (Basel), P. Schanda (Grenoble), and R. Sprangers (Regensburg) for providing NMR pulse sequences. B.M.B. gratefully acknowledges funding from the Swedish Research Council and the Knut och Alice Wallenberg Foundation through a Wallenberg Academy Fellowship as well as through the Wallenberg Centre for Molecular and Translational Medicine, University of Gothenburg, Sweden. The Swedish NMR Centre of the University of Gothenburg is acknowledged for spectrometer time.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}