

Abstract
Genome in a Bottle (GIAB) benchmarks have been widely used to validate clinical sequencing pipelines and develop new variant calling and sequencing methods. Here we use accurate long and linked reads to expand the prior benchmark to include difficult-to-map regions and segmental duplications that are not readily accessible to short reads. Our new benchmark adds more than 300,000 SNVs, 50,000 indels, and 16 % new exonic variants, many in challenging, clinically relevant genes not previously covered (e.g., PMS2). We increase coverage of the GRCh38 assembly from 85 % to 92 %, while excluding problematic regions for benchmarking small variants (e.g., copy number variants and assembly errors) that should not have been in the previous version. Our new benchmark reliably identifies both false positives and false negatives across multiple short-, linked-, and long-read based variant calling methods. As an example of its utility, this benchmark identifies eight times more false negatives in a short read variant call set relative to our previous benchmark, mostly in difficult-to-map regions. To enable robust small variant benchmarking, we still exclude 3.6% of GRCh37 and 5.0% of GRCh38 in (1) highly repetitive regions such as large, highly similar segmental duplications and the centromere not accessible to our data and (2) regions where our sample is highly divergent from the reference due to large indels, structural variation, copy number variation, and/or errors in the reference (e.g., some KIR genes that have duplications in HG002). We have demonstrated the utility of this benchmark to assess performance in more challenging regions, which enables benchmarking in more difficult genes and continued technology and bioinformatics development. The benchmarks are available at: ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/AshkenazimTrio/HG002_NA24385_son/NISTv4.1/ ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/NIST_v4.2_SmallVariantDraftBenchmark_07092020/
Introduction
Advances in genome sequencing technologies have continually transformed biological research and clinical diagnostics, and benchmarks have been critical to ensure the quality of the sequencing results. The Genome in a Bottle Consortium (GIAB) developed extensive data1 and widely used benchmark sets to assess accuracy of variant calls resulting from human genome sequencing.2–4 The Global Alliance for Genomics and Health (GA4GH) Benchmarking Team develop tools and best practices to use these benchmarks.5 These benchmarks and benchmarking tools helped enable the development and optimization of new technologies and bioinformatics approaches, including linked reads,6 highly accurate long reads,7 deep learning-based variant callers,8,9 graph-based variant callers,10 and de novo assembly.11,12 However, these benchmarks did not cover some challenging regions that these new methods could access, including many known medically relevant genes.13,14 This limitation highlighted the need for improved benchmarks covering segmental duplications, the Major Histocompatibility Complex (MHC), and other challenging regions. A separate synthetic diploid benchmark was generated from assemblies of error-prone long reads for two haploid hydatidiform mole cell lines, but this had limitations both in terms of cell line availability and small indel errors due to the high error rate of the long reads.15
Many of the difficult regions of the genome lie in segmental duplications and other repetitive elements. Linked reads were shown to have the potential to expand the GIAB benchmark by 68.9 Mbp to some of these segmental duplications.6 A new circular consensus sequencing (CCS) method was recently developed that enables highly accurate 10 kb to 20 kb long reads.7 This technology identified a few thousand likely errors in the GIAB benchmark, mostly in LINEs. It had >400,000 variants in regions mappable with long reads but outside the benchmark, and it covered many medically relevant genes that are challenging to call using either short reads or lower accuracy long reads. GIAB recently used these data to produce a local diploid assembly-based benchmark for the highly polymorphic MHC region.16
Here, we use linked reads and long reads to expand GIAB’s benchmark to cover challenging genomic regions and better exclude structural and copy number variants for the openly-consented GIAB Ashkenazi trio from the Personal Genome Project.17 We used the diploid assembly-based MHC benchmark16 to cover most of the MHC region in our new benchmark set. We show that the new benchmark reliably identifies false positives and false negatives across a variety of short-, linked-, and long-read technologies.
Results
New benchmark covers more of the reference, including many segmental duplications
GIAB previously developed an integration approach to combine results from different sequencing technologies and analysis methods, using expert-driven heuristics and features of the mapped sequencing reads to determine at which genomic positions each method should be trusted. This integration approach excludes regions where all methods may have systematic errors or locations where methods produce different variants or genotypes and have no evidence of bias or error. While the previous version (v3.3.2) primarily used a variety of short-read sequencing technologies and excluded most segmental duplications,4 our new v4.1 benchmark adds long- and linked-reads to cover 6% more of the autosomal assembled bases for both GRCh37 and GRCh38 than v3.3.2 (Table 1). We also replace the mapping-based benchmark with assembly-based benchmark variants and regions in the MHC.16 v4.1 includes more than 300,000 new SNVs and 50,000 INDELs compared to v3.3.2. In Methods, we detail the creation of the v4.1 benchmark, including using the new long- and linked-read sequencing data in the GIAB small variant integration pipeline, and identifying regions that are difficult to benchmark, including potential large duplications in HG002 relative to the reference as well as problematic regions of GRCh37 or GRCh38.
Summary comparison between v3.3.2 and v4.1 HG002 benchmark set for chromosomes 1 to 22 in GRCh37 and GRCh38, including coverage of segmental duplications (Seg Dups) and regions that appear similar to short reads (i.e., “low mappability” regions where 100 bp read pairs have <=2 mismatches and <=1 indel difference from another region of the genome).
Many of the benchmark regions new to v4.1 are in segmental duplications and other regions with low mappability for short reads (Figure 1 and Table 1). GRCh38 has 270,860,615 bases in segmental duplications and low mappability regions (regions difficult to map with paired 100 bp reads) on chromosomes 1 to 22, including modeled centromeres. v4.1 covers 145,271,904 (53.6%) of those bases while v3.3.2 covers 65,714,199 (24.3%) bases. However, v4.1 still excludes some difficult regions: of the bases in GRCh38 chromosomes 1-22 not covered by v4.1, segmental duplication and low mappability regions account for 57.7% of those bases.

(A) Percent of the genomic region that is covered by v3.3.2 and v4.1 including difficult, non-gap reference, low mappability regions and segmental duplications, medically-relevant genes, and the MHC. (B) The number of unique SNVs by genomic context. Circle size indicates the total number of SNVs in the union of V3.3.2 and V4.1. Circles above the diagonal indicate there is a net gain of SNVs in the newer benchmark, and circles below the diagonal indicate a net loss of SNVs in the newer benchmark.
To identify the types of genomic regions where v4.1 gains and loses benchmark variants relative to v3.3.2, we compared the variant calls and used the v2.0 GA4GH/GIAB stratification files. Figure 1B highlights stratified genomic regions with the largest SNV gains and losses in v4.1 vs. v3.3.2 (the full table is available in Supplementary Table 1). As expected, the inclusion of linked- and long-reads leads to more variants in v4.1 than v3.3.2 in segmental duplications, self chains, the MHC region, as well as other regions that are difficult to map with short reads. The gain in v4.1 relative to v3.3.2 is lower in tandem repeats and homopolymers because v4.1 excludes any tandem repeats and homopolymers not completely covered by the benchmark regions. Inclusion of partially covered tandem repeats and homopolymers in v3.3.2 caused some errors in benchmarking results when only part of a complex variant in these repeats was called in v3.3.2. We show the benchmark overview for GRCh37 in Supplementary Figure 1.
In addition expanding coverage of difficult regions, v4.1 also corrects or excludes errors in v3.3.2. In previous work, variants called from PacBio HiFi were benchmarked against v3.3.2, and 60 SNV and indel putative false positives were manually curated, which identified 20 likely errors in v3.3.2.7 All 20 variants were corrected in the v4.1 benchmark or removed from the v4.1 benchmark regions. Twelve of these errors in v3.3.2 result from short reads that were only from one haplotype, because reads from the other haplotype were not mapped due to a cluster of variants in a LINE; two of these v3.3.2 errors are excluded in v4.1, and 10 variants are correctly called in v4.1 (Supplementary Table 2). In order to verify the new v4.1 variants incorrectly called by v3.3.2 in LINEs, we confirmed all 49 tested variants in 4 LINEs using Long-range PCR followed by Sanger sequencing, as described in Methods.
New benchmark covers additional challenging genes
To focus analysis on potential genes of interest, we analyzed benchmark coverage of genes previously identified to have at least one exon that is difficult to map with short reads, which we call “difficult, medically-relevant genes”.13 v4.1 covers 88 % of the 10,009,480 bp in difficult, medically-relevant genes” on primary assembly chromosomes 1-22 in GRCh38, much larger than the 54% covered by v3.3.2 (Table 2). 3,913,104 bp of the difficult, medically-relevant genes lie in segmental duplication or low mappability regions. The v4.1 benchmark covers 2,917,097 bp (74.5%) of those segmental duplications and low mappability regions while the v3.3.2 benchmark covers 208,882 bp (5.3%). Future work will be needed to cover 22 of the 159 genes on chromosome 1-22 that still have less than 50% of the gene body covered. For example, 5 genes that have potential duplications in HG002 were previously partially covered by v3.3.2 but are excluded in v4.1 because new methods will be needed to resolve and represent benchmark variants in duplicated regions (Figure 2B). For example, the medically-relevant gene KIR2DL1 was partially covered by v3.3.2 but is now completely excluded because v4.1 does a better job excluding duplicated regions, specifically because it excludes regions with higher than normal PacBio HiFi and/or ONT coverage (Figure 3). We summarize the corresponding statistics and summarize results for GRCh37 in Supplementary Table 3 and Supplementary Figure 2.

(A) Cumulative distribution for percent of each gene covered by HG002 v4.1 benchmark regions for difficult, medically-relevant genes. Dashed lines indicate that the number of genes with > 90% coverage increased from 19 in v3.3.2 to 108 in v4.1. (B) Pairwise comparison of medically-relevant gene coverage by benchmark set. Genes falling on the dashed line have similar coverage between the two benchmark sets, whereas genes above (red fill) or below (blue fill) the dashed line have higher coverage in the v4.1 or v3.3.2 benchmark sets, respectively. The genes with higher coverage in v4.1 tend to be in segmental duplications and the few genes with higher coverage in v3.3.2 are mostly genes duplicated in HG002 relative to GRCh38 and should be excluded.

The medically-relevant gene KIR2DL1 was partially covered by v3.3.2 but is correctly excluded by v4.1 due to a likely duplication. The gene has PacBio HiFi and ONT coverage about two times higher than normal coverage, so the region is excluded from all call sets in the v4.1 integration process.
Benchmark coverage of 159 medically relevant genes on chromosomes 1-22 previously identified as difficult for short reads. Coverage is the total bp covered by each benchmark set and percent of bases in the gene set.
As an example of a medically important gene with increased coverage by v4.1, PMS2, a gene involved with DNA mismatch repair, is covered by 2 large and 1 smaller segmental duplication (Figure 4). Variant calling in PMS2 is complicated by the presence of the pseudogene PMS2CL, which contains identical sequences in many of the exons of PMS2 and is within a segmental duplication.18 PMS2 is now covered more by v4.1 (85.6%) than by v3.3.2 (25.9%). Using Long Range PCR followed by Sanger sequencing, we tested 95 of the new v4.1 benchmark variants in PMS2 and 9 other difficult, medically-relevant genes, and all 95 were confirmed. Detailed Sanger sequencing results are in Supplementary Table 4.

The medically-relevant gene PMS2 is 85.6% covered by the v4.1 benchmark regions while it is 25.9% covered by v3.3.2 because segmental duplications (Segdups) were largely excluded in previous benchmark versions.
Comparison to Platinum Genomes identifies fewer potential errors in v4.1
Platinum Genomes found SNVs that were Mendelian inconsistent due to being called heterozygous in all 17 individuals in a pedigree with short read sequencing (“Category 1” errors).19 Some of these heterozygous calls result from regions duplicated in all individuals in the pedigree relative to GRCh37. Therefore, Category 1 SNVs matching SNVs in our benchmarks may identify questionable regions that should be excluded from the benchmark regions. 325 Category 1 SNVs matched HG002 v4.1 SNVs, a decrease relative to the 719 Category 1 SNVs matching HG002 v3.3.2 SNVs. This suggests that v4.1 better excludes duplications in HG002 relative to the reference even as it expands into more challenging segmental duplication regions. However, the remaining 325 matching SNVs may be areas for future improvement in v4.1. Manual curation of 10 random SNVs in HG002 v4.1 that matched Category 1 variants showed 5 were in possible duplications that potentially should be excluded, and 5 were in segmental duplication regions that may have been short read mapping errors or more complex variation in segmental duplications (Supplementary Table 5). Particularly, the v4.1 variants matching Category 1 variants in clusters are likely errors in v4.1. In addition, relative to the short read-based Platinum Genomes benchmark regions, the v4.1 benchmark regions have substantially fewer small gaps that can cause problems when benchmarking,4 so that the NG50 size of benchmark regions in v4.1 is more than two times greater than Platinum Genomes (Supplementary Figure 5).
High Mendelian Consistency in Trio
To further evaluate the accuracy of the benchmark, we formed similar benchmark sets for the HG002’s parents (HG003 and HG004) and performed a trio analysis to identify variants that have a genotype pattern inconsistent with Mendelian inheritance. This identified 2,554 Mendelian inconsistencies out of the 4,984,043 variants in at least one member of the trio and in the intersection of the benchmark regions for the trio (0.05%). We separately analyzed Mendelian inconsistent variants that were potential cell line or germline de novo mutations (that is, the son was heterozygous and both parents were homozygous reference), and those that had any other Mendelian inconsistent pattern (which are unlikely to have a biological origin). Out of 2,554 Mendelian violations in the Ashkenazi son, 1,185 SNVs and 284 INDELs were potential de novo mutations, 75 more SNVs and 71 more INDELs than in v3.3.2.4 Following the manual inspection of ten random de novo SNVs, 10/10 appeared to be true de novo. After manual inspection of ten random de novo indels, 10/10 appeared to be true de novo indels in homopolymers or tandem repeats. The violations that were not heterozygous in the son and homozygous reference in both parents fell in a few categories: (1) clusters of variants in segmental duplications where a variant was missed or incorrectly genotyped in one individual, (2) complex variants in homopolymers and tandem repeats that were incorrectly called or genotyped in one individual, and (3) some overlapping complex variants in the MHC that were correctly called in the trio but the different representations were not reconciled by our method (even though we used a method that is robust to most differences in representation).4,20 We exclude these Mendelian inconsistencies that are unlikely to have a biological origin from the v4.2 benchmark regions of each member of the trio.
Regions excluded from the benchmark
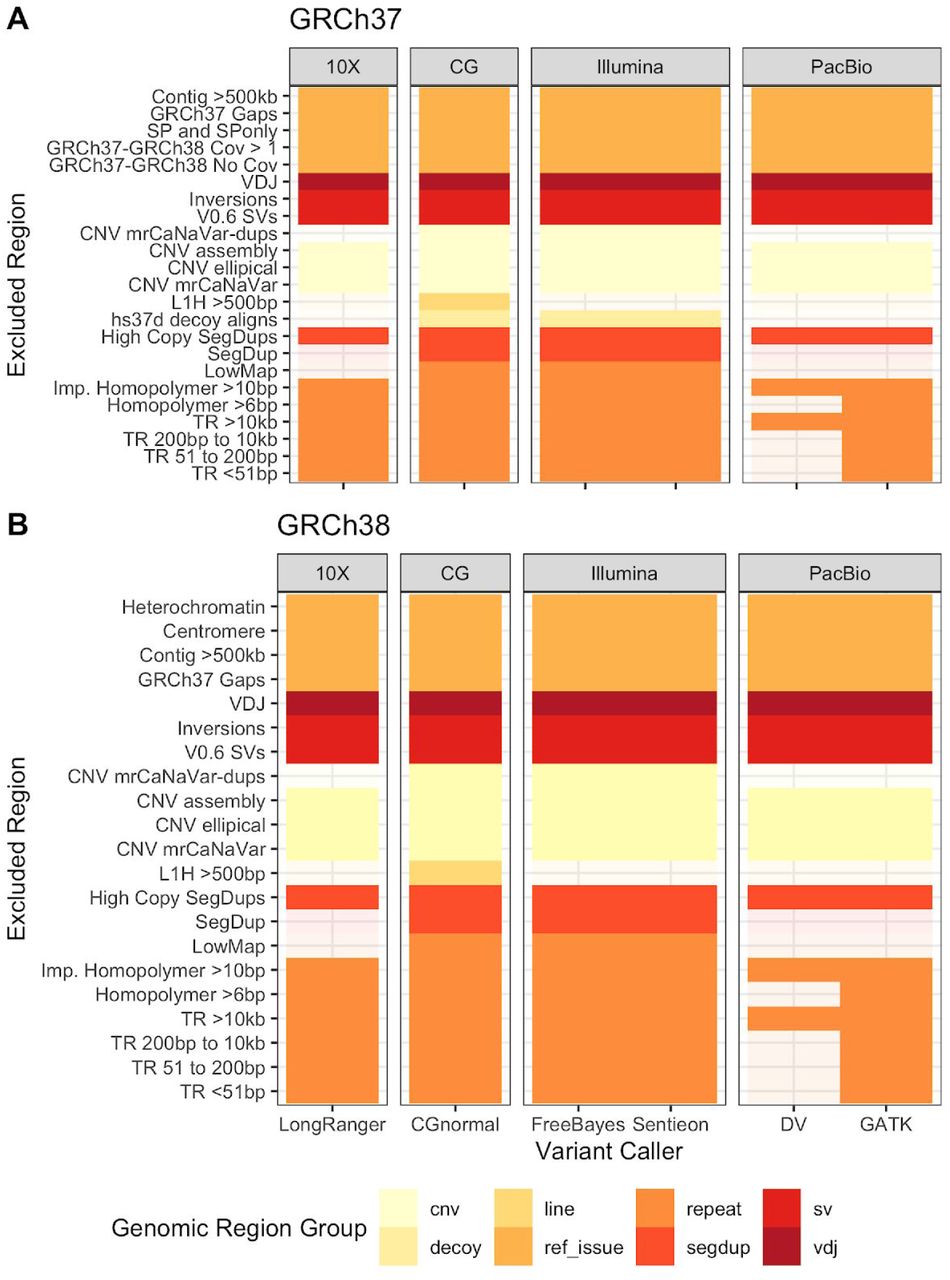
A critical part of forming a reliable v4.1 benchmark was to identify regions that should be excluded from the benchmark. In Table 3 and Supplementary Figure 6, we detail each region type that is excluded, the size of the regions, and reasons for exclusion. We describe how each region is defined in Methods, and Supplementary Note 2 describes refinements to these excluded regions between the initial draft release and the v4.1 benchmark. These excluded regions fall in several categories: (1) the modeled centromere and heterochromatin in GRCh38 because these are highly repetitive regions and identifying biases between technologies is not possible at this time; (2) the VDJ, which encodes immune system components and undergoes somatic recombination in B cells; (3) in GRCh37, regions that are either expanded or collapsed relative to GRCh38; (4) segmental duplications with greater than 5 copies longer than 10 kb and identity greater than 99 %, where errors are likely in mapping and variant calling, e.g., due to structural or copy number variation resulting in calling paralogous sequence variants;21,22 (5) potential large duplications that are in HG002 relative to GRCh37 or GRCh38; (6) inversions identified in HG002 as well as the GIAB v0.6 Structural Variant benchmark Tier1 plus Tier2 regions; (7) tandem repeats larger than 10,000 bp where variants can be difficult to detect accurately given the length of PacBio HiFi reads. As an example of the importance of carefully excluding questionable regions, when comparing variants from ultralong reads to v3.3.2, 74 % of the putative FPs in HG002 on GRCh38 fell outside the v4 benchmark regions (see Supplementary Tables 6-7). Many of these were in centromere regions that have very few benchmark variants but were erroneously included in the v3.3.2 short read-based benchmark, e.g., in chr20. Our new benchmark correctly excludes these regions from the benchmark because they cannot be confidently mapped with short-, linked-, or long-reads used to form the benchmark.
Base pairs covered by different types of difficult regions that are excluded from all input callsets. The table shows progressive subtraction of other difficult regions so each row has all rows above it subtracted before calculating base pairs covered. In non-gap regions on chromosomes 1-22, there are 158,665,407 bp in GRCh37 and 202,792,164 bp in GRCh38 that are not characterized by v4.1 (i.e., outside the v4.1 benchmark regions).
Evaluation and manual curation
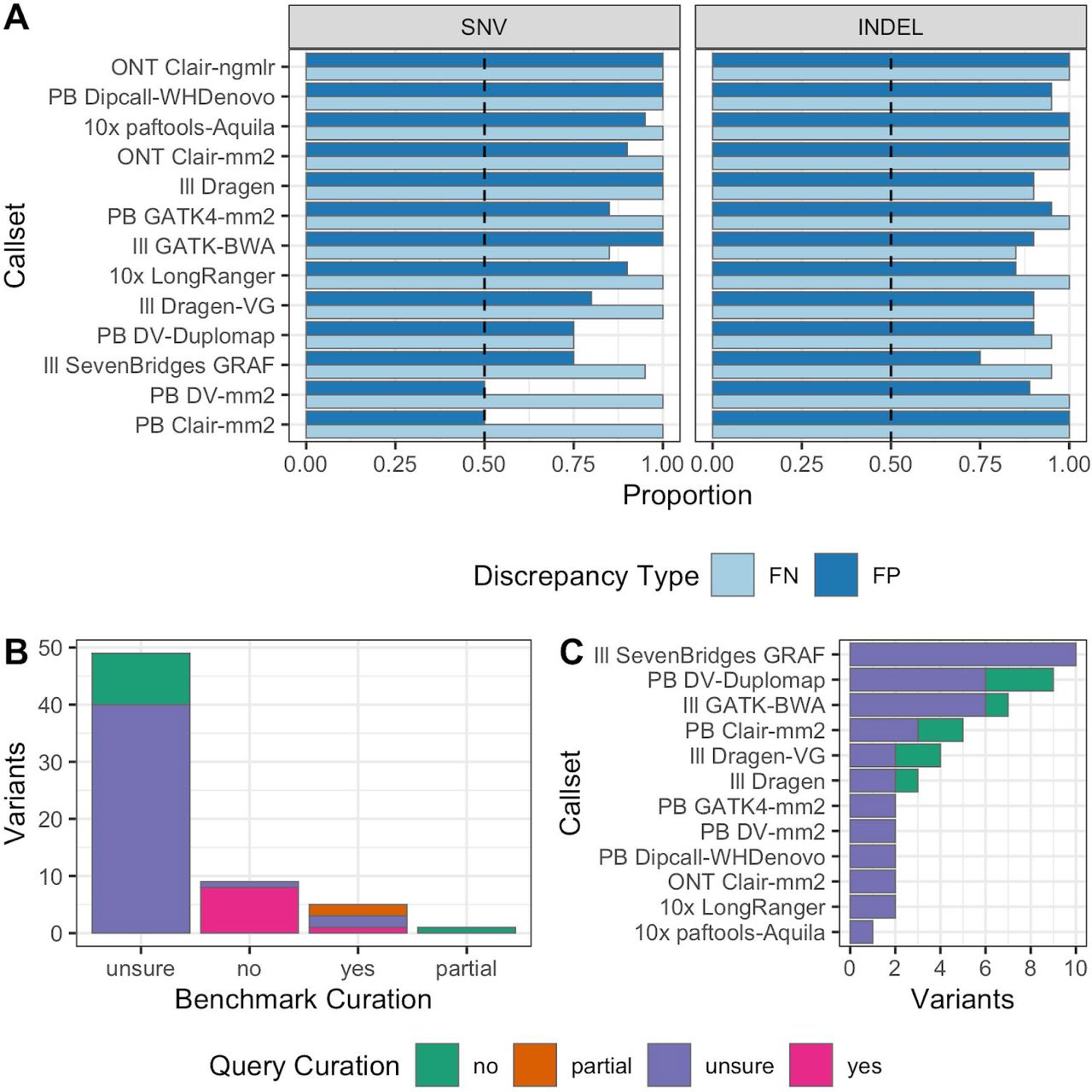
GIAB has established a community evaluation process for draft benchmarks before the official release.3 GIAB recruited volunteer experts in particular variant calling methods to follow the GA4GH Benchmarking Team’s Best Practices5 to compare a variety of query variant call sets to the draft benchmarks. Query call sets, representing a broad range of sequencing technologies and bioinformatics methods, are detailed in Supplementary Table 8 and Supplementary Note 1. Each callset developer curated a random selection of FPs and FNs to ensure the benchmark reliably identifies errors in the query callset. Overall, we found that the benchmark was correct and the query callset was not correct in the majority of FP and FN SNVs and Indels (Figure 5 with all curations in Supplementary Table 9). Some technologies/variant callers, particularly deep learning-based variant callers using HiFi data, had more sites where it was unclear if the benchmark was correct or the query callset was correct. These sites tended to be near regions with complex structural variation or places that appeared to be inside potential large duplications in HG002 but were not identified in our CNV approaches. In general, most sites that were not clearly correct in the benchmark and wrong in the query were in regions where the answer was unclear with current technologies (Figure 5B). Supplementary Figure 7 shows a region for one of these sites that we are unsure which callset is correct. The v4.1 benchmark correctly excludes much of this questionable region, but still includes some small regions where there may be a duplication and the variant calls both in the benchmark and the query are questionable. Future work will be aimed at further refining the benchmark in these questionable regions, but these evaluations demonstrate the v4.1 benchmark reliably identifies both FPs and FNs across a large variety of variant callsets, including those based on short, linked, and long reads, as well as mapping-based, graph-based, and assembly-based variant callers.

(A) The proportion of curated FP and FN variants by callset where the benchmark set was correct and the query callset was incorrect. The dashed black line indicates the majority threshold, 50%. Curated variants from both GRCh37 and GRCh38 (20 total) were used to calculate proportions. (B) Breakdown ofthe total number ofvariants by manual curation category, excluding variants from panel A where the benchmark was deemed correct and query incorrect, showing most of these sites were difficult to curate. (C) Benchmark unsure variants by callset. Technology abbreviations are: ONT=Oxford Nanopore, PB=PacBio HiFi, Ill=Illumina PCR-free, 10x=10x Genomics
More FNs are identified by the new benchmark
We demonstrate the benchmarking utility of v4.1 by comparing an example query call set to the new and old benchmark sets for HG002. For a standard short read-based call set (Ill GATK-BWA in Figure 5), the number of FNs identified by v4.1 was 8.6 times higher than by v3.3.2 (16,780 vs. 1,960). The difference is largely due to FN SNVs in regions of low mappability and segmental duplications with 15,291 in v4.1 vs. 1,381 in v3.3.2. The more challenging variants included in v4.1 will allow further optimization and development of variant callers in segmental duplications and low mappability regions.
Discussion
We present the first diploid small variant benchmark that uses short-, linked-, and long-reads to confidently characterize a broad spectrum of genomic contexts, including non-repetitive regions as well as repetitive regions such as many segmental duplications, difficult to map regions, homopolymers, and tandem repeats. We demonstrated that the benchmark reliably identifies false positives and false negatives in more challenging regions across many short-, linked-, and long-read technologies and variant callers based on traditional methods, deep learning,8,9 graph-based references,10 and diploid assembly.12
We designed this benchmark to cover as much of the human genome as possible with current technologies, as long as the benchmark genome sequence is structurally similar to the GRCh37 or GRCh38 reference. As a linear reference-based benchmark, it has advantages over global de novo assembly-based approaches by using reference information to resolve some of the segmental duplications and other repeats where our samples are similar to the reference assembly. This reference-based approach enables users to take advantage of the suite of benchmarking tools developed by the Global Alliance for Genomics and Health Benchmarking Team, including sophisticated comparison of complex variants, standardized performance metrics, and stratification by variant type and genome context.5 However, our approach necessitates carefully excluding regions where our reference samples differ structurally from GRCh37 or GRCh38 due to errors in the reference, gaps in the reference, CNVs, or SVs. Developing benchmarks in these regions will require the development of methods to characterize these regions with confidence (e.g., using diploid assembly), standards for representing variants in these regions, and benchmarking methodology and tools. For example, for variants inside segmental duplications for which the individual has more copies than the reference, methods are actively being developed to assemble these regions,21,23 but no standards exist for representing which copy the variants fall on or how to compare to a benchmark.
It is critical to understand the limitations of any benchmark. Because our current benchmark excludes regions that structurally differ from GRCh37 or GRCh38, it will not identify deficiencies in mapping-based approaches that rely on these references nor highlight advantages of assembly-based approaches that do not rely on these references. While we have tried to exclude all regions where our samples differ structurally from the reference, some regions with gains in copy number remain, particularly in segmental duplications where these are more challenging to identify. Similarly, we may not exclude all inversions, particularly those mediated by segmental duplications. In addition, the benchmark still excludes many indels between 15 bp and 50 bp in size. Although we have significantly increased our coverage of difficult, medically-relevant genes, more work remains. Many of these genes are excluded due to putative SVs or copy number gains, and future work will be needed to understand whether these are true SVs or copy number gains, and if so, how to fully characterize these regions.
We expect that future benchmarks will increasingly use highly contiguous diploid assembly to access the full range of genomic variation. Our current benchmark is helping enable this transition by identifying opportunities to improve assemblies in the genome regions that are structurally similar to GRCh37 and GRCh38.
Online Methods
Incorporating 10x Genomics and PacBio HiFi reads into small variant integration pipeline
v4.1 uses the same variant call sets as v3.3.2 from Complete Genomics,24 Illumina PCR-free (novoalign, GATK, and freebayes), and Illumina mate-pair (bwa mem, GATK, and freebayes).25–27 v4.1 uses 10x Genomics linked-read data and the variant calls from the LongRanger pipeline6, in place of the conservative, haplotype-separated GATK calls from 10x Genomics used in v3.3.2. Also, v4.1 uses PacBio HiFi data using Sequel II with read lengths of 15kb and 20kb merged into a dataset that has approximately 52x coverage, with variants subsequently called by GATK4 and DeepVariant.7,8 The 10x and PacBio HiFi data provide access to genomic regions that were previously inaccessible to short reads including segmental duplications. As shown in Table 1 the number of base pairs in the benchmark that covers segmental duplications has increased with the incorporation of long- and linked-read data. Table 4 lists the input data sets for the small variant integration pipeline to produce v4.1.
Integration Pipeline Data Set Attributes. Complete Genomics and Illumina data sets were the same as those used in v3.3.2 and were previously described.1 Italicized datasets from 10x Genomics and PacBio HiFi were new for v4.1.
Generating Callable Files with Haplotype-Separated BAMs
We use the CallableLoci utility in GATK3 to find regions with good coverage of high mapping quality reads. For PacBio HiFi and 10x Genomics read data, we use WhatsHap28 haplotag to partition reads by haplotype then use the bamtools filter function to generate separate BAM files for the two haplotypes. For CallableLoci with the unseparated BAM, we set the callable maxDepth threshold to 2 times the median coverage for VCF entries, then the minDepth threshold to 20. For the haplotype separated BAMs, we use median coverage for VCF as the maxDepth and 5 as the minDepth.
For PacBio HiFi, we first remove multi-allelic entries from the VCF and 50 bp on each side of the variant then remove RefCall entries with QUAL value below 40 along with 50 bp on each side of those variants. We then find callable regions for each haplotype BAM and the unseparated BAM then use bedtools multiIntersectBed to find the union of those regions.
For 10x Genomics, we first remove filtered indels along with 50 bp on each side from its callable regions. Then we find callable regions on each haplotype and the unseparated BAM. After using multiIntersectBed to find the union of those callable regions we subtract regions that were callable on one haplotype but not callable on the other haplotype.
Python integration
We implemented the integration pipeline using Python as opposed to the Bash and Perl implementation for v3.3.2. The integration maintains a similar structure and we generated a DNAnexus applet to run on the same platform as v3.3.2. We updated the v4.1 pipeline to exclude all of a tandem repeat that is only partially covered by the benchmark regions. We also provide an option to not provide a callable file for given callsets, which for v4.1 we do not use callable regions for Ion Torrent or SOLiD. This results in a benchmark VCF that includes annotations for those technologies but variants are not excluded based on disagreement with their calls.
Regions excluded from the benchmark
We determined regions to exclude from the benchmark where it was not currently possible to determine which technologies were correct due to the difficulty of resolving variation in that region. The difficult regions included those that had a structural variant identified in the GIAB SV v0.6 Benchmark, regions in which the HG002 sample had a copy variation compared to the reference, high depth and highly similar segmental duplications, tandem repeats > 10kb, regions that are collapsed and expanded from GRCh37/38 Primary Assembly Alignments, modeled centromere and heterochromatin, VDJ, and inversions. We refined these regions with several rounds of internal and external evaluation on intermediate versions of the benchmark. We describe intermediate versions of the benchmark in Supplementary Note 2.
Modeled centromere and heterochromatin
We use the modeled centromere for GRCh38 from ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/NA12878_HG001/NISTv3.3.2/GRCh38/supplementaryFiles/genomic_regions_definitions_modeledcentromere.bed and the heterochromatin region ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/NA12878_HG001/NISTv3.3.2/GRCh38/supplementaryFiles/genomic_regions_definitions_heterochrom.bed.29
VDJ
We downloaded the UCSC genes tracks30 from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/kgXref.txt.gz and selected entries with “abParts”. We then subset to chromosomes 2, 14, and 22 which contain the IGK, IGH, and IGL components that make up the VDJ recombination regions.
Regions that are collapsed and expanded from GRCh37/38 Primary Assembly Alignments
The GRC aligned GRCh37 to GRCh38 (excluding alts) with results available at: ftp://ftp.ncbi.nlm.nih.gov/pub/remap/Homo_sapiens/2.1/GCF_000001405.13_GRCh37/GCF_000001405.26_GRCh38/. We parsed the file GCF_000001405.13.xlxs for two Discrepancy values: (1) SP that denotes collapsed regions and (2) SP-only that denotes a region that was expanded between the reference versions.
Highly similar and high depth segmental duplications longer than 10kb
We begin with the segmental duplications track from UCSC30: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/genomicSuperDups.txt.gz. We filter for entries larger than 10 kb and with identity > 99%. We then use bedtools genomecov to calculate segmental depth and subset to those greater than 5.
Potential copy number variation
We employed several approaches to find potential regions of large duplications in HG002 that are not in GRCh37 and GRCh38:
Short read and Long Read Intersection: We used mosdepth31 to find 1,000 bp windows that have higher than average coverage/2*2.5 in ONT and PacBio HiFi data. We intersected these regions with results from the CNV analysis tool, mrCaNaVar,32 on Illumina HiSeq 300x data (ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/BilkentUni_IlluminaHiSeq_TARDIS_mrCaNaVar_05212019/AJtrio-HG002.hs37d5.300x.bam.bilkentuniv.052119.dups.bed.gz and ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/BilkentUni_mrCaNaVaR_GRCh38_07242019/AJtrio-HG002.hg38.300x.bam.bilkentuniv.072319.dups.bed.gz).
Diploid Assemblies of HG002: We used SVRefine (https://github.com/nhansen/SVanalyzer) to align diploid assemblies to GRCh37/GRCh38 with bedgraph files that denote coverage of the reference by the number of contigs for the maternal and paternal haplotypes. We used bedtools unionBedGraphs and then found reference regions that are covered by 2 or more contigs in the union of haplotypes. We did this separately on a TrioCanu assembly using ONT,33 a Flye assembly using ONT,34 and a TrioCanu assembly of PacBio HiFi 15kb reads.7 We found an intersection across the three assemblies and subset to regions greater than 10 kb.
Elliptical Outlier Boundary with PacBio HiFi and ONT sequencing data: We used mosdepth to calculate coverage in 1,000 bp windows of the PacBio HiFi data and the ONT ultralong data set (ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/Ultralong_OxfordNanopore/guppy-V2.3.4_2019-06-26/ultra-long-ont_hs37d5_phased.bam and ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/Ultralong_OxfordNanopore/guppy-V2.3.4_2019-06-26/ultra-long-ont_GRCh38_reheader.bam). We then found regions that had outlier coverage in PacBio HiFi and/or ONT. To do so, we (1) divided the PacBio HiFi coverage of each window by the median depth HiFi depth and squared it; (2) divided the ONT coverage of each window by the median depth ONT depth and squared it; (3) summed those values; and (4) took the square root of the sum. We found the third quartile and interquartile range for those transformed window coverage values. Finally, we found windows with coverage greater than the third quartile plus 1.5 times the IQR.
Inversions
We used SVrefine (github commit f0fb99969b6e239d1f49bc64a8f6cf5d52a2b88b) to call structural variants with, --maxsize 1000000 option. We then extracted inversions from the call set. Variants were merged with SVmerge (github commit aa8beb6f1cb5c539eea9f980ff30d2648caeee21), default maximum “distances”, which were 0.2 for all. SVrefine and SVmerge were from SVanalyzer (https://github.com/nhansen/SVanalyzer).
v0.6 GIAB Tier1 plus Tier 2 SV Benchmark expanded by 150%
We used bedtools35 slop with params -b -pct .25 to expand the GIAB v0.6 Structural Variant benchmark file: ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6/HG002_SVs_Tier1plusTier2_v0.6.1.bed. This file defines regions in which calls with PASS in the FILTER field as well as regions should contain close to 100% of true insertions and deletions >=50 bp, with variants merged into a single region if they were within 1 kb.
Tandem Repeats greater than 10,000 bp
We took the union of SimpleRepeat dinucleotide, trinucleotide, and tetranucleotide STRs as well as RepeatMasker_LowComplexity, RepeatMasker_SimpleRepeats, and TRF_SimpleRepeats downloaded from UCSC Genome Browser. We then subset to tandem repeats longer than 10,000bp.
Reference assembly contigs shorter than 500,000 bp
We downloaded the gap track from UCSC30: ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/gap.txt.gz. Then subset to regions that are gap. We used bedtools complemented with GRCh37/GRCh38 to find contigs then subset to those less than 500 kb.
Regions excluded for specific technologies
We exclude tandem repeats approximately larger than the read length from each method. Tandem repeats shorter than 51 bp were excluded from all technologies except Illumina PCR-free GATK, Complete Genomics, and PacBio HiFi DeepVariant. We excluded tandem repeats between 51 bp and 200 bp except for Illumina PCR-Free GATK and PacBio HiFi DeepVariant. Tandem repeats between 200 bp and 10,000 bp are excluded from all technologies except PacBio HiFi DeepVariant. Homopolymers greater than 6 bp were excluded from all technologies except Illumina PCR-free GATK, Complete Genomics, Ion Exome, PacBio HiFi DeepVariant. Imperfect homopolymers greater than 10 bp were excluded from all technologies except Illumina PCR-Free GATK. Low mappability regions that are difficult to map for short reads were excluded from all except 10x Genomics and PacBio HiFi. LINE:L1Hs greater than 500 bp were excluded except Illumina MatePair, 10x Genomics, and PacBio HiFi. Segmental duplications were excluded from all technologies except 10x Genomics and PacBio HiFi. Homopolymers were excluded from 10x Genomics and PacBio HiFi. Long homopolymers were included only for GATK based calls for PCR-Free data because GATK gVCF has low genotype quality score if reads do not totally encompass the homopolymer. Overall we trust homopolymers most from PCR-Free short reads. We visualize the regions excluded from each sequencing technology in Figure 6.

The same genomic regions were excluded for both Illumina paired end datasets, high coverage HiSeq 300x, and 2X250 HiSeq. The PacBio HiFi dataset consists of 4 SMRT Cells of 15 kb libraries and 2 SMRT Cells of 20 kb libraries.
Comparing v3.3.2 to v4.1
We subset v3.3.2 variants to v3.3.2 benchmark bed and v4.1 variants to v4.1 benchmark bed and compared the benchmarks using hap.py with v2.0 of the GA4GH benchmarking stratifications (https://github.com/ga4gh/benchmarking-tools).5 To identify the types of genomic regions where v4.1 gains and loses benchmark variants relative to v3.3.2, we subset to stratifications with at least 1000 variants in v4.1, and sorted by the difference between the Precision and Recall metrics, which are measures of the fraction of extra variants in v3.3.2 and v4.1, respectively.
Calculating difficult, medically-relevant genes coverage
We used the 193 clinically-relevant gene names that contained exons that are difficult to map with short reads from 13. We used Ensembl BioMart to retrieve Human Genes Build 99 with Gene Name, Start, End, and Chromosome (http://jan2020.archive.ensembl.org/biomart/martview/2c3a4b803e1a01b3b806829a466b3590). We used those results to find coordinates for the difficult, medically-relevant gene names, subset to genes on chromosomes 1-22, then used bedtools intersect with the v3.3.2 and v4.1 benchmark region files to find overlap.
Evaluation of the benchmark
We used hap.py (https://github.com/Illumina/hap.py) following GA4GH best practices5 with v4.1 benchmark variants as the truth set, v4.1 benchmark bed as confident regions, and each of the 12 call sets as the query. We use the vcfeval engine for comparison.20
To evaluate the utility of the v4.1 benchmark, the GIAB community contributed 13 call sets from short-, linked-, and long-read technologies, and from mapping-, graph-, and assembly-based variant callers. We used hap.py to compare each input callset to v4.1 then asked collaborators to manually curate a small subset of the False Positive and False Negative sites with commands detailed in “Supplementary Materials - Benchmark Evaluations”. Collaborators evaluated 5 False Positive SNVs, 5 False Positive Indels, 5 False Negative SNVs, 5 False Negative Indels both inside and outside v3.3.2 along with 5 False Positive SNVs, 5 False Positive Indels, 5 False Negative SNVs, 5 False Negative Indels in the MHC for GRCh37. We generated IGV sessions with BAM files for Illumina HiSeq, 10x Genomics, PacBio HiFi 15kb & 20 kb merged, and ONT Ultralong11, then asked that the evaluators identify for each site if both alleles in the benchmark were correct and if both alleles in the query call set were correct.
Long Range PCR Confirmation
We performed Long range PCR followed by Sanger sequencing for variants in LINEs and difficult, medically-relevant genes. The difficult genes that were chosen for long-range PCR and Sanger sequence confirmation are potentially medically-relevant and have many characteristics that make them difficult to characterize, especially with short reads. We selected genes with previously published long range PCR assays. The first set of genes make up the RCCX complex, a segmental duplication that includes TNXA, TNXB, C4A, C4B, and CYP21A2.36,37 The similar sequences of these genes in close proximity makes them prone to rearrange, mutate and change the size of the complex as a whole, and they are linked to rare diseases that are inherited together at a higher rate than would be expected by chance. Mutations in the CYP2D6 gene can affect metabolism and bioactivation of many clinical drugs and the gene contains a polymorphic region.38 DMBT1 has been identified as a candidate tumor suppressor for brain, gastrointestinal and lung cancers and contains highly repetitive sequence.39 Rare variants in the HSPG2 gene are linked to cases of idiopathic scoliosis.40 STRC has a pseudogene with high genomic and coding sequence homology making it very difficult to characterize by normal short read sequencing methods.41 The PMS2 gene has multiple pseudogenes, making it difficult to reliably detect mutations or characterize by sequencing.18
Long range PCR was performed to amplify regions with variants in LINEs and difficult medically-relevant genes. Primers for amplification of LINEs were designed with the Primer3Plus software.42 Other primers were sourced from literature. All long range primer sequences and references can be found in Supplementary Table 10. Long range PCR were performed with the PrimerSTAR GXL DNA Polymerase (Takara Bio, Mountain View, CA), and assays specific reaction components can be found in Supplementary Table 11. Long range PCR conditions varied by assay and can be found in Supplementary Table 12.
Sanger primers were designed using the Primer3Plus software.42 Primer sequences can be found in Supplementary Table 10. Long range PCR products were purified with ExoSAP-IT (Applied Biosystems, Foster City, CA). Sanger sequencing was performed with SimpleSeq Premixed Sequencing Kits (Eurofins Genomics, Louisville, KY) using 5 mL of the long range PCR amplicon and 5 mL of 3 mM primer. Sanger sequencing traces were aligned and analyzed with Geneious Prime (Biomatters, Inc., San Diego, CA).
Data availability
Sequence data used is in Table 4, and is in SRA accessions SRX852933, SRX847862, SRX1726841 - SRX1726859, SRX1726861 - SRX1726869, SRX1388733, and SRX7083054-SRX7083059. Aligned reads and other analyses from the GIAB Ashkenazi trio data are available at ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/. The benchmark vcf and bed files are available at: ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/AshkenazimTrio/HG002_NA24385_son/NISTv4.1/ ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/NIST_v4.2_SmallVariantDraftBenchmark_07092020/
Code availability
Scripts for integrating candidate variants to form the benchmark set in this manuscript will be made available in a GitHub repository. Publicly available software used to generate input callsets and evaluation callsets is described in the methods and Supplementary Materials.
Competing Interests
AMW and WJR are employees and shareholders of Pacific Biosciences. AMB and ITF were employees and shareholders of 10X Genomics. FJS has received sponsored travel from Oxford Nanopore and Pacific Biosciences, and received a 2018 sequencing grant from Pacific Biosciences. AS and VK are employees of Seven Bridges. AC is an employee of Google Inc. and is a former employee of DNAnexus. AF and C-SC are employees of DNAnexus. SMES is an employee of Roche.
Acknowledgments
We thank the Genome in a Bottle Consortium for ongoing feedback and discussions about the benchmark. We thank participants in the precisionFDA Truth Challenge V2 for helpful feedback about the v4.2 benchmarks for the trio. We thank Valerie Schneider for advice regarding alignments of GRCh38 to GRCh37. Certain commercial equipment, instruments, or materials are identified to specify adequately experimental conditions or reported results. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the equipment, instruments, or materials identified are necessarily the best available for the purpose.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}