

Abstract
One of the defining features of sexual reproduction is the recombination events that take place during meiosis I. Recombination is both evolutionarily advantageous, but also mechanistically necessary to form the crossovers that link homologous chromosomes. Meiotic recombination is initiated through the placement of programmed double-strand DNA breaks (DSBs) mediated by the protein Spo11. The timing, number, and physical placement of DSBs are carefully controlled through a variety of protein machinery. Previous work has implicated Mer2(IHO1 in mammals) to be involved in both the placement of breaks, and their timing. In this study we use a combination of protein biochemistry and biophysics to extensively characterise various roles of the Mer2. We gain further insights into the details of Mer2 interaction with the PHD protein Spp1, reveal that Mer2 is a novel nucleosome binder, and suggest how Mer2’s interaction with the HORMA domain protein Hop1 (HORMAD1/2 in mammals) is controlled.
Introduction
The starting point of meiotic recombination is the creation of programmed double-strand DNA breaks (DSBs) by the transesterase Spo111. DSBs are preferentially repaired via recombination from the homologous chromosome which, depending on how recombination intermediates are processed, can yield crossovers. Together with distal cohesin, crossovers provide the physical linkage between homologous chromosomes. In order to both control DSB formation, and to prepare the chromosomes for synapsis, meiotic chromosomes are organised into a distinctive loop-axis architecture (Figure 1a). In the budding yeast S. cerevisiae the proteinacious axis contains Red1, the HORMA domain protein Hop1, and Rec8 cohesin2. The Spo11 machinery associates physically with the axis, but makes breaks in nucleosome depleted regions of the loop defined by H3K4me3 marks3,4.

Spp1 binds to Mer2 tetramerisation domain with a 2:4 stoichiometry. A) Cartoon of meiotic loop-axis architecture and the role of Mer2. During meiosis proteins Red1 and Hop1 form a protein:DNA (coloured black) axis together with cohesin (not shown for clarity). Loops of chromatin are extruded from the axis, where DNA breaks are made by Spo11 complex (magenta). Breaks are directed to the proximity of H3K4me3 nucleosomes (green) through the combined activity of Mer2 and Spp1. Mer2 is also thought to interact with additional Spo11 accessory proteins (Rec114 and Mei4, orange) through phosphorylation (P labels on Mer2). B) Domain diagram of Mer2 and Spp1. The four principal Mer2 constructs used throughout this study are shown. The only clear feature of Mer2 (and its orthologs IHO1 in mammals, Rec15 in fission yeast and Prd3 in plants) is the central coiled-coil motif. Spp1 is shown for comparison with its N-terminal PHD domain and C-terminal Mer2 interaction domain which is predicted to contain a coiled-coil. Purification of Mer2 and Spp1. A complex of Mer2 and Spp1 were purified to homogeneity. In this case we show a double MBP-tagged complex. Molecular weight markers are shown in grey. The relative absorbance of the complex at 280 nm and 260 nm shows that it is free of any significant nucleic acid contamination. C) SEC-MALS of different complexes. Three illustrative SEC-MALS experiments are shown for MBP-Spp1 (yellow) Mer2 core (peach) and a complex of Mer2core-Spp1 (green). The table below summarizes additional SEC-MALS experiments. The coloured circles refer to the SEC-MALS chromatogram above. D) Microscale thermophoresis of Mer2-Spp1. Using Red-NHS labelled untagged Spp1 at a constant concentration of 20 nM two different Mer2 constructs were titrated against it, and the change in thermophoresis measured. Experiments were carried out in triplicate and the Kd determined from the fitting curve.
Mer2 (also known as Rec107) was originally identified as a high-copy number suppressor of the mer1 phenotype (Mer1 later shown to be a co-factor for the splicing of various meiotic mRNAs, including Mer25), where it was also shown to be essential for meiosis6. Mer2 is central to both the temporal and spatial control of breaks. In temporal control Mer2 is a target of S-Cdk and DDK phosphorylation that presumably allows the binding of the Spo11 though the associated factors Rec114 and Mei47–9. In spatial control, Mer2 interacts directly with the PHD domain containing protein Spp1. Spp1 interacts with nucleosomes tri- (or di-) methylated on H3K4 (H3K4me3 nucleosomes)10,11, and as such Spp1 and Mer2 link the DSB forming machinery with H3K4me3 marks 12,13. Spp1 is canonically part of the COMPASS (a.k.a. Set1 complex) and Spp1’s interaction with Mer2 is mutually exclusive with the remainder of the COMPASS complex14. Furthemore, Spp1 associated with Mer2 has a longer residence time on nucleosomes when compared with Spp1 when part of COMPASS15.
The reciprocal interaction domains between Spp1 and Mer2 have been previously identified. The C-term of Spp1 interacts with a central, predicted coiled-coil, region of Mer212,13. A single amino acid substitution in the Mer2 (V185D) is sufficient to disrupt the interaction with Spp114. Deletion of Set1 or Spp1 severely reduces the number of DSBs, but cells still form viable tetrads, whereas a deletion of Mer2 completely eliminates break formation.
Given the central role of Mer2 in DSB formation we set out to more completely biochemically characterise both Mer2. We examine the interaction of Mer2 with Spp1, nucleosomes, and with the meiotic axis. Our results give a more complete picture of Mer2, including novel functions, and provide mechanistic explanations for a number of previously observed phenomena.
Results
Mer2-Spp1 is a constitutive complex with a 2:4 stoichiometry
We started out by producing constructs of both Spp1 and Mer2 to determine their stoichiometry and confirm their mutual interaction regions (Figure 1B). Using our inhouse expression system, InteBac16, we were able to produce all our Spp1 and Mer2 proteins in E.coli with N-terminal MBP tags. In co-expression and co-lysis experiments we discovered that we could purify a complex of Mer2 and Spp1 to homogeneity, and free of nucleic acid contamination (Figure 1C), thus indicating that the interaction between Mer2 and Spp1 does not require any post-translational modifications or additional cellular co-factors.
It has previously been reported that the Spp1 interaction domain of Mer2 lies within a central region comprising residues 140-256 of Mer212,13 which we from now on refer to as Mer2 “core” (Figure 1B). We measured the molecular mass of Mer2core by size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) and concluded that the Mer2 core is a tetramer (Figure 1D), consistent with recently published data17. We also find that while most Mer2 constructs lacking the core are monomeric, there is also a dimerisation region in the C-terminal region between residues 255 and 314 (Figure 1D). We measured the binding affinity of Spp1 to the Mer2 core versus the Mer2 full length, both bound at mid-nanomolar affinity (Figure 1D), whereas Mer2 constructs lacking the core showed weak binding, which is most likely experimental noise (Supplementary Figure 1). Thus we confirm that the majority of the Spp1 binding interface is indeed within the core of Mer2.
Given that the tetramerisation region of Mer2 is also the principal Spp1 binding region, we determined the stoichiometry of the Mer2-Spp1 complex. Using SEC-MALS we determined that the complex of Mer2-Spp1 has a 4:2 stoichiometry, whereas Spp1 alone is a monomer (Figure 1D). Thus at least one function of Mer2 appears to be the dimerisation Spp1.
Dimeric Spp1 binds tightly to H3K4me3
We hypothesised that dimeric Spp1 should bind to H3K4me3 nucleosomes more tightly than monomeric Spp1, and set out to test this hypothesis. We constructed a synthetic dimer of Spp1 using a GST-fusion. We compared the binding of Spp1, GST-Spp1 and Spp1-Mer2 to nucleosomes in both EMSAs (Figure 2A) and via pulldowns on H3 and H3K4me3 biotinylated nucleosomes (Figure 2B). We observed that Spp1 binds relatively weakly to nucleosomes, consistent with the reported 1 μM affinity of the PHD domain with H3K4me3 peptide11. Spp1-GST bound more tightly, and Spp1-Mer2 bound apparently the tightest. We also tested Mer2-Spp1 binding to H3K4me3 mononucleosomes via analytical SEC and found we could make a stable complex (Figure 2C).

Dimeric Spp1 binds tightly to nucleosomes. A) EMSA of different Spp1s on H3K4me3 nucleosomes. 0.2 μM of H3K4me3 nucleosomes were incubated with 0.33, 1 and 3 μM protein (Spp1, Spp1 with a C-terminal GST fusion and Mer2-Spp1). Gel was post-stained with SybrGold. B) Biotinylated-Mononucleosome pull-down of different Spp1. 0.5 M nucleosomes wrapped with 167 bp of biotinylated DNA (either with or without the H3K4me3 modification) were incubated with 1.5 M protein (same proteins as in a)). Samples were taken for the input before and incubation with streptavidin beads. The beads were then washed and eluted with 1x Laemmli buffer. Input and elution samples were run on a 10-20% SDS-PAGE gel and stained with InstantBlue. C) SEC analysis of Spp1-Mer2-MN complex. H3K4me3 mononucleosomes (blue), Mer2-Spp1 complex (peach) and the mixture (green) were run on a Superose 6 5/150 column. The same fractions were loaded in each case (magenta line) onto an SDS-PAGE gel and stained with InstantBlue.
Mer2 binds directly to nucleosomes with a 4:1 stoichiometry
Our pulldown data suggest that Mer2 might be binding to mononucleosomes (Figure 2B), a previously unreported function of Mer2. We tested whether Mer2 could bind to nucleosomes alone, and found remarkably that it formed a SEC stable complex, on unmodified nucleosomes (Figure 3A). We tested the molecular mass, and thus the stoichiometry, of the Mer2-mononucleosomes complex using Mass Photometry (MP), a new technique that determines molecular mass in solution at low concentrations based on the intensity of scattered light on a solid surface18. In MP we observe a mix of three species, free Mer2 tetramer (measured at 127 kDa), free mononucleosomes (measured at 187 kDa) and a complex at 303 kDa (Figure 3b). The experiment was carried out at a protein concentration of 60 nM, which explains the mix of species seen.
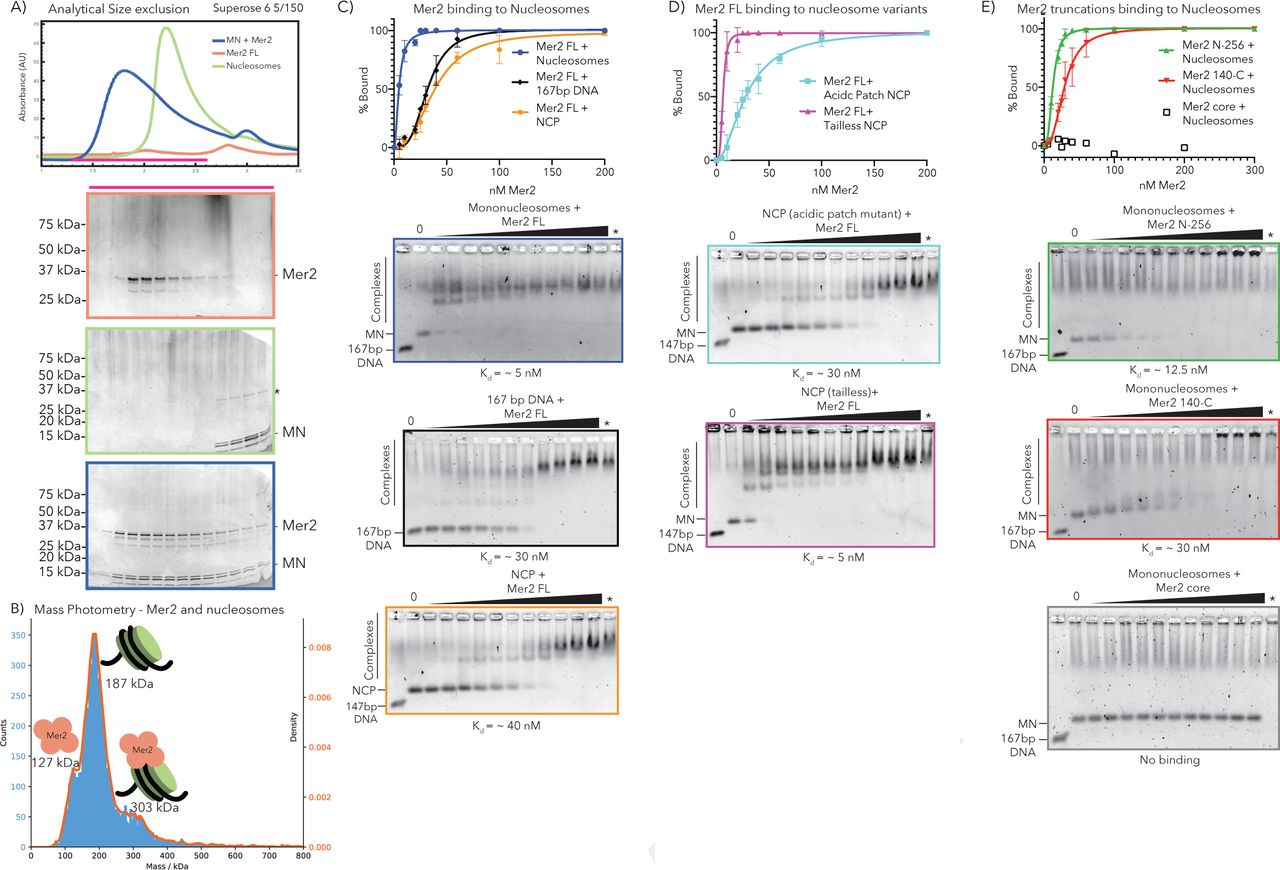
Mer2 N-terminus binds directly to nucleosomes. A) SEC analysis of Mer2-MN complex. Mononucleosomes (grey), full length untagged Mer2 (peach) and a mixture of Mer2 and mononucleosomes (green) were run on a Superose 6 5/150 column. The same fractions were loaded in each case (magenta line) onto an SDS-PAGE gel and stained with InstantBlue. B) Mass Photometry of Mer2 and mononucleosomes. 60 nM of Mer2 and mononucleosomes were mixed and analyzed using a Refeyn One mass photometer. Three separate species were identified and the molecular mass determined using a molecular mass standard curve created under identical buffer conditions. Negative data points (i.e. unbinding events) were excluded. C) EMSAs of untagged Mer2 FL. Mer2 was titrated against a constant 5 nM concentration of mononucleosomes (blue), 167 bp 601 DNA (black), or nucleosome core particle (orange). Binding curves were derived based on the Mer2 dependent depletion of free nucleosomes, DNA or NCP, and based on four independent experiments. D) EMSAs of full-length Mer2 on acidic patch nucleosome core particles (NCP) (cyan trace) or tailless NCPs (magenta trace). E) EMSAs of Mer2 constructs on mononucleosomes. Mer2 constructs (N-256; green, 140-C; red or core; grey) were titrated against a constant 5 nM concentration of mononucleosomes. Binding curves were derived based on the Mer2 dependent depletion of free nucleosomes, and based on four independent experiments..
It has been reported that Mer2 binds to DNA, therefore we tested whether Mer2 might simply be binding the free DNA on mononucleosomes. Using analytical EMSAs we found that Mer2 binds with a 6-fold higher apparent affinity to nucleosomes (5 nM vs. 30 nM), when compared to free DNA (Figure 3C, blue vs. black trace). The discrepancy between Kd determined by EMSA and the apparent Kd from MP is presumably because EMSAs are non-equilibrium experiments, carried out by necessity at low salt19. We also tested Mer2 binding on the nucleosome core particle (NCP) that lacks free DNA ends with a 40 nM apparent Kd (Figure 3C orange trace).
Next we tested the binding of Mer2 FL to both nucleosomes containing the acidic patch mutation (H2A E56T-E61T-E64T-D90S-E91T-E92T)20 and to “tailless” nucleosomes made up of just the histone cores (see materials and methods). In order to try to emphasise any differences in binding affinity we reconstituted these histones with 147 bp DNA, forming a NCP, and carried out EMSAs with full-length Mer2 (Figure 3D). In both cases we observed no weakened binding of Mer2 when compared to the wild-type NCP (Figure 3C orange trace), and in the case of tailless nucleosomes Mer2 appeared to bind even more tightly (Figure 3D, purple trace).
Using Mer2 truncation constructs we determined that the binding to nucleosomes lies in the N- and C- termini (mostly in the N-terminus) whereas the tetramerisation core shows no binding (Figure 3E). Given that the N-term of Mer2 seems to be playing a greater role in nucleosome binding. We looked for conserved residues the N-terminus and identified a conserved patch of residues, found in Rec15 and IHO1 (Supplementary Figure 2). In EMSAs mutation of three conserved residues to alanine (3A) resulted in weaker binding as determined by EMSA (Supplementary Figure 2, purple vs. blue trace), but this mutation does not completely ablate the binding.
Mer2 binds to open Hop1 on the meiotic axis
Finally we tested the ability of Mer2 to interact with the proteins of the meiotic axis Hop1 and Red1 (Figure 4A). We purified Hop1 out of E.coli using an N-terminal Twin-Strep-II tag and used this to pulldown Mer2. We observed a faint band in the pulldown, indicative of a weak interaction (Figure 4A and B). We then co-expressed MBP-Red1 containing a I743R mutation with Hop1. The I743R mutation that should abolish filament formation21 and remain a tetramer. However MBP-Red1 I743R but in our hands is a monomer, with no evidence of Hop1 oligomerisation on Red1 (Supplementary Figure 3), although this could be a feature of the low protein concentrations used in the MP experiment.

Mer2 binds to the axis via open Hop1 A) Pulldowns of Strep-Hop1 (left) and MBP-Red1I743R/Strep-Hop1 (right). 1 μM Strep-Hop1 or Red1-Hop1 complex was used as bait against 3 μM full length, untagged Mer2. B) Quantification of three separate experiments as in A). C) Pulldown of Strep-Hop1 with MBP-Red1I743R either co-expressed and purified with the Hop1 (left lanes) or MBP-Red1I743R expressed and purified separately and added to Hop1 for the pulldown. D) Cartoon model of possible mechanism for Mer2 Hop1 interaction. Closed Hop1 binds to its own closure motif (left) which sterically hinders Mer2 interaction. Then action of Pch2 (pink, centre) is required to facilitate the interaction of the HORMA domain of Hop1 with the closure motif (CM) of Red1. The Red1-Hop1 complex is then functional for Mer2 binding (right), possibly via a motif in the C-terminal region of Hop1.
Given that we have an excess of Hop1 in our Hop1-Red1 purification (Supplementary Figure 3), we carried out a pulldown on the MBP-tag of Red1 with Mer2 as bait. In this case we observed considerably more Mer2 binding (Figure 4A). We quantitated the Mer2 intensity relative to the Hop1 band in both pulldowns, and observe a several-fold increase in Mer2 binding (Figure 4B). In order to determine which protein, Hop1 or Red1, is responsible for the interaction with Mer2, we purified MBP-Red11743R separately from Hop1. We then tried to reconstitute the interaction between Hop1 and Red1 in a Strep-Hop1 pulldown (Figure 4C). Under these conditions, while we observed a complex of Hop1 and Red1, when the proteins had been co-expressed, we were not able to capture any Red1 I743R when added in vitro. We conclude that an additional cellular factor, presumably a AAA+ ATPase (see below) is required to facilitate the Red1-Hop1 interaction.
Discussion
We have biochemically dissected the function of Mer2 in vitro to reveal several novel features. Firstly, the interaction between Spp1 and Mer2 is tight (~ 25 nM) and not dependent on PTMs or additional cofactors. As such we conclude that Spp1-Mer2 is a constitutive interaction. We also find that Mer2 serves as a dimerisation platform for Spp1, thus increasing its affinity for H3K4me3 nucleosomes. Presumably this is essential given that the interaction between COMPASS bound Spp1 and H3K4me3 is transient, whereas the association of the DSB forming machinery is an apparently more stable event15. Given that the interaction domain is the same as the tetramerisation domain we speculate that the antiparallel arrangement of coiled-coils of Mer2core form two mirrored binding sites for Spp1, with V195 at the centre14,17.
Surprisingly we find that Mer2 itself is a bona-fide nucleosome binder. This interaction occurs at high affinity, though we assume that the true affinity is less than what we measure from EMSA titrations. Given that Mer2 has been previously shown to bind DNA, yet still binds to the nucleosome core particle (with 147 bp DNA and no DNA overhangs), and that neither the loss of histone tails, nor the acidic patch mutant disrupted the interaction, we suggest that it binds to the nucleosomal DNA. Indeed The nucleosome binding ability of Mer2 provides a molecular basis for the observation that neither Spp1, nor H3K4me3 is required to make some meiotic DSBs. As such we speculate that under these circumstances Mer2 binds stochastically to loop nucleosomes. Such a model speculates that some of these binding events present a nucleosome depleted loop region to Spo11, but many do not, explaining also that meiotic DSBs are severely reduced in number in an Spp1Δ or Set1Δ background. On the other hand if Mer2 preferentially bound free-DNA, there might not be such a severe DSB phenotype in an Spp1Δ or Set1Δ.
Our observation of the direct interaction between Hop1 and Mer2 offers a tantalising glimpse into how axial proteins may be regulating DSB formation. We only observe binding between Mer2 and Hop1 in the presence of Red1. This could of course be explained through direct binding to Red1, but Hop1 orthologs have been implicated as the direct interactor of Mer2 orthologs22,23. We propose a model whereby Hop1 that is bound to its own closure motif is not competent for Mer2 binding (Figure 4D, left). Only once Hop1 is bound to Red1 is the binding site for Mer2 then exposed. Intriguingly, in fission yeast, the zinc-finger of Hop1 has been suggested to be required Mer2 binding22, which would be consistent with our model and our data. We were unable to obtain an interaction between Hop1 and Red1I743R in vitro. We propose that Hop1 needs to be “opened” by the AAA+ ATPase Pch224,25, which facilitates the binding of the HORMA domain to the closure motif of Red126 (Figure 4D, middle). This is in line with recent models that show there is a balance between Pch2 and Hop1 levels, and that Pch2 is both required for checkpoint establishment (loading open Hop1 onto the axis) and silencing (removing Hop1 from the axis in a Zip1 dependent fashion) 27–29.
Taken together our data show that Mer2 forms the keystone of meiotic recombination, binding directly to both the axis via Hop1 and the loop via nucleosomes. Presumably once assembled on the loop-axis Mer2 is then able to interact with Rec114 and Mei4 in phospho-dependent manner (likely via the PH domains of Rec11430–32), which may also be further controlled by liquid-liquid phase separation17. In organisms with a synaptonemal complex (SC), shutting meiotic DSB formation is associated with synapsis. It has been shown that synapsis results in the Pch2 mediated displacement of Hop1 from the axis28,33,34). We expect that this action would also result in the displacement of Mer2 as Hop1 would “snap shut” and further interaction with Hop1 would be prevented via steric hindrance of the binding site.
Materials and Methods
Protein expression and Purification
Sequences of Saccharomyces cerevisiae Spp1, Mer2, Hop1 and Red1 were derived from SK1 strain. All Mer2 constructs were produced as an 3C HRV cleavable N-terminal MBP fusion in competent C41 E. coli cells. Protein expression was induced by addition of 0.25 mM IPTG and the expression continued in 18°C overnight. Cells were washed with PBS and resuspended in lysis buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 5 % glycerol, 0.1 % Triton, 1 mM MgCl2, 5 mM beta-mercaptoethanol. Resuspended cells were lysed using EmulsiFlex in presence of DNAse and AEBSF before clearance at 20,000g at 4C for 30 min. Cleared lysate was applied on MBP-trap (GE Healthcare) and extensively washed with a lysis buffer. Mer2 constructs were eluted with a lysis buffer with 1 mM maltose and passed through ResourceQ column (GE Healthcare). The proteins were eluted by increasing salt gradient to 1M NaCl. Protein containing elution fractions were concentrated on Amicon concentrator (100 kDa cutoff) and loaded on pre-equilibrated Superose 6 16/600 (GE Healthcare) in SEC buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM TCEP). Untagged Mer2_FL was prepared likewise until concentration of protein eluted from ResourceQ. The concentrated eluent was mixed with 3C HRV protease in molar ratio of 50:1 and incubated at 4C for 6 hours. Afterwards, the cleaved protein was loaded on Superose 6 16/600 pre-equilibrated in SEC buffer for cleaved Mer2 (20 mM HEPES pH 7.5, 500 mM NaCl, 10% glycerol, 1 mM TCEP, 1 mM EDTA, AEBSF).
Spp1 constructs were produced as an 3C HRV cleavable N-terminal MBP or GST fusion in like manner as MBP-Mer2. To purify GST-Spp1, the cleared lysate was applied on GST-Trap (GE Healthcare) before extensive washing with lysis buffer. The protein was eluted with a lysis buffer with 40 mM reduced glutathione and passed through ResourceQ. Both GST and MBP could be cleaved by adding 3C HRV protease to concentrated protein (using an Amicon concentrator with 30 kDa cutoff) in 1:50 molar ratio. After app. 6 hour incubation in 4C, the cleaved protein was loaded on Superdex 200 16/600 pre-equilibrated in SEC buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM TCEP).
Hop1 constructs were produced as 3C HRV cleavable N-terminal Twin-StrepII tag in BL21* E.coli cells. The expression was induced by addition of 0.25 mM IPTG and the expression continued at 18C ON. Cleared lysate was applied on Strep-Tactin Superflow Cartridge (Quiagen) before extensive washing. The bound protein was eluted with a lysis buffer containing 2.5 mM desthiobiotin and loaded on HiTrap Heparin HP column (GE Healthcare) and subsequently eluted with increasing salt gradient to 1M NaCl. Eluted Strep-Hop1 constructs were concentrated using Amicon concentrator with 30 kDa cutoff and loaded on Superdex 200 16/600 pre-equilibrated in SEC buffer.
Red1 was produced in insect cells as a C-terminal MBP-fusion either alone or in co-expressed with Strep-Hop1. Bacmids were in both cases produced in EmBacY cells and subsequently used to transfect Sf9 cells to produce baculovirus. Amplified baculovirus was used to infect Sf9 cells in 1:100 dilution prior to 72 hour cultivation and harvest. Cells were extensively washed and resuspended in Red1 lysis buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM MgCl2, 5 mM beta-mercaptoethanol, 0.1% Triton-100). Resuspended cells were lysed by sonication in presence of Benzonase and a protease inhibitor cocktail (Serva) before clearance at 40,000g at 4C for 1h. Cleared lysate was loaded on Strep-Tactin Superflow Cartridge (in case of Red1-Hop1 complex) or MBP-trap column (in case of Red1 alone). Proteins were eluted using a lysis buffer containing 2.5 mM desthiobiotin and 1 mM maltose, respectively. Partially purified proteins were further passed through HiTrap Heparin HP column and eluted with increasing salt gradient to 1M NaCl. Purified proteins were subsequently concentrated using Pierce concentrator with 30 kDa cutoff in 50 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, 1 mM TCEP. Because of small yield of the proteins, the SEC purification step was neglected and the purity of the proteins was checked using Refeyn One mass photometer.
SEC-MALS analysis
50 μL samples at 5-10 μM concentration were loaded onto a Superose 6 5/150 analytical size exclusion column (GE Healthcare) equilibrated in buffer containing 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM TCEP attached to an 1260 Infinity II LC System (Agilent). MALS was carried out using a Wyatt DAWN detector attached in line with the size exclusion column.
Microscale thermophoresis
Triplicates of MST analysis were performed in 50 mM HEPES pH 7.5, 300 mM NaCl, 5% glycerol, 1 mM TCEP, 0,005% Tween-20 in 20C. The final reaction included 20 μM RED-NHS labelled untagged Spp1 (labelling was performed as in manufacturer’s protocol-Nanotemper) and titration series of MBP-Mer2 constructs (concentrations calculated based on oligomerization stage of Mer2). The final curves were automatically fitted in Nanotemper analysis software.
Recombinant Nucleosome production
Recombinant X. laevis histones were purchased from “The Histone Source” (Colorado State) with the exception of H3-C110A_K4C cloned into pET3, which was kindly gifted by Francesca Matirolli. The trimethylated H3 in C110A background was prepared as previously described 35. X. laevis histone expression, purification, octamer refolding and mononucleosome reconstitution were performed as described 36. Plasmids for the production of 601-147 (pUC19) and 601-167 (pUC18) DNA were kindly gifted by Francesca Matirolli and Andrea Musacchio, respectively. DNA production was performed as previously described. Reconstituted mononucleosomes were shifted to 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM TCEP with addition of 20% glycerol prior to freezing in −80°C.
Electrophoretic mobility shift assays
Quadruplate EMSAs were carried out as previously described 37, with the DNA being post-stained with SybrGold (Invitrogen). Gels were imaged using a ChemiDocMP (Bio-Rad Inc). Nucleosome depletion in each lane was quantitated using ImageJ, using triplicate nucleosome alone measurements for each individual gel. Binding curves were fitted using Prism software and the following algorithm Y = Bmax*Xh/(Kdh + Xh). It was necessary in each Mer2 case to add a Hill coefficient to obtain the best fit.
Protein streptavidin pulldowns
Streptavidin pulldowns were performed using pre-blocked Strep-Tactin Sepharose beads in pulldown buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 5% glycerol, 1 mM TCEP). 1 μM Strep-Hop1 as a bait was incubated with 3 μM Mer2 as a prey in 40 μL reaction for 2 hours on ice without beads and another 30 min after addition of 10 μL of pre-blocked beads. After incubation, the beads were washed twice with 250 μL of buffer before elution of the proteins with a buffer containing 2.5 mM desthiobiotin. Samples were loaded on 10% SDS-PAGE gel and afterwards stained with InstantBlue.
Biotinylated nucleosome pulldown
Biotinylated nucleosomes (0.5 μM) of NCP (0.4 μM) were incubated with prey proteins (1.5 μM) for 30 min on ice in buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol, 1 mM EDTA, 0.05% Triton-X100, 1 mM TCEP in a reaction volume of 40 μL. 10 μL of protein mix were taken as an input before adding 10 μL of pre-equilibrated magnetic Dynabeads M 270 streptavidin beads (Thermo Fisher Scientific) to the reaction. The samples with beads were incubated on ice for 2 min before applying magnet and removing the supernatant. The beads were washed twice with 200 μL of buffer. To release the streptavidin from the beads, Laemmli buffer (1x) was added to the beads and incubated for 10 min. Samples were analyzed on 10-20% SDS-PAGE gel and stained by InstantBlue.
Analytical Size exclusion chromatography
Analytical SEC was performed using Superose 6 5/150 GL column (GE Healthcare) in a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol, 1 mM TCEP, 1 mM EDTA. All samples were eluted under isocratic elution at a flow rate of 0.15 ml/min. Protein elution was monitored at 280 nm. Fractions were subsequently analysed by SDS-PAGE and InstantBlue staining. To detect complex formation, proteins were mixed at 5 μM concentration in 50 μL and incubated on ice for 1 hour prior to SEC analysis.
Mass Photometry
Mass Photometry was performed in 20 mM HEPES pH 7.5, 150 mM NaCl, 5% glycerol, 1 mM TCEP, 1 mM EDTA. 600 nM Mer2 and mononucleosomes were mixed and incubated for 1 hour on ice prior to analysis using Refeyn One mass photometer. Immediately before analysis, the sample was diluted 1:10 with the aforementioned buffer. Molecular mass was determined in Analysis software provided by the manufacturer using a NativeMark (Invitrogen) based standard curve created under the identical buffer composition.
Author Contributions
Cloning, expression and purification: DR, SKF, HR; Recombinant nucleosome production: DR; Biochemical and biophysical assays: DR; Data analysis: DR, SKF, JRW; Supervision: JRW; Drafting of manuscript: JRW, DR; Funding acquisition: JRW
Supplementary Data

MBP-Mer2 255-314 (green trace), MBP-Mer2 1-139 (black trace) and MBP alone (grey trace) were titrated against labelled Spp1. Results are based on triplicate experiments. The large error bars, and the high similarity to MBP alone suggest that there is no binding to Spp1 for either of the Mer2 constructs.

A) conserved N-terminal region was idenfitied in Mer2, Rec15 and IHO1 (Red stars). We mutated the three conserved residues to alanine (3A mutation). B) Mer2 FL 3A (purple trace) shows weakened binding on mononucleosomes when compared with Mer2 WT (blue trace).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
~30 nM MBP-Red1 Strep-Hop1 complex was analysed on a Refeyn MP as described. Molecular mass shown is as determined by the software based on standard curves. Theoretical molecular masses are as follows: Strep-Hop1 77.6 kDa MBP-Red1 137.7 kDa; Strep-Hop1/MBP-Red1 complex (1:1 stoichiometry) 215.3 kDa
Acknowledgements
We are extremely grateful to Francesca Mattiroli (Hubrecht Institute, Utrecht), for assistance with producing mononucleosomes. The histone H3 expression plasmid pET3 and the plasmid pUC19_601-147 DNA were a kind gift from Francesca Mattiroli. The plasmid pUC18_601-167 DNA was a kind gift from Andrea Musacchio (MPI Dortmund). We thank Christopher Heim (MPI Developmental Biology) for technical help with SEC-MALS and MST measurements. We thank Andreas Blaha, David Liedke and Constanze Gremmelmaier for their technical support. Thanks to Gerben Vader (MPI Dortmund) for insightful discussions in to Hop1 and Pch2. Work in the Weir lab is funded by the Max Planck Society. SKF is funded by a studentship from the International Max Planck Research School (IMPRS) “From Molecules to Organisms”.
Footnotes
Small alteration to the colouring on Figure 2 and Figure 3
References




