

Abstract
Subunit vaccines induce immunity to a pathogen by presenting a component of the pathogen and thus inherently limit the representation of pathogen peptides for cellular immunity based memory. We find that SARS-CoV-2 subunit peptides may not be robustly displayed by the Major Histocompatibility Complex (MHC) molecules in certain individuals. We introduce an augmentation strategy for subunit vaccines that adds a small number of peptides to a vaccine to improve the population coverage of pathogen peptide display. We augment a subunit vaccine by selecting additional pathogen peptides to maximize the total number of vaccine peptide hits against the distribution of MHC haplotypes in a population. For each subunit we design independent MHC class I and MHC class II peptide sets for augmentation, and alternatively design a combined set of peptides for MHC class I and class II display. We evaluate the population coverage of 9 different subunits of SARS-CoV-2, including 5 functional domains and 4 full proteins, and augment each of them to fill a predicted coverage gap. We predict that a SARS-CoV-2 receptor binding domain subunit vaccine will have fewer than six peptide-HLA hits with ≤ 50 nM binding affinity per individual in 51.31% (class I) and 32.99% (class II) of the population, and with augmentation, the uncovered population is predicted to be reduced to 0.54% (class I) and 1.46% (class II). We find that a joint set of pathogen peptides for MHC class I and class II display is predicted to produce a more compact vaccine design than using independent sets for MHC class I and class II. We provide an open source implementation of our design methods (OptiVax), vaccine evaluation tool (EvalVax), as well as the data used in our design efforts here: https://github.com/gifford-lab/optivax/tree/master/augmentation.
1 Introduction
Subunit vaccines seek to reduce the safety risks of attenuated or inactivated pathogen vaccines by optimizing the portion of a pathogen that is necessary to produce durable immune memory (Moyle and Toth, 2013). Early vaccines that consisted of attenuated or inactivated pathogens provided robust protection to disease, but came with risks associated with the potential reversion of inactivation, harmful components, and the potential for pathogenic infection of attenuated vaccines in immunocom-promised individuals. Thus, only including the non-infective components of a pathogen necessary to induce durable immunity is a natural goal. Such subunit vaccines have been enabled by our ability to engineer and express pathogen surface components that retain their three-dimensional structure. The retention of three-dimensional structure is important to induce neutralizing antibodies and a corresponding B cell memory. Neutralizing antibodies bind to a specific epitope on a pathogen and disable it, and choosing such an epitope as a subunit vaccine can productively focus an antibody-based immune response. However, the production of durable immune memory rests in part upon help from T cells, which get their cues from peptides displayed by Human Leukocyte Antigen (HLA) molecules encoded by the Major Histocompatibility Complex (MHC) of genes.
Subunit vaccines for coronaviruses have been proposed that utilize a diverse set of coronavirus proteins and domains. Suggested components include the spike (S) protein, the receptor binding domain (RBD), the S1 domain, the S2 domain, the nucleocapsid (N), the membrane (M), the envelope (E), the N-terminal domain (NTD), and the fusion domain (FD) (Wang et al., 2020; Yu et al., 2020). A universal RBD subunit design that can be adapted to COVID-19, MERS, and SARS has been proposed showing the versatility of a subunit approach (Dai et al., 2020). Here we examine how subsetting a pathogen for vaccination may influence the induction of cellular immunity.
Since a subunit vaccine does not fully represent a pathogen, vaccine excluded pathogen peptides will not be observed during vaccination by an individual’s T cells. Subunit vaccine-based simulation of a T cell response is further limited because each individual’s HLA molecules have specific preferences for the peptides they will bind and display. Cytotoxic CD8+ T cells observe peptides displayed by molecules encoded by an individual’s classical class I loci (HLA-A, HLA-B, and HLA-C), and helper CD4+ T cells observe peptides displayed by molecules encoded by an individual’s classical class II loci (HLA-DR, HLA-DQ, and HLA-DP). Computational methods can be used to predict which peptides will be displayed by a given set of HLA alleles. Furthermore, given a set of peptides and the distribution of HLA alleles present in a population, the number of expected peptide-HLA hits that each individual in the population will display can be computed (Liu et al., 2020). A peptide-HLA hit is a predicted binding instance of a peptide to an HLA allele, and thus a given peptide can have multiple hits in a given individual. Since HLA loci exhibit linkage disequilibrium, we use the frequencies of population haplotypes in our coverage computations. Each haplotype describes the joint appearance of HLA alleles. It is important to note that a peptide’s display does not guarantee that it will be immunogenic and stimulate a T cell response. However, it has been observed that most displayed peptides are immunogenic in a mouse model, with individual to individual variation in peptide immunogenicity (Croft et al., 2019).
One proposed solution to improve the response of helper T cells to subunit vaccines is including universal CD4+ peptide epitopes that are pathogen independent (Alexander et al., 2000). Universal peptides are selected to bind a large fraction of human HLA-DR alleles, and can be derived from example pathogens (Falugi et al., 2001) or be synthetic sequences (Alexander et al., 2000). While these approaches have proven to be useful in promoting CD4+ T cell responses at vaccination time, CD4+ memory T cells created at vaccination time will not be activated upon pathogen infection because the peptides they recognize will not be present. This is an issue because pathogen specific memory CD4+ T cells are thought to be important for durable immunity (Gasper et al., 2014).
We evaluate the population coverage of proposed subunit vaccines for SARS-CoV-2 based upon the predicted display of their component peptides across different populations of HLA haplotype frequencies. We use a conservative 50 nM threshold for peptide binding to an HLA molecule. Thus, it is possible in practice that the gaps in population coverage will be smaller than our predictions. However, our population coverage calculations assume that every peptide displayed is immunogenic. This is the best approximation we are able to make given that present methods for predicting immunogenicity of a peptide are inaccurate and limited to a few HLA alleles (Calis et al., 2013; Ogishi and Yotsuyanagi, 2019; Riley et al., 2019). Thus, we compensate for our inability to model immunogenicity by predicting the number of expected peptide-HLA hits in each individual. This is based upon our assumption that the probability of inducing cellular immune memory in an individual will in general increase with the number of viral peptides their immune system observes.
We present vaccine design methods that produce either two sets of peptides or a single set of peptides. In the separate design method we design MHC class I and MHC class II pathogen peptide sets that are predicted to be displayed to meet population coverage criteria (Figure 1A). In the joint design method we design a single set of peptides that are designed to meet both MHC class I and class II population coverage criteria (Figure 1B). The use of separate vs. joint peptide sets may depend upon the vaccine delivery platform utilized.

A) In the separate method, windowed pathogen proteomes are filtered for acceptable peptides and MHC class I and class II vaccine designs are chosen to optimize population coverage at specified levels of peptide-HLA hits. B) In the joint method, 25-mer pathogen peptides are annotated with their MHC class I and class II peptides, which are filtered, scored, evaluated for population coverage, and used to optimize the selection of their parent 25-mers into a joint vaccine.
Using our analysis and design methods we present methods for augmenting subunit vaccines to increase their predicted population coverage and specific augmentation designs for SARS-CoV-2. The augmentation methods we introduce compute the relationship between augmentation peptide count and predicted increased population coverage. This permits a flexible selection of augmentation peptide count based upon their delivery. We provide an analysis of the population coverage gaps for SARS-CoV-2 subunit vaccines, showing the relationship between augmentation peptide count and population coverage for all subunits being considered for vaccines. Our predictions suggest that certain subunit vaccines, such as RBD, will have gaps in population coverage that can be addressed by vaccine augmentation with supplemental peptides. For example, we predict that an RBD subunit vaccine will have fewer than six peptides displayed in 51.31% (class I) and 32.99% (class II) of the population, and with augmentation the uncovered population is predicted to be reduced to 0.54% (class I) and 1.46% (class II).
2 Methods
2.1 SARS-CoV-2 proteome and candidate peptides
The SARS-CoV-2 proteome is comprised of four structural proteins (E, M, N, and S) and open reading frames (ORFs) encoding nonstructural proteins (Srinivasan et al., 2020). We obtained the SARS-CoV-2 viral proteome from GISAID (Elbe and Buckland-Merrett, 2017) sequence entry Wuhan/IPBCAMS-WH-01/2019, the first documented case, as provided by Liu et al. (2020). Nextstrain (Hadfield et al., 2018) was used to identify ORFs and translate the sequence. We use sliding windows to extract all peptides of length 8–10 (MHC class I) and 13–25 (MHC class II) inclusive from the SARS-CoV-2 proteome, resulting in 29,403 peptides for MHC class I and 125,593 peptides for MHC class II. For vaccine augmentation, we use the same filtered candidate peptide set as Liu et al. (2020), in which peptides with mutation rate > 0.001 or non-zero glycosylation probability predicted by NetNGlyc (Gupta et al., 2004) are filtered.
Candidate peptides are scored by their predicted binding affinity (IC50) to MHC molecules. For MHC class I, we use an ensemble that outputs the mean predicted binding affinity of NetMHCpan-4.0 (Jurtz et al., 2017) and MHCflurry 1.6.0 (O’Donnell et al., 2020, 2018). For MHC class II, we use NetMHCIIpan-4.0 (Reynisson et al., 2020b). We conservatively use a 50 nM threshold of predicted binding affinity to classify peptides as binders, which has been shown to improve precision (Liu et al., 2020).
We use HLA class I and class II haplotype frequencies provided by Liu et al. (2020). For the HLA class I locus, this dataset contains 2,138 distinct haplotypes spanning 230 HLA-A, HLA-B, and HLA-C alleles. For HLA class II, this dataset contains 1,711 distinct haplotypes spanning 280 HLA-DP, HLA-DQ, and HLA-DR alleles. Population frequencies are provided for three populations self-reporting as having White, Black, or Asian ancestry.
We consider nine subunit vaccines: the full envelope (E), membrane (M), nucleocapsid (N), and spike (S) proteins as well as the S1, S2, receptor binding domain (RBD), N-terminal domain (NTD), and fusion peptide (FP) domains from S. The amino acid positions for each of the S protein subunits are shown in Figure 2. When evaluating these subunit vaccines we include all peptides of length 8–10 (MHC class I) and 13–25 (MHC class II) spanning the corresponding regions of the proteome.
Illustration of functional domains on SARS-CoV-2 S protein.
2.2 EvalVax subunit vaccine evaluation
We evaluate population coverage of SARS-CoV-2 subunit vaccines using EvalVax-Robust (Liu et al., 2020). EvalVax-Robust computes population coverage of a given peptide set using the HLA haplotype frequencies in each population of individuals self-reporting as having Black, Asian, or White ancestry (Section 2.1). Population coverage P(n) is defined as the fraction of individuals predicted to have ≥ n peptide-HLA binding hits with ≤ 50 nM predicted binding affinity. EvalVax-Robust computes the frequency of diploid HLA genotypes, and accounts for both homozygous and heterozygous HLA loci. We compute the average population coverage as an unweighted average of population coverage over the three populations. Insufficient coverage of ≤ n hits is defined as 100% P(n + 1).
Our subunit population coverage estimates are not lowered by discarding subunit peptides as unsuitable. We consider all peptides that result from a windowing of the subunit proteome, and include the redundant peptides caused by using varying window sizes at the same proteome start position. In addition, we do not filter peptides for mutation rate or glycosylation during evaluation.
2.3 Design of separate MHC class I and II peptide sets to augment subunit vaccine population coverage
In the separate design method we use OptiVax-Robust (Liu et al., 2020) to augment subunit vaccines with additional peptides to produce separate sets of peptides for class I and class II augmentation (Figure 1A). These additional peptides are selected from the remaining SARS-CoV-2 proteome (all peptides except those spanning the subunit), excluding peptides that are likely to mutate (have mutation rate > 0.001) or have non-zero predicted probability of glycosylation (Section 2.1). All candidate peptides considered during augmentation have ≤ 50 nM predicted binding affinity (Section 2.1).
The augmentation algorithm uses a starting peptide set which is extracted from the subunit vaccine to maximize the coverage of the subunit while removing redundant peptides resulting from overlapping sliding windows using the redundancy elimination algorithm found in Liu et al. (2020). Using a non-redundant starting peptide set ensures that augmentation does not depend upon redundant peptides for population coverage support. OptiVax-Robust performs vaccine augmentation by adding peptides to this starting set to improve the population coverage at each peptide-HLA hits cutoff n. At each iteration redundant peptides are removed from consideration, and redundancy is defined with an edit distance metric (Liu et al., 2020). OptiVax-Robust uses a beam search algorithm that iteratively expands the solution by one peptide and gradually optimizes population coverage from n = 1 to the targeting level of per-individual peptide-HLA hits (Liu et al., 2020). We use a beam size of 5 for the augmentation of subunit vaccines.
For each desired budget of augmentation peptides, OptiVax produces an augmentation set. Larger augmentation sets are not necessarily supersets of smaller augmentation sets, as the underlying combinatorial optimization problem is complex. A vaccine designer can evaluate how many peptides they wish to use to realize a predicted population coverage. For the augmentation sets in Table 1 we targeted 99% coverage at n = 7 for MHC class I augmentation (except for S which we target at 99.5% coverage) and 97% coverage at n = 7 for MHC class II augmentation. These population coverage targets are achieved during augmentation of the non-redundant subunit. For the evaluation of original and augmented subunit vaccines in Table 1, we provide results for all window derived subunit peptides and the non-redundant set of subunit peptides. All window peptides can include the same HLA binding epitope multiple times from its sampling by multiple windows, and thus serves as the predicted lower bound on population insufficient coverage. The non-redundant results are the predicted upper bound of population insufficient coverage.
Percentage of a population that is insufficiently covered by subunit vaccines and the improvement after adding MHC class I and MHC class II augmentation peptides. Results are shown for both separate and joint designs of augmentation peptides. The list is sorted by decreasing insufficient coverage of unaugmented subunits.
2.4 Design of a single set of peptides to maximize MHC class I and II population coverage
We developed the OptiVax-Joint method to produce a minimal set of 25-mer peptides to reach a target population coverage probability at a threshold of n predicted hits for each individual for both MHC class I and class II (Figure 1B). The 25-mer candidate peptides are produced by windowing the pathogen proteome that is not part of a selected subunit, using a window step size of 8 amino acids between candidate peptides. Each of the candidate 25-mer peptides is annotated with its non-redundant peptides of length 8–10 (MHC class I) and 13–25 (MHC class II) and the HLA alleles they bind with ≤ 50 nM predicted binding affinity. Peptide redundancy is defined with an edit distance metric for the elimination of overlapping peptides (Liu et al., 2020).
OptiVax-Joint begins with the non-redundant set of peptides from a selected subunit or an empty set, and performs vaccine augmentation by adding candidate 25-mer peptides to this starting set to improve both MHC class I and class II population coverage at a target number of peptide-HLA hits n. When OptiVax-Joint is started with an empty set of peptides it produces a de novo peptide vaccine design without an associated subunit component. Each 25-mer is scored based on its contained annotated class I and class II peptides for its improvement in the number of per-individual peptide-HLA hits (Liu et al., 2020) over the haplotypes of the target population. Contained peptides are not counted towards population coverage if they have an observed mutation rate > 0.001 or have a non-zero predicted probability of glycosylation. OptiVax-Joint uses a beam search algorithm that iteratively expands the solution by one 25-mer peptide and gradually optimizes population coverage from n = 1 peptide hit to the targeted level of per-individual peptide-HLA hits for both MHC class I and class II (Liu et al., 2020). We use a beam size of 5 for the augmentation of subunit vaccines.
For each desired budget of augmentation peptides, OptiVax-Joint produces an augmentation set. Larger augmentation sets are not necessarily supersets of smaller augmentation sets, as the underlying combinatorial optimization problem is complex. A vaccine designer can evaluate how many peptides they wish to use to realize a predicted population coverage. For the joint augmentation sets in Table 1, we targeted 99% coverage at n = 7 for MHC class I augmentation and 97% coverage at n = 7 for MHC class II augmentation jointly.
3 Results
3.1 SARS-CoV-2 subunit population coverage analysis
We first computed the predicted number of peptide-HLA hits that would result from an infection by the SARS-CoV-2 virus as a baseline. With a redundant sampling of the SARS-CoV-2 proteome we predict SARS-CoV-2 will have 342 (White), 330 (Black), and 426 (Asian) peptide-HLA hits for MHC class I on average in the respective self-reporting populations. For an MHC class II redundant sampling we predict SARS-CoV-2 will have 5108 (White), 3822 (Black), and 2041 (Asian) peptide-MHC hits. Thus the average number of predicted SARS-CoV-2 peptide-HLA hits for MHC class I is 366 and for MHC class II 3557.
We found that all subunits of SARS-CoV-2 have gaps in their predicted population coverage for robust peptide MHC display, either by having no of peptide-HLA hits or fewer than six peptide-HLA hits. We computed the predicted uncovered percentage of S subunit variants as a function of the minimum required predicted number of peptide-HLA hits displayed by an individual (Figure 3). Results for other subunits are shown in Figure S1. As shown in Table 1, the predicted percentage of the uncovered population increases for smaller subunits. The two subunits with the least coverage are the fusion peptide (FP) domain which comprises 40 amino acids and the E protein which comprises 75 amino acids. The receptor binding domain (RBD) subunit is predicted to have no MHC class II peptides displayed in 23.68% of the population (averaged across Asian, Black, and White self-reporting individuals) with ≤ 50 nM affinity. We note that the uncovered population of RBD with no predicted display of MHC class II peptides ranges from 5.81% for the population self-reporting as White, to a high of 51.54% for the population self-reporting as Asian. Thus, clinical trials need to carefully consider ancestry in their study designs to ensure that efficacy is measured across an appropriate population. For the RBD subunit, 32.99% of the population had fewer than 6 MHC class II peptide-HLA hits. For RBD MHC class I, the coverage gap is 1.19% for no hits and 51.31% for fewer than six hits. EvalVax predicted that on average for an S subunit vaccine the uncovered population would be 0.004% (class I) and 1.24% (class II) for no display, and 1.002% (class I) and 4.22% (class II) for fewer than 6 peptide-HLA hits.

Predicted uncovered percentage of populations as a function of the minimum number of peptide-HLA hits in an individual. Annotated percentages are the average across populations self-reporting as Asian, Black, and White. A redundant sampling of peptides is depicted by solid lines for populations self-reporting as Asian, Black, and White as well their average. A non-redundant sampling of peptides is depicted by dotted lines.
3.2 SARS-CoV-2 subunit augmentation with separate peptide sets for MHC class I and II
We found augmentation peptides to improve the population coverage of MHC class I and class II peptide display for all of the SARS-CoV-2 subunit vaccines that we considered (Table 1; peptides given in Table S4). In the first design, we produced separate sets of peptides for display by MHC class I and class II molecules. We predicted the uncovered fraction of the population as a function of the additional peptides added to the vaccine subunits (Figure 4). We chose the augmentation set with minimal number of peptides that achieves the targeting criteria specified in Section 2.3. With the augmentation peptides, EvalVax predicted that on average for an S subunit vaccine the percentage of the population not displaying any vaccine peptides is reduced to 0.00% (class I) and 0.33% (class II), and for an RBD subunit vaccine it is reduced to 0.00% (class I) and 0.33% (class II). We then predicted the ability of the augmented vaccines to achieve more than five peptide-HLA hits and found the S subunit population coverage gap is reduced to 0.26% (class I) and 0.68% (class II), and 0.45% (class I) and 1.39% (class II) for RBD. The augmentation algorithm is also able to reduce the percentage of the population not displaying any vaccine peptides to 0.00% (class I) and less than 0.33% (class II), and reduce the population percentage displaying fewer than 6 vaccine peptides to less than 0.53% (class I) and less than 1.39% (class II) for all subunits by adding 16–42 additional peptides (Tables 1 and 2).

Predicted uncovered percentage of the population for MHC peptide display using separate sets for MHC class I and class II as a function of the number of augmentation peptides at different predicted peptide-HLA hit thresholds in each individual. The dotted vertical line shows the peptide count used in Table 1.
Number of peptides used in augmentation set listed in Table 1 and total number of amino acids required to build the full construct, including 10 amino acid linkers between every peptide.
We used stringent criteria for predicting peptide-MHC hits (Section 2.1) to produce conservative metrics for population coverage, and thus alternative binding criteria may show smaller gaps in population coverage. Additional evaluation of subunits and augmentations using NetMHCpan-4.1 (Reynisson et al., 2020a) (MHC class I) and NetMHCIIpan-4.0 (Reynisson et al., 2020b) (MHC class II) predicted eluted ligand (EL) percentile ranking for selecting binders are shown in Table S3.
3.3 SARS-CoV-2 subunit augmentation with a joint peptide set for MHC class I and II
We used Optivax-Joint to compute a joint set of SARS-CoV-2 peptides to maximize the predicted population coverage for a target number of MHC class I and class II peptide hits in every individual. We computed joint peptide sets to augment 9 SARS-CoV-2 subunits and predicted the uncovered fraction of the population as a function of joint peptide set size (Table 1, Figure 5). We chose the set with the minimal number of peptides that achieves the targeting criteria specified in Section 2.4 for Table 1. We show that joint design simultaneously increases the predicted population coverage for both MHC class I and class II display (Figure 5).

Predicted uncovered percentage of the population for MHC peptide display using a joint set as a function of the number of augmentation peptides at different predicted peptide-HLA hit thresholds in each individual. The dotted vertical line shows the peptide count used in Table 1.
3.4 A peptide-only SARS-CoV-2 vaccine design with a joint peptide set for MHC class I and II
We designed a standalone peptide vaccine that did not assume an associated subunit component and found that it was predicted to have greater than 94% population coverage with n ≥ 5 hits with 9 25-mer peptides (Table 3). We explored the predicted decrease of the uncovered population as a function of peptide count and found that joint vaccine designs are predicted to simultaneously produce a large number of predicted hits for both MHC class I and class II display (Figure 5). The predicted population coverage of a 24 25-mer peptide design exhibits a diverse display of peptides across populations self-reporting as Black, White, and Asian (Figure 6). Peptide-only designs have been found to be effective (Herst et al., 2020), and our joint design of a peptide-only vaccine is more compact than a separate design at equivalent levels of population coverage (Figure 7).
Predicted population coverage of a peptide-only vaccine jointly optimized for MHC class I and class II coverage with 4, 9, 14, 19, and 24 25-mer peptides.
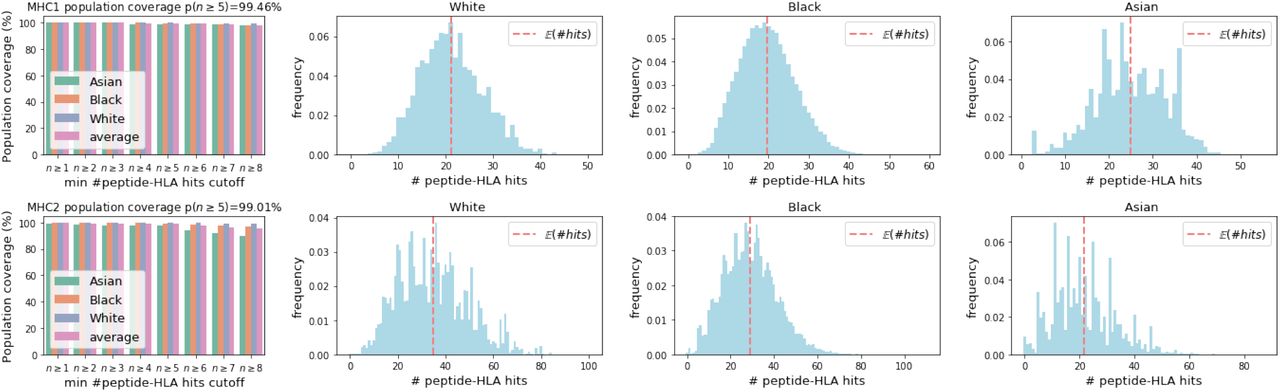
Predicted coverage in populations self-reporting as White, Black, and Asian with a peptide-only vaccine comprising 24 25-mer peptides jointly optimized for MHC class I and MHC class II coverage. The red dotted vertical line shows the expected number of hits.
Comparison of the number of amino acids used by separate and joint designs and their respective predicted population coverage with more than 7 peptide-HLA hits per individual. The predicted population coverage is shown for the augmentation of nine subunits and for a peptide-only de novo vaccine design.
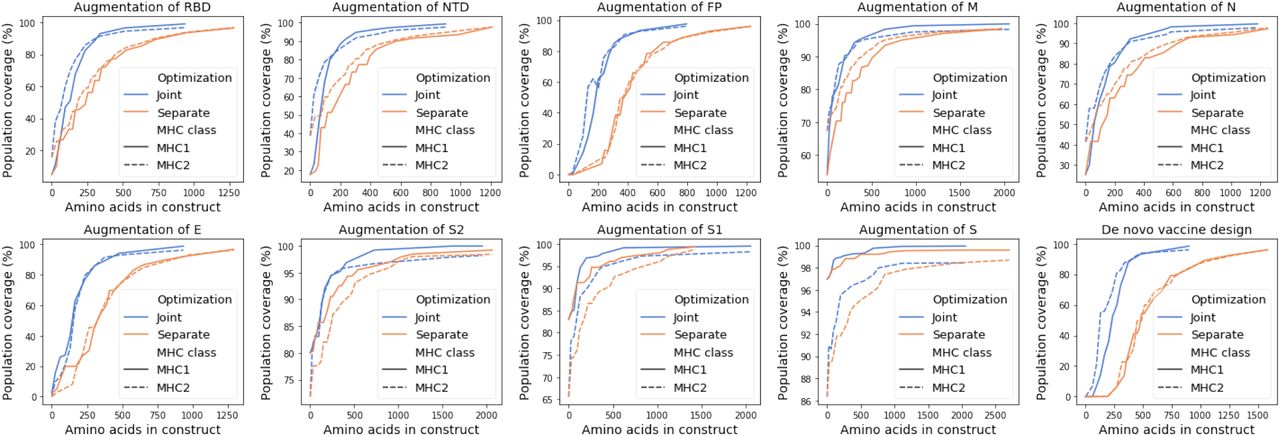
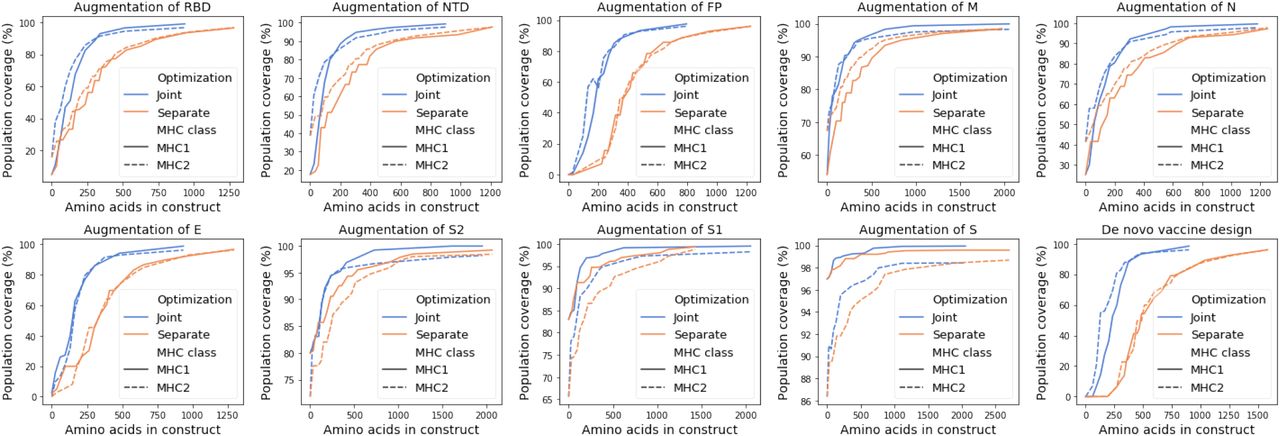
3.5 SARS-CoV-2 joint designs are more compact than separate designs
To compare the efficiency of joint optimization and separate optimization, we set a series of population coverage goals for MHC class I and MHC class II coverage with more than 7 peptide-HLA hits per individual. We considered 25 evenly spaced coverage levels between the unannotated subunit coverage and 100% coverage. We computed the total number of peptides needed to reach each set of MHC class I and MHC class II coverage goals simultaneously, where the number of MHC class I and MHC class II peptides are summed for separately designed peptide sets. We computed the total number of amino acids needed for a construct with a typical mRNA delivery platform with 10 amino acid linkers (Sahin et al., 2017) (Table 2, Section 4). A single mRNA delivery construct has been demonstrated to work for both MHC class I and class II peptides (Kreiter et al., 2008; Sahin et al., 2017). As shown in Figure 7 and Figure 8, the joint designs reduce the total number of required peptides and amino acids to achieve each level of population coverage.

Comparison of the number of peptides used by separate and joint designs and their respective predicted population coverage with more than 7 peptide-HLA hits per individual. The predicted population coverage is shown for the augmentation of nine subunits and for a peptide-only de novo vaccine design.
4 Discussion
We augment subunit vaccines with a compact set of peptides to improve the display of a vaccine on HLA class I and II molecules across a population of people. Subunit vaccines offer safety advantages over inactivated or attenuated pathogen vaccines, but their ability to fully mimic a pathogenic infection to train cellular immunity is limited. Immunity to a pathogen may rest in part upon T cell based adaptive immunity and corresponding T memory cells. We expect that a vaccine that provides a diverse display of a pathogen’s peptides will create reservoirs of CD4+ and CD8+ memory cells that will assist in establishing immunity to the pathogen.
We found that for SARS-CoV-2 the joint optimization of predicted MHC class I and class II pathogen peptide display achieves population coverage criteria with a more compact vaccine design than designing separate peptide sets for MHC class I and class II. Using a simpler design with shorter constructs may contribute to the effectiveness of a vaccine by providing an equivalent diversity of peptide display in a population with a less complex mixture of vaccine peptides.
Augmentation peptides can be delivered using the same vehicle as their associated subunit vaccine or they can be delivered separately. Nucleic acid based vaccines can incorporate RNA or DNA sequences that encode class I and class II augmentation peptides with desired signal sequences, linkers, and protease cleavage sites (Kreiter et al., 2008; Sahin et al., 2017) (examples in Supplementary Information, Tables S1 and S2). The peptides can be expressed as part of the subunit or separately, and can be encoded on the same or different molecules as the primary subunit. When augmentation peptides are added as a new subunit domain a vaccine designer can trade-off domain complexity for additional coverage using Figures 7 and 8. Nucleic acid constructs carrying augmentation peptides can be delivered by injection in lipid nanoparticle particle carriers or directly (Dowdy, 2017; Wolff et al., 1990). Protein based vaccines can include independent augmentation peptides into the vaccine formulation. The delivery of independent augmentation peptides can be accomplished using nanoparticles (Herst et al., 2020).
Our computational objective function encodes the two key goals of our augmentation strategy: population coverage and the display of a highly diverse set of peptides in each individual. Our population coverage goal is ensured by optimizing predicted display coverage over population haplotype frequencies. The display of a diverse set of peptides is established by setting augmentation design goals for the number of peptides that need to be displayed by each individual.
Early results from clinical studies of subunit vaccines for SARS-CoV-2 show that some vaccine recipients did not develop positive T cell responses. It is difficult to fully evaluate these results because the HLA types of study participants are not provided by these early studies. Thus these study populations may not be reflective of HLA types in the general world population. The BNT162b1 RBD subunit vaccine produced a less robust CD8+ response than CD4+ response (Sahin et al., 2020), and this was also noted in the mRNA-1273 S subunit vaccine results (Jackson et al., 2020). Further clinical data is required to fully assess the the T cell immunogenicity of various subunits and delivery methods. Clinical trials should select their participants to have representative HLA type distributions to test for population coverage. We note that in a study of SARS-CoV-2 subunit vaccines in non-human primates the level of neutralizing antibodies was observed to correlate with SARS-CoV-2 protection shortly after vaccination, but T cell response was not observed to correlate with protection (Yu et al., 2020). In this study rhesus macaques were vaccinated with an S subunit vaccine at week 0 and 3, T cell activity was measured at week 5, animals were challenged with SARS-CoV-2 at week 6, and then it was observed that week 5 T cell activity was uncorrelated with viral load post challenge. Future studies will need to examine the durability of immunity in individuals with minimal T cell response.
By simultaneously achieving the twin goals of coverage and diversity with peptides derived from a pathogen, we effectively compress the cellular immunologic fingerprint of a pathogen into a vaccine. To produce an antibody response, the subunit component of a vaccine can encode a three-dimensional epitope to stimulate neutralizing antibody production by B cells. Taken together, these two designed components, a pathogen subunit and its augmentation, will provide both B cell and T cell epitopes of a pathogen while permitting epitope selection to mitigate deleterious effects and improve population coverage.
All of our software and data are freely available as open source to allow others to use and extend our methods.
Supplementary Information
Sequence Construct Examples
Here we provide example construct designs optimized to augment RBD subunit vaccines. The constructs in Table S1 contain 29 peptides and 36 peptides independently optimized for either MHC class I and MHC class II, respectively (Table 1). Table S2 contains a construct with 31 25-mer peptides jointly optimized for both MHC class I and MHC class II. Peptides are prepended with a secretion signal sequence at the N-terminus and followed by an MHC class I trafficking signal (MITD) (Kreiter et al., 2008; Sahin et al., 2017). The MITD has been shown to route antigens to pathways for HLA class I and class II presentation (Kreiter et al., 2008). Here we combine all peptides of each MHC class into a single construct using a non-immunogenic glycine/serine linkers from Sahin et al. (2017), though it is also plausible to construct individual constructs containing single peptides with the same secretion and MITD signals as demonstrated by Kreiter et al. (2008).
Example protein constructs for augmentations to RBD subunit vaccines optimized for either MHC class I or MHC class II by OptiVax-Robust. Constructs contain a secretion signal sequence (red), peptides (bold) joined by non-immunogenic glycine/serine linkers, and an MHC class I trafficking signal (blue). The augmentation peptides encoded are the same as those evaluated in Table 1.
Example protein construct for augmentations to RBD subunit vaccines jointly optimized for both MHC class I and class II by OptiVax-Joint. Constructs contain a secretion signal sequence (red), 31 25-mer peptides (bold) joined by non-immunogenic glycine/serine linkers, and an MHC class I trafficking signal (blue). The augmentation peptides encoded are the same as those evaluated in Table 1.
Percentage of population that is insufficiently covered by subunit vaccines and the improvement after adding augmented peptides evaluated using eluted ligand (EL) ranking. Augmented peptides and the non-redundant subunit peptide sets are the same as in Table 1. The list is sorted by decreasing insufficient coverage of unaugmented subunits. Here, for evaluation, peptides are considered binders using recommended EL percentile rank strong binder thresholds by NetMHCpan-4.1 (≤ 0.5%, MHC class I) and NetMHCIIpan-4.0 (≤ 2%, MHC class II). The list is sorted by decreasing insufficient coverage of unaugmented subunits.
Table S4. Detailed augmentation designs for optimized peptide sets in Table 1. See AugmentationPeptides.xlsx.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Predicted uncovered percentage of populations as a function of the minimum number of peptide-HLA hits in an individual for E, M, N protein and fusion peptide (FP). Annotated percentages are the average across populations self-reporting as Asian, Black, and White. A redundant sampling of peptides is depicted by solid lines for populations self-reporting as Asian, Black, and White as well their average. A non-redundant sampling of peptides is depicted by dotted lines.
Acknowledgements
This work was supported in part by Schmidt Futures, a Google Cloud Platform grant, and NIH grant R01CA218094 to D.K.G. We benefited from thoughtful comments from Michael Birnbaum and Brooke Huisman.
References




