

ABSTRACT
Fibroblast growth factor 23 (FGF23) is a bone-derived hormone that binds to binary FGFR/α-Klotho receptor complexes in the kidney tubules to inhibit phosphate reabsorption and 1,25(OH)2D production. Excess FGF23 causes X-linked hypophosphatemia (XLH) and tumor induced osteomalacia (TIO). Recently, Burosumab, an FGF23 blocking antibody, was approved for treating these hypophosphatemic disorders. A small molecule inhibitor that specifically binds to FGF23 to prevent activation of FGFR/α-Klotho complexes has potential advantages over a systemically administered FGF23 blocking antibody. We previously identified the small molecule ZINC13407541 (N-[[2-(2-phenylethenyl)cyclopenten-1-yl]methylidene]hydroxylamine) as a therapeutic lead FGF23 antagonist. Additional structure-activity studies developed a series of ZINC13407541 analogues with enhanced drug-like properties. In this study, we tested in a pre-clinical Hyp mouse homologue of XLH a direct connect analogue (8n) [(E)-2-(4-(tert-butyl)phenyl)cyclopent-1-ene-1-carbaldehyde oxime] that exhibited the greatest stability in microsomal assays, and 13a [(E)-2-((E)-4-methylstyryl)benzaldehyde oxime] that exhibited increased in vitro potency. We found that pharmacological inhibition of FGF23 with either of these compounds blocked FGF23 signaling and significantly increased serum phosphate and 1,25(OH)2D concentrations in Hyp mice. Long-term parenteral treatment with 8n or 13a also enhanced linear bone growth, increased mineralization of bone, and narrowed the growth plate in Hyp mice. The more potent 13a compound showed greater therapeutic efficacy in Hyp mice. Further optimization of these FGF23 inhibitors may lead to versatile drugs to treat FGF23-mediated disorders.
INTRODUCTION
Fibroblast Growth Factor 23 (FGF23) is a hormone produced by osteoblasts/osteocytes in the bone that activates FGF receptor/α-Klotho (Kl) binary complexes in renal proximal tubules to regulate phosphate reabsorption and 1,25(OH)2D metabolism and in distal tubules to adjust sodium and calcium reabsorption (1, 2). FGF23 plays a causal role in hereditary hypophosphatemic disorders, such as X-linked (XLH) and autosomal recessive (ARH) hypophosphatemic rickets and acquired tumor-induced osteomalacia (TIO). Elevated FGF23 plays an adaptive role in maintaining phosphate homeostasis in chronic kidney disease and is associated with left ventricular hypertrophy and increased cardiovascular mortality in this setting (3–7).
Until recently, treatment of these hypophosphatemic disorders consisted of 1,25(OH)2D and phosphate supplements. This approach does not cure the disease and is associated with toxicities related to excess phosphate and 1,25(OH)2D, including nephrocalcinosis (8, 9). TIO can be cured by resection of the tumor that is producing FGF23, but surgical removal of the tumor is not possible in approximately 50% of these patients (10, 11).
Recent therapeutic advances use FGF23 blocking antibodies to treat disorders of FGF23 excess. An FGF23 blocking antibody KRN23 (Burosumab) has been recently approved for the treatment of XLH and TIO by the FDA(12, 13). KRN23 subcutaneously administered at an average dose of 0.98 mg/kg every two weeks improves rickets and increases serum phosphate levels in XLH (14), and was superior to conventional treatment with phosphate and 1,25(OH)2D supplements. To date, KRN23 has not been associated with toxicity at the doses used to treat hypophosphatemia, but the initial use of a high affinity FGF23 blocking antibody in pre-clinical rodent models resulted in excess mortality (15), principally due to over-suppression of FGF23 resulting in hyperphosphatemia and 1,25(OH)2D toxicity (16–18). Additional disadvantages of KRN23 include the need for systemic administration, long-half life (elimination half-life of 18 days), and cost.
Developing an orally, bioavailable small molecule drug to block FGF23 would have several potential advantages over KRN23. To this end, using homology modeling, molecular dynamics simulation, and virtual high-throughput screening we identified ZINC13407541 (N-[[2-(2-phenylethenyl)cyclopenten-1-yl]methylidene]hydroxylamine) which was experimentally validated to bind to FGF23 and inhibit its interaction with FGFRs (19). Subsequent medicinal chemistry investigation of structure-activity relationships based on ZINC13407541 produced 36 analogues with different stabilities and potencies. The core ZINC13407541 structure contains two ring systems, bridged with an ethylene, and a carbaldehyde oxime functional group on the ortho-position of ring A that is essential for activity (Figure 1). Replacing a five-membered aliphatic ring A with a six-membered ring or an aromatic system markedly increases potency of the FGF23 antagonists. A substitution of the electron-donating groups (CH3 or OCH3) on para-position of aryl ring B increased the in vitro FGF23 inhibitor effects. Removal of the ethylene bridge to create a direct connect derivative decreased efficacy, but increased metabolic stability (20).

Structure, in vitro efficacy, and metabolic stability of ZINC13407541 and its analogues 8n and 13a.
In this study, we tested the in vivo efficacy of the direct connect analogue (8n) [(E)-2-(4-(tert-butyl)phenyl)cyclopent-1-ene-1-carbaldehyde oxime], eliminating the ethylene bridge between the two ring systems of ZINC13407541, and the most potent compound (13a) [(E)-2-((E)-4-methylstyryl)benzaldehyde oxime] in the preclinical Hyp mouse model of XLH.
RESULTS
Pharmacokinetics, in vitro metabolic stability and IC50
We performed murine pharmacokinetics of the lead compound ZINC13407541. ZINC13407541 was administered as a single intravenous dose (1 mg/kg) and plasma concentrations were determined by LC-MS/MS. ZINC13407541 was found to have a half-life of ≈2 hours (Figure 1S).
Structure activity relationship (SAR) studies generated 36 compounds with different stability and potency in the in vitro assays (20). Compound (8n) [(E)-2-(4-(tert-butyl)phenyl)cyclopent-1-ene-1-carbaldehyde oxime], a direct connect analogue of ZINC13407541showed the greatest stability and 13a [(E)-2-((E)-4-methylstyryl)benzaldehyde oxime] the greatest potency of the 36 compounds generated.
As shown in Figure 1, compound 8n has eliminated the ethylene bridge to form a directly connected two-ring scaffold, which decreased maximum inhibition activity (% Maximum inhibition 84%), but showed increased IC50 (2.8 μM) and metabolic stability (31 minutes) compared to ZINC13407541 (100%, 5.0 μM, and 8 min). In contrast, compound 13a has exchanged the cyclopentene with a phenyl group, showing no changes in metabolic stability (9 minutes) and similar maximum inhibition activity (% Maximum inhibition 100%), but 36-fold higher potency for inhibiting FGF23 activity (13a, IC50= 0.14 μM) compared with ZINC13407541 (IC50 = 5.0 μM).
Molecular docking of experimentally verified hits on the N-terminal domain of FGF23
Analysis of our structural model of FGF23 shows that Gln156 is a consensus binding site on the N-terminal domain of FGF23 for ZINC13407541 and its two analogs, 13a and 8n (Figure 2). ZINC13407541 and 13a adopt similar conformations in silico. Further, ZINC13407541 (Figure 2B) and 13a (Figure 2C) each have two hydrogen bonds interactions with Gln156 and Asn122, and compound 8n (Figure 2D) has one hydrogen bond interaction with Gln156. Table 1 shows the estimated free energy of binding (ΔG) of the three compounds on FGF23 using two different methods: AutoDock VinaMPI (21, 22) and KDEEP (23). The differences in ΔG between the compounds are within the uncertainties associated with the methods used (22,23). Taken together, our computational results show that these three compounds are likely to bind to Gln156 on FGF23 with fairly similar binding affinities.

The computationally predicted interaction of ZINC13407541 and its two analogs, 13a and 8n, with the N-terminal domain of FGF23 (PDB code: 5W21) shown in A: 3D structure; in B, C, and D: 2D residue-contacting map for ZINC13407541, 13a, and 8n, respectively. Hydrogen bonds are shown in red dash lines with donor-acceptor distances in Å. Hydrophobic interactions are shown in gray.
Mineral ion homeostasis changes in serum of Hyp mice treated with single-dose or short-term FGF23 inhibitors
Next, we examined the in vivo efficacy of both 8n and 13a analogues compared to the parent molecule ZINC13407541 in preclinical animal studies. In Hyp mice a single intraperitoneal (IP) injection of ZINC13407541 resulted in increasing serum phosphate levels 2 hours after administration, peaked 4 hours after treatment, and returned to original levels by 24 hours post injection (Figure 3A and 3B). Also, the single IP injection of ZINC13407541 increased Hyp mice serum phosphate levels in a dose-dependent manner, the maximum effect achieved in the dose of 100 mg/kg ZINC13407541 (Figure 3A and 3B). In contrast, the ZINC13407541 treatment did not affect serum calcium levels (Figure 3C and 3D). Unexpectedly, Hyp mice with 100 mg/kg dosing of ZINC13407541 after 24 hours or 200 mg/kg dosing of ZINC13407541 after 4 hours exhibited a 2-fold increase in serum FGF23 levels (Figure 3E and 3F). Compound 13a showed same time- and dose-dependent responses similar to what we observed with ZINC13407541, but had a greater magnitude in the increase in serum phosphate levels in Hyp mice 4 hours after dosing, consistent with its improved IC50 (Figure 4A and 4B). At 100 mg/kg 13a almost completely corrected the phosphate levels of Hyp mice. No changes were observed in serum calcium and FGF23 levels in Hyp mice after administration (Figure 4C-4F). Subsequently, we performed short-term treatments with ZINC13407541 (100 mg/kg), 8n (100 mg/kg), or 13a (100 mg/kg) IP injection twice a day for 3 days. We obtained similar effects of these three compounds in increasing serum phosphate levels in Hyp mice (Figure 5A and 5B). No changes in serum calcium or FGF23 levels were observed with this short-term treatment regimen in Hyp mice (Figure 5C-5F). In long-term exposure studies, we decided to use half the effective dose (50 mg/kg) of ZINC13407541, 8n, and 13a IP injection twice a day for 4-weeks in Hyp mice to assess potential skeletal effects.
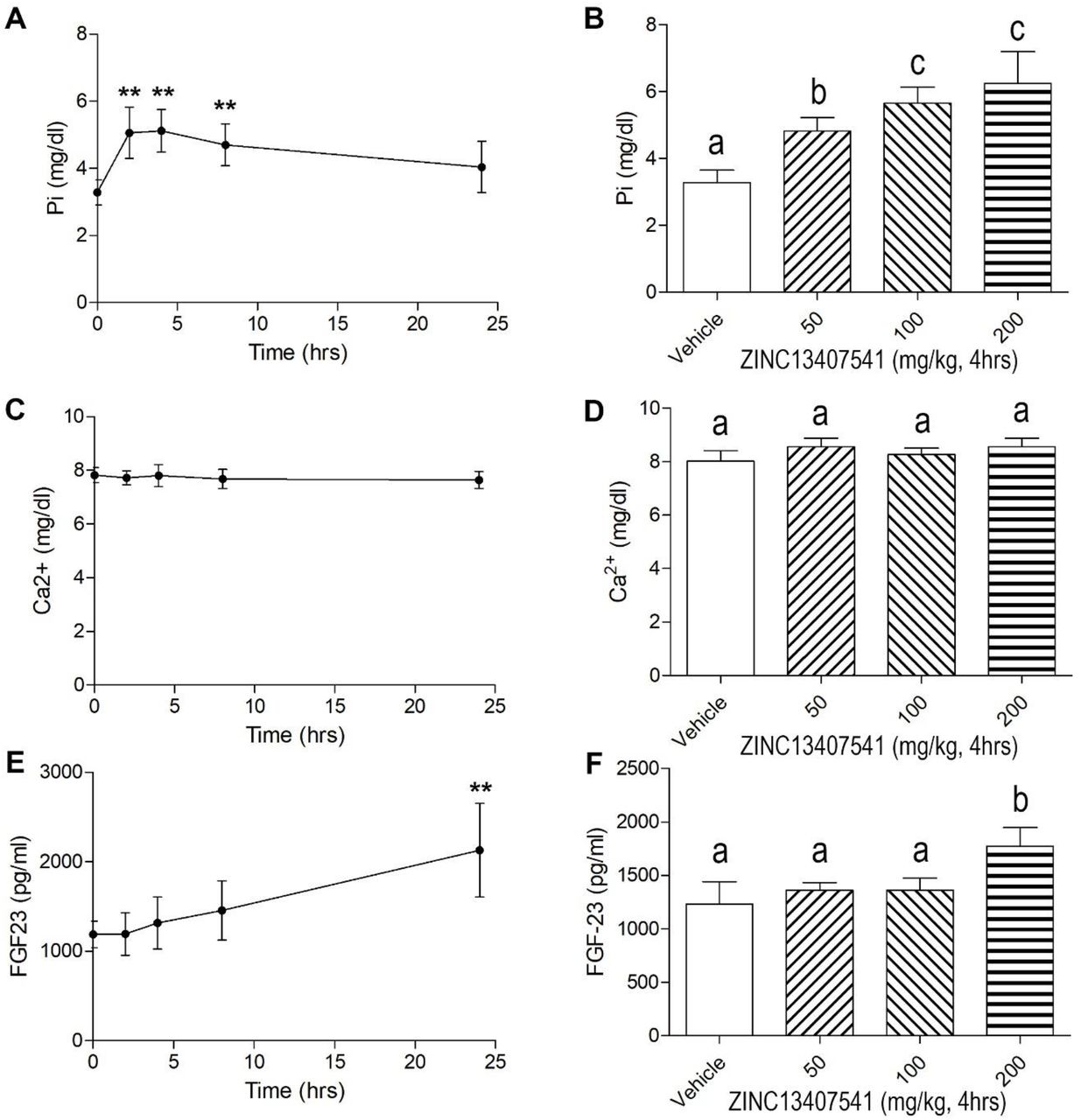
Time and dose dependent effects of ZINC13407541 on mineral ion homeostasis and FGF23 levels in Hyp mice. *, **, *** indicates significant difference from vehicle control group. Values (mean ± SD, n=5-6) with the different superscript are significantly different at P<0.05.

Time and dose dependent effects of 13a on mineral ion homeostasis and FGF23 levels in Hyp mice. *, **, *** indicates significant difference from vehicle control group. Values (mean ± SD, n=5-6) with the different superscript are significantly different at P<0.05.

Short-term effects of ZINC13407541, 8n, and 13a on mineral ion homeostasis and FGF23 levels in Hyp mice. Data are expressed as the mean ± S.D. from 5-6 mice. *, **, *** indicates significant difference from vehicle control group.
Serum biochemistry and skeletal phenotype changes of Hyp mice treated long-term with FGF23 inhibitors
We treated 4- to 8-week-old Hyp mice with either vehicle control, ZINC13407541 (50mg/kg), 8n (50mg/kg), or 13a (50mg/kg) for 4 weeks followed by measurement of serum biochemistry and assessment of skeletal features. Serum phosphate levels were significantly elevated in all treated groups compared to controls. The increase in serum phosphate was significantly greater in 13a compared to ZINC13407541 and 8n treated mice. No significant changes of FGF23 levels were observed in 8n and 13a treated groups compared to vehicle treated mice (Table 2). There was a 1.5-fold increase of FGF23 in the ZINC13407541 treated group, similar to previous observations (24). FGF23 is known to suppress parathyroid hormone (PTH) and active form (1,25(OH)2D) of vitamin D levels. Consistent with inhibition of FGF23 signaling, both PTH and 1,25(OH)2D levels were markedly increased in ZINC13407541, 8n and 13a treated mice. FGF23 also has distal tubular effects, increasing sodium reabsorption and suppressing aldosterone levels (25, 26). ZINC13407541, 8n, and 13a resulted in significant stimulation in aldosterone levels compared to vehicle treated mice (Table 2).
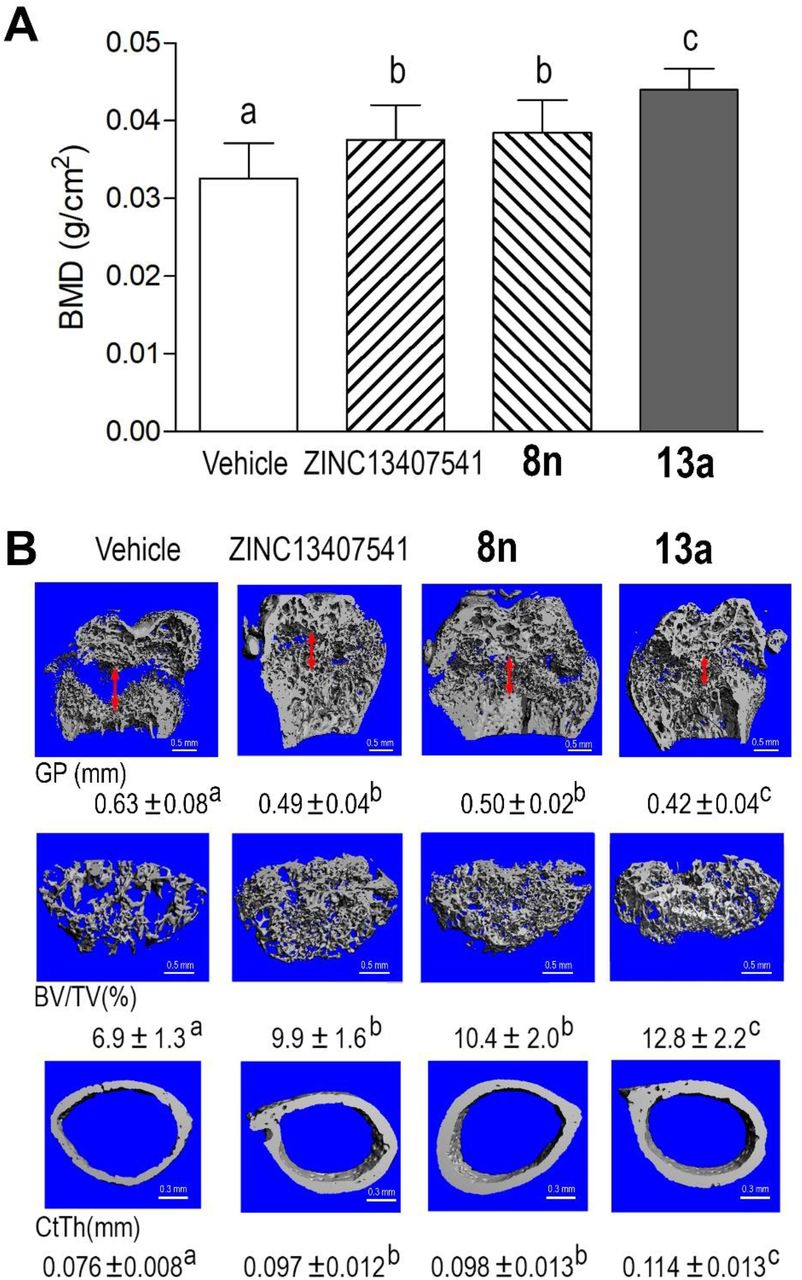
With regards to skeletal effects, we observed that body weight, body length, femur length, and tail length were significantly increased in ZINC13407541, 8n, or 13a treated groups compared to vehicle controls at the end of the treatment period (Figure 6). Hyp mice treated with ZINC13407541, 8n, or 13a showed 15%, 18%, and 30% increments in femoral bone mineral density (BMD), respectively, compared to vehicle controls (Figure 7A). Micro-CT 3D images revealed that both ZINC13407541 and 8n treated groups had similar increases in trabecular bone volume (TBV, 51%) and cortical bone thickness (Ct.Th, 30%). The Hyp mice treated with 13a had greater increases in both TBV (78%) and Ct.Th (44%) than either ZINC13407541 or 8n group (Figure 7B). The width of the growth plate was significantly decreased following treatment with ZINC13407541, 8n, or 13a. In summary, 13a had greater therapeutic effects on the healing of the growth plate in Hyp mice (Figure 7B).

Long-term effects of ZINC13407541, 8n, and 13a on body length (BL) (A), body weight (B), tail length (C), and femur length (D) in Hyp mice. Data are expressed as the mean ± S.D. from 6-10 mice. Values with the different superscript are significantly different at P<0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Long-term treatment of ZINC13407541, 8n, and 13a on bone mineral density (BMD), width of the growth plate (GP, double red arrow), femoral bone volume (BV/TV), and cortical thickness (CtTh) in Hyp mice. (A) BMD, (B) Micro-CT 3D images. Data are expressed as the mean ± S.D. from 6-10 mice. Values with the different superscript are significantly different at P<0.05.
Gene expression profiles changes in bone of Hyp mice treated long-term with FGF23 inhibitors
Hyp mice are known to have increased FGFR1 signaling and Wnt/β-catenin signaling, and a characteristic gene expression profile in bone associated with the presence of rickets/ osteomalacia (27–29). Real-time RT-PCR revealed that the antagonist-treated groups significantly reduced FGFR1 expression and attenuated Wnt/β-catenin signaling, as evidence by reductions in Wnt10b and Axin2. Reduction in expression of osteoblast message levels for Type 1 collagen, ALP, and Dmp1 were observed (Table 3). In contrast, mature Ob markers such as MEPE and Osteocalcin were significantly upregulated in femurs from the antagonists treated groups. In addition, adipocyte markers Pparγ2, aP2, and lipoprotein lipase (Lpl) were significantly downregulated in all antagonists treated groups (Table 3). The group treated with 13a had greater effects in all gene expressed markers than did either ZINC13407541 or the 8n group. However, there was no obvious changes in osteoclast markers including OPG, RankL, Mmp9, and Trap as well as Fgf23 and Fgf2 transcripts in the treated groups as compared to vehicle treated controls (Table 3). Compound 13a had greater therapeutic effects on Hyp mouse homologue of XLH.
Renal FGF23 signaling changes in kidney of Hyp mice treated long-term with FGF23 inhibitors
Four weeks of treatment with FGF23 inhibitors significantly altered FGF23 responsive gene expression in kidney of Hyp mice (Table 4). Compared with ZINC13407541 and 8n groups, 13a exhibited greater effects to increase Npt2a and Npt2c message expression, consistent with higher serum phosphate levels in the 13a treated groups. Consistent with FGF23 stimulation of Cyp27b1, and inhibition of Cyp24a1, leading to reductions in circulating 1,25(OH)2D, the administration of ZINC13407541 and its analogues to Hyp mice increased the serum concentration of 1,25(OH)2D (Table 2), in association with increased Cyp27b1 and decreased Cy24a1 message expression (Table 4). FGF23 also stimulates NCC expression in the distal renal tubule cells leading to increased Na+ and K+ channel activity, sodium retention, and suppression of serum aldosterone. ZINC13407541, 8n, and 13a treatment suppressed NCC expression (Table 4), in association with increased circulating aldosterone levels in Hyp mice. In contrast, treatment with ZINC13407541, 8n, and 13a had no effects on Fgfr1 expression, but significantly increased Klotho transcripts in kidney of Hyp mice (Table 2). These data indicate that ZINC13407541 and its analogues may have clinical utility in blocking the effects on the kidney of excess FGF23.
Potential off-target effects of compound 8n
In contrast to the specificity of blocking antibodies, small molecules have a greater potential for off-target effects. Indeed, radioligand binding assays revealed that compound 8n has greater than 50% inhibition of androgen, serotonin, dopamine, and norepinephrine receptors and activation of histamine receptor (Table 5 and Table 1S).
DISCUSSION
We are pursuing the development of small molecules that bind to FGF23 and inhibit its activation of the FGFR/α-Klotho binary receptor complex. Herein we report on the efficacy of ZINC13407541 derivatives that displayed either greater stability in in vitro microsomal assays or greater potency in in vitro functional assays. Compound 8n and 13a (most potent) both demonstrated short- and long-term efficacy in blocking FGF23 end-organ effects in Hyp mice, a human homologue of XLH, used in pre-clinical assessment of KRN23. We found that pharmacological inhibition of FGF23 efficiently abrogates aberrant FGF23 signaling and corrects the hypophosphatemia caused by elevated FGF23 levels in Hyp mice. Increases in serum phosphate levels were observed as early as 3 to 5 hours after intraperitoneal injections of these compounds. The minimal effective dose was 50 mg/kg body weight.
Long-term FGF23 inhibition with 8n and 13a treatment of Hyp mice for 4 weeks resulted in sustained increases in serum phosphate and 1,25(OH)2D levels, enhanced bone growth, increased bone mineralization, and improvement of the rachitic growth plate abnormalities. 13a had greater therapeutic effects on FGF23-induced abnormalities in Hyp mice, consistent with its lower IC50. The improvement in biochemistries in Hyp mice after treatment with these chemical FGF23 inhibitors was associated with reversal of FGF23’s renal effects. Elevated FGF23 in Hyp mice suppresses the phosphate transporters Npt2a and Npt2c and Cyp27b1 the enzyme that 1-hydroxylates 25(OH)D, and suppresses Cyp24a1, which degrades 1,25(OH)2D in the proximal tubule. Consistent with the increase in serum phosphate and 1,25(OH)2D levels, 8n and 13a treatment increased Npt2 and Cyp27b1 expression and decreased Cyp24a1 message expression.
Excess FGF23 in Hyp mice also leads to stimulation of the sodium chloride co-transporter in the distal tubule that leads to sodium retention and suppression of aldosterone (30). Treatment with 8n and 13a also increased aldosterone levels and decreased expression of NCC, the sodium chloride transporter in the distal tubule. FGF23 also suppresses α-Klotho message expression in the distal tubule in Hyp mice. Treatment with 8n and 13a also increased α-Klotho transcript levels in Hyp mice. Thus, these small molecule FGF23 inhibitors antagonized both the proximal and distal tubular effects of FGF23 excess.
Excess FGF23 has both indirect effects on bone and cartilage mineralization mediated by the reductions in serum phosphate and 1,25(OH)2D (31, 32), as well as possible direct effects due to FGF23 activation of FGFRs in osteoblasts (33, 34). We observed substantial improvement in bone abnormalities after only 4 weeks of treatment as well as alteration in gene expression profiles in bone. Treatment with 8n and 13a altered osteoblast function as evidence by decreased Runx2-II, Alp, Col1, Opn (35), and Dmp1 and increased Mepe, Bsp, and Osteocalcin expressions. FGF23, may also have effects on adipocytes to modulate adipogenesis (30). This may explain the effects of FGF23 antagonists in reducing adipogenic markers in Hyp bone. Treatment with 8n and 13a significantly reduced Fgfr1, Wnt10b, Fzd2, and Axin2 gene transcripts, consistent with our previous findings that Hyp mice exhibited higher Fgfr1 and Wnt signaling (27) (28). In addition, treatment with 8n and 13a did not increase the FGF23 transcripts and serum FGF23 levels in Hyp mice, which differs from increased serum total FGF23 levels following treatment of XLH patients with FGF23 blocking antibodies (https://www.ultragenyx.com/file.cfm/95/docs/Crysvita_Full_Prescribing_Information.pdf)
Off-target effects of small molecule inhibitors are a potential liability compared to the high target specificity of blocking antibodies. In this regard, 8n blocked the androgen receptor, as well as dopamine, norepinephrine, and serotonin receptors and activated the histamine receptor in vitro. Additional chemical modification may be needed to further increase the specificity of these leads for FGF23.
The short half-life of small molecules may be of considerable clinical benefit in titrating and reversing drug effects, which may be particularly important given the narrow therapeutic window for FGF23 suppression, particularly in chronic kidney disease (CKD). Preclinical CKD models show that inhibiting FGF23 with a blocking antibody increases mortality due to oversuppression of FGF23 (15). Drawing analogies to treatment of secondary hyperparathyroidism with short acting calcimimetics in CKD to lower PTH levels, and pointing to data that even partial reductions of FGF23 may be sufficient to reduce cardiovascular effects, a short-acting, titratable FGF23 antagonist might reduce FGF23 dose-dependent cardiotoxicity with acceptable side effects in CKD (1, 36, 37). Further studies are needed to test the effects of these small molecules in models of CKD.
In conclusion, these studies further advance the premise that the development of FGF23 small molecule inhibitors is a novel approach for treatment of FGF23-mediated hypophosphatemic diseases. Small molecules are generally more cost-effective, have a longer shelf life, and are more easily distributed. In addition, an oral anti-FGF23 therapy with small molecule inhibitors has the potential to expand the current treatment repertoire by increasing patient satisfaction and adherence.
MATERIALS AND METHODS
Molecular docking
To computationally analyze the binding conformation and affinity of ZINC13407541 and its two analogs, 13a and 8n, to FGF23, molecular docking of the three compounds on FGF23 was performed using AutoDock VinaMPI (21, 22). The crystal structure of FGF23 [PDB code: 5W21(38)] was used as the receptor, and a pose-searching box of 40 x 30 x 40 Å was centered at the geometric center of Gly38 to Arg161 to include the whole N-terminal domain of FGF23. An exhaustiveness of 30 was used for adequate sampling of ligand conformations within the box. Following docking, the pose with the best score (i.e., the lowest value indicating estimated free energy of binding, ΔG) for each compound was plotted in 3D and 2D using VMD (39) and LigPlot+ (40), respectively. For hydrogen-bond calculation in LigPlot+, the maximum hydrogen-acceptor and maximum donor-acceptor distances were set at 3.0 and 4.0 Å, respectively. For non-bonded contact calculation, the minimum and maximum contact distances were set at 2.0 and 4.0 Å, respectively. KDEEP (23), a protein-ligand binding affinity predictor based on deep convolutional neural networks, was also used to obtain ΔG for each compound as a comparison with AutoDock VinaMPI (21, 22).
Chemicals and reagents
Synthetic preparation of new ZINC13407541 analogues was conductend in the medicinal chemistry lab at UTHSC College of Pharmacy and Tennessee Technological University. All reagents used for organic synthesis were sourced from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Fairfield, NJ), and stored in accordance with manufacturer recommendations. Each test compound was fully characterized by mass spectrometry and NMR (1H and 13C), with a >95% chemical purity which was determined with HPLC. The medicinal chemistry group also generated gram-scale quantitates for in vivo animal studies as we previously reported (24). The compounds were stored at −20 °C freezer and were tested both in vitro assays and in vivo in the laboratories.
Recombinant human FGF23 and FGF2 were purchased from R&D Systems (Minneapolis, MN, USA). Synthetic human C-terminal FGF23 (residues 180-251) peptide (FGF23CT), which binds to α-KL and blocks full-length FGF23 binding to the FGFR/α-KL binary complex, was obtained from Phoenix Pharmaceuticals, Inc. (Burlingame, CA, USA). Recombinant human FGF-, FGF-19, and FGF-21 were purchased from PeproTech (Rocky Hill, NJ, USA).
Cell culture and in vitro functional assays
HEK293T cells were cultured in DMEM containing 10% FBS and 1% penicillin and streptomycin (P/S).To test the effects of the novel compounds on FGF23-mediated activation of FGFR1/α-KL complex, HEK293T cells were transiently transfected with either empty expression vector or full-length human α-KL along with the ERK luciferase reporter system (41) and Renilla luciferase-null as internal control plasmid. Transfections were performed by electroporation using Cell Line Nucleofector Kit R according to the manufacturer’s protocol (Amaxa, Inc., Gaithersburg, MD). Thirty-six hours after transfection, the transfected cells were treated with the test compound with a range of 10−9~10−4 M in the presence or absence of 1 nM FGF23. After 5 hours, the cells were lysed and luciferase activities measured using a Synergy H4 Hybrid Multi-Mode Microplate Reader (Winooski, VT, USA) and Promega Dual-Luciferase Reporter Assay System (Madison, WI, USA).
In vitro ADME and in vivo pharmacokinetics (PK) studies
We contracted the in vitro ADME screening and in vivo PK studies with Eurofins Pharma Discovery Services. Microsomal stability of ZINC13407541 and ZINC13407541 analogues was determined in liver microsomes, including human liver microsomes (HLM) and mouse liver microsomes (MLM) to determine potential species differences in metabolism (42). Aqueous solubility was assessed in PBS (pH 7.4), simulated intestinal fluid, and simulated gastric fluid. We also extended our assessment of compound in vitro ADME by assessing protein binding, permeability, and metabolism. We assessed protein binding using rapid equilibrium dialysis, as previously described (43). A study on single dose pharmacokinetics after intravenous (i.v.) and oral administration determined basic PK parameters including clearance, volume of distribution, elimination half-life, systemic exposure, residence time and oral bioavailability. Mice (24/route of administration) received a single oral or i.v. (1~10 mg/kg) dose of nominated analogues. Mice (n=3/time point) were sacrificed at 0, 5, 15, 30, 60, 90, 120, 240, 360 and 1440 min. Plasma were stored at −80°C until analysis by LC-MS/MS. PK parameters (bioavailability, clearance, and half-life) were determined using non-compartmental methods (44).
RNA purification and quantitative real-time RT-PCR
For quantitative real-time RT-PCR, 1.0 μg total RNA isolated from heart, kidney or long bone of 8-week-old mice was reverse transcribed as previously described (45, 46). PCR reactions contained 100 ηg template (cDNA or RNA), 300 ηM each forward and reverse primers, and 1XqPCR Supermix (Bio-Rad, Hercules, CA) in 50 μl. The threshold cycle (Ct) of tested-gene product from the indicated genotype was normalized to the Ct for cyclophilin A. Expression of total Klotho isoform transcripts was performed using the following Klotho-isoform-specific primers: Forward primer of mouse m-KL135 transcript: 5’-CAT TTC CCT GTG ACT TTG CTT G-3’, and reverse primer: 5’-ATG CAC ATC CCA CAG ATA GAC-3’. Forward primer of mouse s-KL70 transcript: 5’-GAG TCA GGA CAA GGA GCT GT-3’, and reverse primer: 5’-GGC CGA CAC TGG GTT TTG-3’. The sequences of primers for kidney and bone gene transcripts were previously reported (45, 46). In addition, the fold change of tested gene transcripts was calculated from the relative levels of the normal gene transcripts in wild-type mice.
Specificity binding assays by radioligand binding assays of small molecule 8n
We contracted the specificity binding assays of compound 8n with Eurofins Panlabs Discovery Services Taiwan, Ltd. Radioligand binding assays were performed using LeadProfilingScreen (Total number of Assays: 67) and compound 8n at a concentration of 10 μM. Biochemical assay results presented as the percent inhibition of specific binding or activity throughout the report. Significant responses (≥ 50% inhibition or stimulation for biochemical assays) were noted in the primary assays listed (47, 48).
Animal experiments
All animal research was conducted according to guidelines provided by the National Institutes of Health and the Institute of Laboratory Animal Resources, National Research Council. The University of Tennessee Health Science Center’s Animal Care and Use Committee approved all animal studies (Protocol number: 18-111.0). All mice were maintained in our vivarium on a standard diet (7912; Harlan Teklad, Madison, WI, USA). To generate hemizygous Hyp mice, we crossed male hemizygous with female wild-type to obtain both male and female hemizygous Hyp mice as previously described (28). At 4 weeks of age, hemizygous Hyp mice were randomly assigned to the following experiments. For single-dose experiment, the Hyp mice received a single IP dose of ZINC13407541 (0, 50, 100, 200 mg/kg) or 13a (0, 10, 50, 100 mg/kg) at time 0 and were collected serum samples 0, 2, 4, 8, and 24 hours after administration. For short-term efficacy studies, the Hyp mice were divided into four different groups: (1) Vehicle control; (2) ZINC13407541 (100 mg/kg); (3) 8n (100 mg/kg); and (4) 13a (100 mg/kg). The mice were treated with intraperitoneal injection of either vehicle control (5% DMSO in corn oil), or ZINC13407541, or 8n, or 13a twice a day for 3 days. For long-term treatments, the Hyp mice were divided into four different groups: (1) Vehicle control; (2) ZINC13407541 (50 mg/kg); (3) 8n (50 mg/kg); and (4) 13a (50 mg/kg). The mice were treated with intraperitoneal injection of either vehicle control (5% DMSO in corn oil), or ZINC13407541, or 8n, or 13a twice a day for four weeks. We assessed the effects of the compounds on the skeletal phenotype at 8 weeks of age (after 4 weeks treatment) using the methods previously described in studies that characterize the in vivo phenotypes of Hyp mice (28). The serum samples were collected 4 hours after last dose administration. Serum FGF23 levels were measured using the FGF23 ELISA kit (Kainos Laboratories, Tokyo, Japan). Serum phosphorus levels were measured using a Phosphorus Liqui-UV kit (Stanbio Laboratories, Boerne, TX, USA) and serum calcium levels were measured using a Calcium (CPC) Liquicolor kit (Stanbio Laboratories, Boerne, TX, USA). Serum PTH levels were measured using the Mouse Intact PTH ELISA kit (Immutopics, Carlsbad, CA, USA). Serum aldosterone levels were measured using aldosterone ELISA Kit (Cayman Chemical, MI, USA). Serum 1,25(OH)2D levels were measured using the 1,25-dihydroxy-vitamin D EIA Kit (Immunodiagnostic Systems, Fountain Hills, AZ, USA) as previously escribed (28).
Bone densitometry, histomorphometric, and micro-CT analysis
Bone mineral density (BMD) of femurs was assessed at 8 weeks of age using a small animal bone densitometer (Lunar Corp, Madison, WI). Calcein (Sigma-Aldrich, St. Louise, MO) double labeling of bone and histomorphometric analyses of periosteal mineral apposition rate (MAR) in tibias were performed using the osteomeasure analysis system (Osteometrics, Inc., Decatur, GA). The distal femoral metaphyses were also scanned using a micro-CT 40 scanner (Scanco Medical AG, Brüttisellen, Switzerland). A 3D images analysis was done to determine width of growth plate (GP), bone volume (BV/TV) and cortical thickness (Ct.Th) as previously described (49, 50).
Statistical analysis
We evaluated differences between two groups by unpaired t-test, multiple groups by one-way analysis of variance, and two groups over time by two-way analysis of variance with interactions (Table S3). All values are expressed as means ± S.D. All computations were performed using a commercial biostatistics software (GraphPad Software Inc. La Jolla, CA).
Acknowledgement
This work was supported by grant R01-DK121132 to LDQ from the National Institutes of Health. K.L. and L.P. were sponsored by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory. This research also used resources of the Compute and Data Environment for Science (CADES) at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725.
REFERENCES




