

ABSTRACT
Microglial TYROBP (also known as DAP12) has been identified by computational transcriptomics as a network hub and driver in late-onset sporadic Alzheimer’s disease (AD) and as an important regulator of the microglial environmental sensing function. TYROBP is the transmembrane adaptor of AD-related receptors TREM2 and CR3, but importantly, TYROBP interacts with many other receptors, and little is known about its roles in microglial action and/or in the pathogenesis of AD. Herein, using dual RNA in situ hybridization and immunohistochemistry, we demonstrate that endogenous Tyrobp transcription is increased specifically in recruited microglia in the brains of wild-type and AD-related mouse models. To determine whether chronically elevated TYROBP might modify microglial phenotype and/or progression of AD pathogenesis, we generated a novel transgenic mouse overexpressing TYROBP in microglia. TYROBP-overexpressing mice were crossed with either APP/PSEN1 or MAPTP301S mice, resulting in a decrease of the amyloid burden in the former and an increase of TAU phosphorylation in the latter. Apolipoprotein E (Apoe) transcription was upregulated in MAPTP301S mice overexpressing TYROBP and transcription of genes previously associated with Apoe, including Axl, Ccl2, Tgfβ and Il6, was altered in both APP/PSEN1 and MAPTP301S mice overexpressing TYROBP. Lastly, Tyrobp and Apoe mRNAs were clearly increased in Trem2-null mice in microglia recruited around a cortical stab injury or amyloid-β (Aβ) deposits. Conversely, microglial Apoe transcription was dramatically diminished when Tyrobp was absent. Our results provide compelling evidence that TYROBP-APOE signaling in the microglial sensome does not require TREM2. We propose that activation of a TREM2-independent TYROBP-APOE signaling could be an early or even initiating step in the transformation of microglia from the homeostatic phenotype to the Disease-Associated Microglia (DAM) phenotype.
INTRODUCTION
Microglia play a sentinel role in the brain, capable of detecting a wide variety of environmental stimuli, including microbial pathogens, aggregated proteins (such as amyloid-β; Aβ) and cellular debris (such as membrane fragments). This sensing activity is an essential part of the host response and is broad in its scope, sometimes triggering homeostatic adjustment, while, at other times, host defense. Microglia are also of interest in neurodegenerative diseases due to proteinopathies, e.g. Alzheimer’s disease (AD), in which large genetic studies have reported increased disease risk linked to many loci associated with microglial genes implicated in clearance of Aβ peptides1–7. More recently, transcriptomic analyses have revealed distinct profiles and signatures for microglia associated with aging and age-related diseases, indicating that a wide range of specific proteins in microglia underlies sensing, activation, and/or other cellular responses. Using RNA sequencing, Hickman et al. (2013) identified 100 transcripts highly enriched in microglia and coined the term “sensome” to describe this class of microglial transcripts8. Network analysis of this list identified a TYROBP (for tyrosine kinase binding protein; also known as DAP12, for DNAX activating protein-12)-centered pathway with 24 of these 100 genes interacting directly with TYROBP and 20 interacting indirectly with TYROBP. Concurrently, members of our team employed an integrative network-based approach and identified TYROBP as a key network driver in sporadic late-onset Alzheimer’s disease (AD)9. More recently, Keren-Shaul et al.10 used single-cell RNA sequencing in mouse to define a specific microglial phenotype that they termed “Disease-Associated Microglia” (DAM). Tyrobp was one of the genes most robustly upregulated in the proposed earliest stage of transition of microglia from the basal “homeostatic state” into the DAM phenotype.
TYROBP is a transmembrane signaling polypeptide that contains an immunoreceptor phospho-tyrosine-based activation motif (ITAM) in its cytoplasmic domain. TYROBP is expressed in microglia and serves as an adaptor for a variety of immune receptors, including two molecules closely linked to AD pathogenesis: TREM2 (triggering receptor expressed on myeloid cells 2) and CR3 (complement receptor 3). TREM2 is expressed at the plasma membrane of microglia in the brain and some mutations and polymorphisms of TREM2 are linked to autosomal dominant AD or sporadic late-onset AD11. Other TREM2 mutations can also cause a polycystic leukoencephalopathy osteodystrophy also known by the eponym Nasu-Hakola disease12. Most TYROBP mutations represent loss-of-function mutations and also result in Nasu-Hakola disease13. Similarly, TYROBP genetic variants have also been identified in early-onset AD14.
TREM2 is required in order for microglia to limit growth of amyloid-β (Aβ) deposits and is essential for a full transition of homeostatic microglia to a DAM state. Keren-Shaul et al.10described a two-stage program for DAM transition with a Trem2-independent step (stage 1) where Tyrobp and other genes are upregulated, followed by a Trem2-dependent step during which both Tyrobp and Trem2 are upregulated (stage 2). Krasemann et al.15described a very similar microglial signature associated with neurodegenerative diseases, designated the “MGnD” phenotype, and showed that the transition from homeostatic to MGnD microglia was both TREM2- and APOE-dependent with a TREM2-APOE signaling pathway driving the transition from homeostatic microglia to MGnD. Notably, when DAM and MGnD are compared, Keren-Shaul et al10. also observed by single-cell RNA sequencing an apparent sequence of events whereby Tyrobp was upregulated prior to the upregulation of Trem2. For clarity, since DAM and MGnD microglia appear to share key features of the phenomena described here, we will refer only to DAM for the remainder of this report. Insofar as we are aware, principles established here underpin both DAM and MGnD.
In light of the central role of TYROBP in the microglial sensome8, its key role as adaptor for multiple microglial receptors16, its upregulation in the early Trem2-independent DAM stage 110, and its upregulation in AD9, we employed a number of strategies aimed at interrogation of the causes and consequences of TYROBP upregulation. Using dual RNA in situ hybridization and immunohistochemistry, we found that Tyrobp mRNA level is significantly increased when microglia are recruited, including in wild-type (WT) mouse, in an APP/PSEN1 transgenic mouse model of cerebral Aβ amyloidosis, and in a MAPTP301S transgenic mouse model of tauopathy. To determine whether elevated TYROBP can modify microglial phenotype and AD pathogenesis, we generated a novel transgenic mouse, designated the Iba1Tyrobpmouse, wherein the Iba1 promoter was used to drive overexpression of a mouse Tyrobp transgene in microglia. The APP/PSEN1 and MAPTP301S phenotypes were both altered in the setting of a constitutive increase in TYROBP, as was the transcription of Apoe and associated genes. Finally, using two mouse models of cerebral Aβ amyloidosis and a mouse model of penetrating cortical stab injury, we show that upregulation of Tyrobp and Apoe does not require Trem2, but that upregulation of microglial Apoe requires Tyrobp to reach normal levels.
Our results provide compelling evidence that TYROBP-APOE signaling in the microglial sensome is independent of Trem2, and we propose that activation of this pathway could be an early or even initiating step in the transformation of microglia from the homeostatic phenotype to the DAM phenotype.
MATERIALS & METHODS
Animals
All experimental procedures were conducted in accordance with the NIH Guidelines for Animal Research and were approved by the Institutional Animal Care and Use Committee (IACUC) at Icahn School of Medicine at Mount Sinai. TYROBP-overexpressing mice, named after Iba1Tyrobp mice, were constructed by cloning the mouse Tyrobp and Egfp sequences separated by an internal ribosome entry site (IRES) under the mouse Iba1 promoter in a pBluescript II SK(+) vector17. GFP protein was not detected by immunohistochemistry. Microinjection were performed in C57BL/6J mice by Dr Kevin Kelly here at Mount Sinai. APPKM670/671NL/PSEN1∆exon9 (APP/PSEN1) and MAPTP301S (PS19) mice were obtained from Jackson Laboratories. Tyrobp knockout (Tyrobp−/−) mice were obtained from Taconic/Merck Laboratory. TgCRND8 were obtained from Dr Paul Fraser. Trem2 knockout (Trem2-/-) mice were constructed by use of targeted homologous recombination to remove exons 1 and 2, thereby deleting the start codon and the major extracellular IgG domain. In contrast to the reported Velocigene construct, the direction of the Hygromycin cassette was “reversed”. Crucially, in agreement with two other models, but in contrast to the Velocigene construct, RT-qPCR analyses confirmed (data not shown) specific loss of Trem2 expression without the perturbation of Treml1 expression observed in the Velocigene construct18. For the penetrating cortical stab-injured mice, WT, Tyrobp−/− and Trem2−/− mice were anesthetized by an intraperitoneal injection of ketamine/xylazine (80/16 mg/kg body weight) and placed in a stereotactic frame (Stoelting, Wood Dale, IL, USA). The skull was drilled and an empty Hamilton syringe was introduced for 1 minute at the following coordinates (relative to bregma): anteroposterior, −0.3 mm; mediolateral, +3 mm; dorsoventral, −2mm. Mice were killed 3 days after.
Tissue collection and sample preparation
Mice were anesthetized in a CO2 chamber then transcardially perfused with 20 ml ice-cold phosphate buffered saline (PBS). One hemisphere was post-fixed by incubation for 48 h in 4% paraformaldehyde (PFA) and cut into 35 μm sections with a vibratome (Leica) for histological analyses (brains used for RNAscope were perfused with both PBS and 4% PFA prior post-fixation and cut in 10 μm sections with a cryostat). The contralateral hemisphere was dissected for isolation of the hippocampus and cortex. Hippocampus was used for RT-qPCR and RNA sequencing, whereas cortical samples were homogenized in a RIPA buffer (Thermo Scientific, #89901) containing phosphatase and protease inhibitors (Thermo Scientific, #78442), centrifuged for 20 min at 15,000 × g and the supernatant was used.
Immunohistochemistry
Sections were washed with 0.1% Triton in PBS, non-specific binding was eliminated by incubation with 0.1% Triton in PBS/5% goat serum, and then each slice was incubated with one of a panel of primary antibodies as follows: AT8 anti-pTAU pSer202/Thr205 (1:1000, Thermo Scientific, #MN1020); anti-pTAU Thr205 (1:1000, Invitrogen, #44-738G), anti-IBA1 (1:1000, Wako, #019-19741), anti-CD68 (1:500, Bio-Rad, #MCA1957); anti-6E10 (1:1000, Covance, #9320500), anti-TYROBP (1:500, antibody provided in collaboration with Richard W. Cho at Cell Signaling Technology). For fluorescent immunostaining, sections were incubated with the appropriate secondary antibody: anti-rabbit IgG Alexa Fluor 488 or 568 (1:2000, Invitrogen); anti-mouse IgG Alexa Fluor 488 or 568 (1:2000, Invitrogen); anti-mouse IgG Alexa 350 (1:2000, Invitrogen). For immunoperoxidase immunostaining, endogenous peroxidase was quenched with pre-incubation with PBS containing 3% H2O2 for 15 min. Sections were then incubated with the ABC system (Vectastain Elite ABC HRP Kit, Vector Laboratories, #PK6100). Horseradish peroxidase conjugates and 3,3′-diaminobenzidine were used for visualization of immune complexes according to the manufacturer’s manual (SeraCare, #5510-0031). Images were obtained with an Olympus BX61 microscope and analyzed with Fiji software (ImageJ). For Thioflavin-S (ThioS) labeling, sections were first mounted on slides and rehydrated prior to incubation with freshly prepared 1% Thioflavin-S (Sigma-Aldrich, #T1892) for 10 minutes at room temperature, under standard photoprotection conditions. Sections were washed 2×3 min with 80% ethanol, then 2×3 min with 95% ethanol. The final washes were performed in water 3×5 min per wash, and coverslips were placed prior to visualization.
Cell culture
Murine primary microglia were isolated from cerebral cortices, dissected from postnatal day P0-P3 WT mice (C57BL/6 J background). Briefly, tissue was homogenized in ice-cold PBS then centrifuged at 300 x g for 5 min. The pellet was resuspended in DMEM supplemented with 10% heat-inactivated FBS (Gibco), 2 mM glutamine and penicillin/streptomycin (100 U/ml and 0.1 mg/ml respectively) and cells were seeded in poly-L-lysine T75 precoated flasks. Cultures were maintained at 37°C in humidified 95/5% air/CO2 incubators for 2 weeks, during which time, media change was performed twice a week. After 2 weeks, cultures were agitated at 180 rpm for 30 min to detach microglial cells from the astrocytic monolayer for collection. Isolated primary microglia were seeded at 500,000 cells per well (24 wells plate) and treated one day after with 2 μg/ml of Lipopolysaccharide (LPS, Sigma, #L3024-5MG) for 1h. Post-LPS treatment, cells were washed with PBS and RNA was extracted.
RNA in situ hybridization
Mice used for RNA in situ hybridization (RNAscope®) were anesthetized in a CO2 chamber and transcardially perfused with 20 ml ice-cold PBS followed by 20 ml ice-cold 4% paraformaldehyde (PFA). Brains were then post-fixed in 4% PFA for 24h at 4ºC followed by equilibration in several sucrose gradients (10%, then 20%, finally 30%). Tissue samples were embedded in optimal cutting temperature compound (OCT), frozen on dry ice, and 10 µm-thick sections were cut using a cryostat (Leica). Fluorescent in situ hybridization (FISH) was performed using RNAscope® according to the manufacturer’s instructions (ACD). Briefly, the mounted frozen sections were washed in PBS then baked at 60ºC for 30 min (Lab-Line Instruments Inc., Melrose Park, IL, USA). Slides were then postfixed in 4 % PFA for 1h at room temperature (RT). After fixation, the sections were dehydrated in ethanol (incubated serially in 50%, then 70%, then 100% ethanol for 5 min at each concentration) and permitted to dry at RT for 45 min. Sections were treated with H2O2 for 10 min and washed twice with distilled water. Subsequently, target retrieval was performed by boiling the slides for 5 min in retrieval reagent. Slides were then washed in distilled water, immersed in 100% ethanol, and finally subjected to air-drying for 5 min at RT. Sections were then treated with protease III for 20 min at 40ºC in the pre-warmed ACD HybEZ II Hybridization System (Cat. No. 321721, ACD) inside the HybEZ Humidity Control Tray (Cat. No. 310012, ACD). After this step, sections were washed twice with distilled water. The following probes from ACD were used and diluted at 1:50 in the RNAscope® Probe Diluent (Cat No. 300041): Tyrobp-C3 (NM_011662.2, bp8-580, Cat. No. 408191-C3), Trem2-C2 (NM_031254.3, bp2-1081, Cat. No. 404111-C2) and Apoe-C4 (NM_009696.3, bp83-1245, Cat. No. 313271-C4). Sections were then hybridized with one corresponding probe at 40ºC for 2 h in the HybEZ Oven (ACD), washed twice with 1× wash buffer, and stored overnight at RT in 5x SSC buffer (Thermo Fisher Scientific, Fair Lawn, NJ, USA). The next day, slides were rinsed 2×2 min with wash buffer (2 min per wash), followed by the three amplification steps (AMP 1, AMP 2, and AMP 3 at 40ºC for 30, 30, and 15 min respectively, with two washes with wash buffer after each amplification step). The signal was developed by treating the sections with the HRP reagent corresponding to each probe channel (e.g. HRP-C2, HRP-C3 or HRP-C4) at 40ºC for 15 min, followed by the TSA Plus fluorophore Opal 690 (dilution of 1:750 in RNAscope® Multiplex TSA Buffer [Cat. No 322809]), at 40ºC for 30 min, and HRP blocker at 40ºC for 15 min, with two wash steps after each of the incubation steps. Finally, the slides were counterstained or not with DAPI for 30 seconds. Fluorescent immunohistochemistry was performed by incubating the slides with the primary antibody diluted in PBS for 2h at RT. Slides were washed twice for 5 min per wash with 1X wash buffer, and then incubated with the secondary antibody diluted in PBS for 1h30. Immunostained slides were then washed twice with 1X wash buffer and mounted.
Western blotting
Equal amounts of protein from (30 μg, mouse cortex was used) were separated by electrophoresis in precast 4-12% Bis-Tris Gels (Bio-Rad) and transferred to nitrocellulose membranes. The membranes were hybridized with the following primary antibodies as indicated: TYROBP (1:500, antibody provided in collaboration with Dr Richard W. Cho at Cell Signaling Technology); AT8 anti-pTAU pSer202/Thr205 (1:1000, Thermo Scientific, #MN1020); anti-GAPDH (1:2000, Santa Cruz, #SC-32233), anti-p-TAU Thr205 (1:1000, Invitrogen, #44-738G), anti-TAU HT7 (1:1000, Invitrogen, #MN1000). Secondary antibodies included: peroxidase-labeled anti-mouse IgG (1:2000, Vector Laboratories); peroxidase-labeled anti-rabbit IgG (1:2000, Vector Laboratories); peroxidase-labeled anti-rat IgG (1:2000, Vector Laboratories). ECL (Pierce, #32106) was used to reveal the immunoreactive proteins, and images were acquired using a Fujifilm ImageReader LAS-4000. Membranes were stripped using a stripping buffer (Thermo Scientific, #46430) when required. Luminescent immunoreactive protein bands were quantified using Fiji software (ImageJ).
Aβ assays
To quantify Aβ levels, human/rat Aβ 1–40/1–42 ELISA kits (Wako, #294-64701, #290-6260) were used according to the manufacturer’s instructions. Absolute concentrations of Aβ were normalized to initial tissue weight. Supernatants from the RIPA-extracted cortices were used.
RNA extraction and RT-qPCR analysis
RNAs were isolated from mice hippocampi using the QIAzol Lysis Reagent (Qiagen) and the miRNeasy Micro Kit (Qiagen) following the manufacturer instructions. For the qPCR analyses, 1 μg of RNA was reversed transcribed using the High Capacity RNA-to-cDNA kit (ThermoFisher, #4387406). The All-in-One qPCR Mix (GeneCopoeia, #QP001-01) was used to perform RT-qPCR. 40 cycles were done and the abundance of each transcript was normalized to the abundance of GAPDH with the ∆∆Ct method. The sequences of oligonucleotides used can be found in Litvinchuk et al. 201819 except for mouse IL6 and TGFβ where TaqMan® probes were used.
RNA sequencing
RNA sequencing on mice hippocampi was performed by Novogene (https://en.novogene.com) using Illumina Novaseq 6000 S4 flow cells. Only samples with RNA integrity number (RIN) > 9 were included. Non-directional libraries were constructed with a NEB kit using the manufacturer’s protocol. RNA sequencing assays were performed after ribosomal RNA depletion by Ribo-Zero. For the data QC, four main steps were implemented including determination of the (1) distribution of sequencing quality; (2) distribution of sequencing error rate; (3) distribution of A/T/G/C bases; and (4) results of raw data filtering. The filtering process included: (1) removal of reads containing adapters, (2) removal of reads containing N > 10% (N represents bases that cannot be determined), and (3) removal of reads containing low quality bases (Qscore ≤5) that are over 50% of the total bases contained in the read. Sequencing reads were aligned to mouse reference genome mm10 (GRCm38.90) using STAR aligner guided by mouse GENCODE gene model release v15. Accepted mapped reads were summarized to gene levels using the featureCounts program. Raw count data were normalized by the voom function in the R limma package, after which differential expression was identified by the moderated t-test implemented in limma. Differentially expressed genes (DEGs) were defined to have at least 1.2-fold change in expression and Benjamini– Hochberg adjusted p ≤ 0.1 comparing different genotypes.
Statistics
The non-genomic data were analyzed with GraphPad Prism 8. Graphs represent the mean of all samples in each group ± SEM. Sample sizes (n values) and statistical tests are indicated in the figure legends. One-way or two-way ANOVA were used for multiple comparisons. A Student’s t-test was used for simple comparisons. Significance is reported at *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. Grubb’s test for outliers was used with α = 0.05.
RESULTS
Tyrobp transcription is increased in recruited microglia
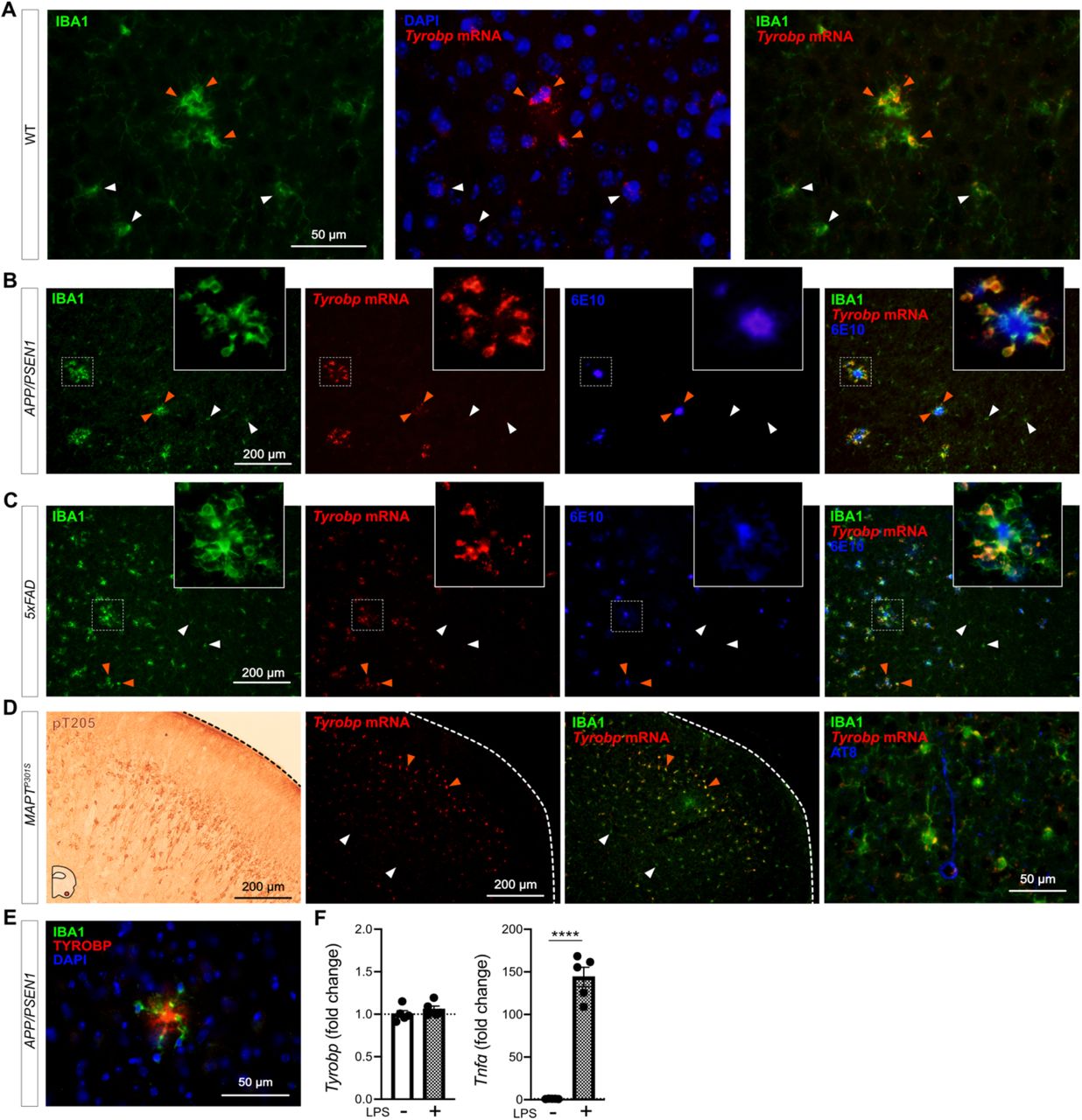
Microglia continuously sense changes in the brain environment and are recruited to sites of injury, microbial invasion, or where abnormal folding or modification of cellular constituents are detected, as with the accumulation of aggregated Aβ. We performed dual RNA in situ hybridization (RNAscope®) and immunohistochemistry for Tyrobp and IBA1 respectively in WT mice and observed increased levels of Tyrobp mRNA in areas exhibiting clustered microglia (Fig 1A). Using the same experimental approach in two independent mouse models of cerebral amyloidosis (APP/PSEN120 and 5xFAD21), we observed a similar pattern in that the Tyrobp mRNA level is extensively and selectively increased in microglia recruited in close proximity to amyloid plaques as compared to microglia that are more distant from the plaques (Fig. 1B-C). We similarly assayed Tyrobp mRNA and IBA1 protein in the MAPTP301S mice22 (also known as PS19), a mutant tauopathy mouse model. We previously described an elevated number of anti-phosphorylated-TAU immunostained neurons in the piriform cortex23, and we detected increased amounts of Tyrobp mRNA in microglia in this same region (Fig. 1D). We confirmed the increase of TYROBP at the protein level in microglia around amyloid plaques (Fig. 1E) as previously reported24. To discriminate between the role of TYROBP in activated vs recruited microglia, we isolated primary microglia from WT mice and exposed them to the gram-negative bacterial endotoxin lipopolysaccharide (LPS) to induce microglial activation25, the status of which we established by quantifying the robust increase of Tnfα mRNA following LPS treatment. Interestingly, Tyrobp mRNA level was unchanged, suggesting that Tyrobp transcription may be increased only when microglia are both recruited and activated but not in resident microglia despite evidence that these residents are also activated (Fig. 1F).

(A) Dual RNA fluorescent in situ hybridization (RNAscope®) and immunohistochemistry for Tyrobp mRNA (red) and IBA1 protein (green) respectively in WT mice (DAPI in blue). Scale bar = 50 μm. (B-C) Dual RNA in situ hybridization and immunohistochemistry for Tyrobp (red), IBA1 (Green) and Aβ (antibody 6E10) (blue) in APP/PSEN1 (B) and 5xFAD (C) mice. Scale bar = 200 μm. (D) Left panel: representative image of immunohistochemistry with antibody pT205 in the piriform cortex of MAPTP301S (PS19) mice. Scale bar = 200 μm. Right panels: dual RNA in situ hybridization and immunohistochemistry for Tyrobp (red), IBA1 (green) and pTau (antibody AT8) (blue) in the piriform cortex of MAPTP301S mice. Scale bars = 200 and 50 μm. (E) Co-immunohistochemistry for TYROBP (green) and human Aβ (antibody 6E10) (red) in APP/PSEN1 mice (DAPI in blue). Scale bar = 50μm. (F) RT-qPCR analyses of Tyrobp and TNFα mRNAs in WT primary microglia with and without LPS. Mice were either 4 (A) or 8 (B-E) months of age and were all WT for Tyrobp. White and orange arrows indicate examples of non-recruited and recruited microglia respectively. Slice thickness = 10μm.
Microglia are normal in Iba1Tyrobp mice
To determine whether constitutive elevation of TYROBP via transgenesis may influence microglial phenotype and progression of AD pathology, we generated a novel transgenic mouse overexpressing Tyrobp selectively in microglia in the central nervous system. We used the mouse Tyrobp and Enhanced Green Fluorescent Protein (EGFP) sequences separated by an Internal Ribosome Entry Site (IRES) under the control of the mouse Iba1 regulatory sequences (Supp Fig. 117). Microinjections were performed in C57BL/6J mice and one line was selected for further use based on expression level of the transgene, now referred to Iba1Tyrobp. We first assessed the overexpression of Tyrobp mRNA by RT-qPCR and measured a ≈ 2.5-fold increase (Fig. 2A). Despite this elevated mRNA level, western blot analyses of protein extracts from the cortex did not reveal a significant overexpression at the protein level (Fig. 2B). Using combined RNA in situ hybridization and immunohistochemistry for Tyrobp and IBA1, respectively, in mice deficient (Tyrobp−/−), WT or overexpressing (Iba1Tyrobp) Tyrobp, we confirmed the 2-fold increase in Tyrobp mRNA in Iba1Tyrobp mice compared to WT (Fig. 2C-D). We observed that only a subset of microglia was overexpressing Tyrobp mRNA in Iba1Tyrobp mice. This selectivity is likely due to the use of the Iba1 promoter in a WT background without extensive microglia activation, thereby also accounting for the lack of a significant increase of TYROBP at the protein level in the resting state. RNA sequencing of hippocampi from Iba1Tyrobp mice did not reveal any differentially expressed genes (DEGs) other than Aif1 (= Iba1), which is increased due to the inclusion of the first two exons in the transgenic vector (Supp Fig. 1). These data indicate that Iba1Tyrobp microglia do not display molecular and phenotypic changes in WT mice.

(A) Hippocampi from 4-month old Tyrobp−/−, WT and Iba1Tyrobp mice were assayed for Tyrobp and Gfp mRNAs by RT-qPCR (n = 3-4 mice per group). (B) Representative western blot and quantification of TYROBP and GAPDH in the cortex of the same groups used in (A) (n = 2-6 mice per group). (C) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Tyrobp mRNA (red) and IBA1 (green) respectively (DAPI in blue) in Tyrobp−/−, WT and Iba1Tyrobp mice. Scale bar = 50 μm and slice thickness = 10μm. (D) Quantification of Tyrobp mRNA intensity from the experiment described in (C). n = 4, 17 and 17 slices per group (from N = 1 mouse per genotype) for Tyrobp−/−, WT and Iba1Tyrobp mice respectively. (E) Volcano plot representation of the whole hippocampal DEGs in Iba1Tyrobp vs WT mice (n = 4 4-month old males per genotype). Error bars represent means ± SEM. Statistical analyses were performed using a Student t-test (A) or a One-Way ANOVA followed by a Tukey’s post-hoc test (B, D), *p < 0.05, **p < 0.01, ****p < 0.0001. na = not applicable; ns = non-significant.
TYROBP overexpression in microglia decreases amyloid plaque load in APP/PSEN1 mice
To assess whether TYROBP overexpression in microglia modulates Aβ deposition in APP/PSEN1 mice, double-heterozygous APP/PSEN1;Iba1Tyrobp mice were generated and studied at 4 months of age. We measured a ≈50% decrease of the plaque density in the cerebral cortices of both male and female APP/PSEN1;Iba1Tyrobp mice compared to sex-matched APP/PSEN1 mice (Fig. 3A-B) in sections stained for amyloid plaques using Thioflavin S (ThioS). This observation was supported by measuring levels of human Aβ42 and Aβ40 by ELISA, both of which were apparently associated with a trend toward decreases in the cortex with TYROBP overexpression, mostly among male APP/PSEN1;Iba1Tyrobp mice (Fig. 3C). There was no genotype-dependent difference in the number of plaque-associated microglia (Fig. 3D-E), unlike what has been reported in 5xFAD mice in the presence of a transgenic increase in TREM226. To evaluate microglia activation, we probed both groups with anti-IBA1 antibody and observed a weaker staining in APP/PSEN1;Iba1Tyrobp mice (Supp Fig. 2A). We next performed RT-qPCR on a group of microglial genes previously described in homeostatic or activated microglia. There was a significant increase of Axl and Ccl2 and a decrease of Cd68 and Tgfβ in brains of APP/PSEN1;Iba1Tyrobp mice (Fig. 3F). Finally, we observed that the decrease of amyloid plaques persisted in 8-month-old APP/PSEN1;Iba1Tyrobp mice as shown by the decreased percentage of ThioS positive areas in somatomotor, hippocampus, and visual areas (Fig. 3G-H).

(A) Representative images of Thioflavine-S (ThioS) staining in APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. Scale bar = 500 μm. (B) Quantification of the number of ThioS-positive plaques per hemibrain in APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. N = 4-5 mice per genotype and sex with 3 slices per animal. (C) Human Aβ42 and Aβ40 concentrations measured by ELISA in the cortex of the same groups described in (B). (D) Representative images of double-label immunohistochemistry with anti-IBA1 and anti-6E10 antibodies in APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. Scale bar = 100 μm. (E) Quantification of the number of plaque-associated microglia in the 4 groups described in (B). N = 10-24 plaques from 4-5 mice per group. (F) RT-qPCR analyses of microglial gene mRNAs in the hippocampus of APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 4 months of age. N = 7-9 mice per group, females and males were pooled. (G) Representative images of ThioS staining in male APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 8 months of age. Scale bar = 500 μm. (H) Quantification of the ThioS immunoreactive area in male APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice at 8 months of age (somatomotor and visual areas of the cortex, and hippocampus were quantified). N = 3-4 mice per group. Error bars represent means ± SEM. Statistical analyses were performed using a Two-Way ANOVA followed by a Sidak post-hoc test for (B,C and E) or a Student t-test for (C) when *t is indicated and (F-H), *p<0.05, **p<0.01, ***p<0.001.
TYROBP overexpression in MAPTP301S mice increases TAU phosphorylation and microglia activation
We previously reported that deletion of Tyrobp altered both mouse amyloid and tauopathy phenotypes and the microglial response to these pathologies23, 27, 28. In MAPTP301S;Iba1Tyrobp double heterozygous mice, western blot analyses using AT8 and T205 antibodies revealed increased levels of phosphorylated-TAU (pTAU) in the cortex of both male and female mice compared to MAPTP301S mice at 4 months of age, whereas total human TAU levels detected with the HT7 antibody were unchanged (Fig. 4A-B). Increased pTAU within brains from MAPTP301S;Iba1Tyrobp mice was further confirmed immunohistochemically (Fig. 4C). We also observed increased IBA1 intensity in MAPTP301S;Iba1Tyrobp compared to MAPTP301S mice that correlated with the increased pTAU (Fig. 4D, Supp Fig. 2B). We confirmed an increased microglial activation state by double-label immunohistochemistry with anti-IBA1 and anti-CD68 in the piriform cortex (Fig. 4E). Using RT-qPCR, we measured increases of Tyrobp, P2ry12, Apoe, Axl, Itgax, Iba1, Tgfβ and Il6 mRNAs in MAPTP301S;Iba1Tyrobp mice compared to MAPTP301S mice (Fig. 4F).

(A) Western blot analyses of phosphorylated-TAU on S202 or T205 epitopes (AT8 and pT205 antibodies) and total human TAU (HT7 antibody) in cortical homogenates from WT, MAPTP301S (PS19) and MAPTP301S;Iba1Tyrobp mice at 4 months-old. n = 4-9 mice per group. (B) Densitometric analyses of western blots presented in (A) standardized to GAPDH or HT7. (C) Representative images of DAB-immunohistochemistry with antibody pT205 in 4 month-old MAPTP301S and MAPTP301S;Iba1Tyrobp mice. Scale bar = 200 μm. (D) Left panel: representative images of anti-IBA1 immunohistochemistry on the same groups described in (C). Scale bar = 200 μm. Additional representative pictures are presented in Supplementary Figure 2. Right panel: western blot-AT8/GAPDH quantification plotted against anti-IBA1 immunoreactivity in the cortex. Linear regression with trend line (red line) and 95% confidence intervals (black lines) are indicated. (E) Representative images of double-label immunofluorescence with anti-IBA1 and anti-CD68 antibodies in the piriform cortex on the same groups described in (C). Scale bar = 500 μm. (F) RT-qPCR analyses of microglial gene mRNAs in the hippocampus of MAPTP301S and MAPTP301S;Iba1Tyrobp mice at 4 months of age. N = 7-11 per group. Error bars represent means ± SEM. Statistical analyses were performed using a One-Way ANOVA followed by a Tukey’s post-hoc test for (B) or a Student t-test for (B) when *t is indicated and (F), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.

The insert was linearized after digestion with PacI.

(A) DAB-immunohistochemistry with anti-IBA1 in 4-month-old APP/PSEN1 and APP/PSEN1;Iba1Tyrobp mice. (B) DAB-immunohistochemistry with anti-IBA1 in 4-month-old MAPTP301S and MAPTP301S;Iba1Tyrobp mice. N = 3 males and 3 females per genotype. Scale bar = 200μm.
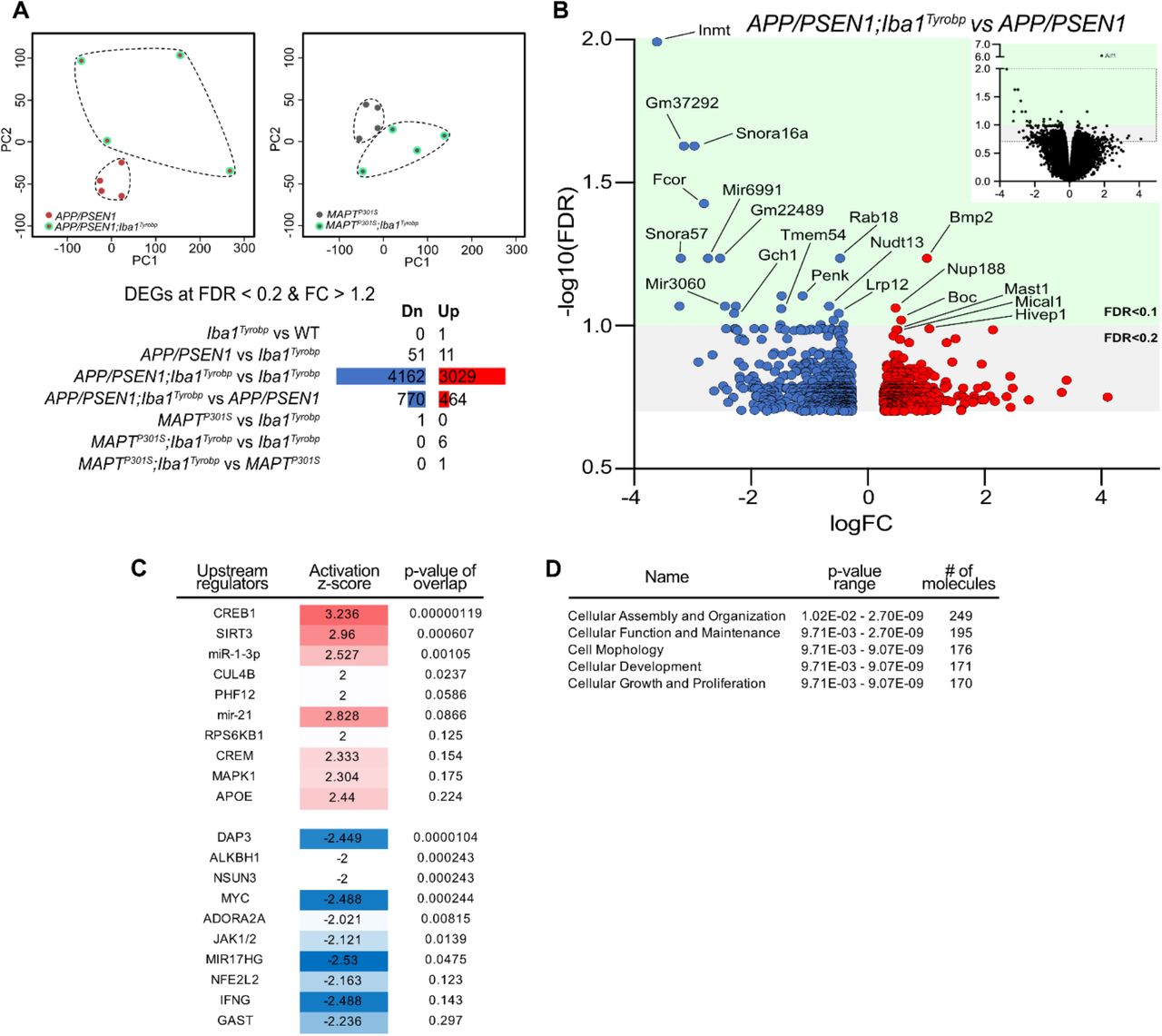
(A) RNA sequencing was performed on hippocampi from 4-month-old male WT, Iba1Tyrobp, APP/PSEN1, APP/PSEN1;Iba1Tyrobp, MAPTP301S or MAPTP301S;Iba1Tyrobpmice, N = 4/genotype. Top: Principal Component Analysis (PCA) of the APP/PSEN1 vs APP/PSEN1;Iba1Tyrobp RNA sequencing samples (left) and MAPTP301S vs MAPTP301S;Iba1Tyrobp RNA sequencing samples (right). Bottom: Numbers of DEGs (FDR <0.2 and FC > 1.2) in the genotype comparisons. Blue and red bars represent the number of significantly down- and up-regulated genes, respectively. (B) Volcano plot representation of the DEGs (FDR <0.2 and FC > 1.2) from the APP/PSEN1;Iba1Tyrobp vs APP/PSEN1 comparison. The top right quadrant represents the DEGs that have been graphed (all genes with FDR < 0.2 except Aif1). (C-D) Ingenuity Pathway Analysis (IPA) was used to identify the top upstream regulators and predicted activation states (C) as well as the top molecular and cellular functions (D) in the APP/PSEN1;Iba1Tyrobp vs APP/PSEN1 comparison.

RNA fluorescent in situ hybridization for Trem2 or Tyrobp or Apoe mRNA double-labeled with an anti-IBA1 antibody (green) or DAPI (blue) in Trem2−/− mice injured as described. The red dot line indicates the needle track. Scale bar = 200 μm.
Iba1Tyrobp mice reveal a likely relationship between Tyrobp and Apoe
Despite the obvious differences across APP/PSEN1 and MAPTP301S mouse models and the diverse consequences of Tyrobp upregulation in each of these mice, there are shared changes in Axl, Ccl2, Tgfβ and Il6 mRNAs in both APP/PSEN1 or MAPTP301S mice overexpressing TYROBP (Fig. 3F and 4F). These genes have been recently associated with Apoe in microglia, macrophages and mononuclear phagocytes. AXL has been identified as a regulator of APOE29 and accumulation of IL6 and CCL2 have been associated with APOE overexpression30,31. Similarly, reciprocal suppression of TGFβ and induction of APOE have been described in DAM microglia15. Apoe is indeed significantly upregulated in MAPTP301S;Iba1Tyrobp mice as compared to MAPTP301S mice but not in APP/PSEN1;Iba1Tyrobp mice compared to APP/PSEN1 mice (Fig. 3F and 4F). However, in bulk RNA sequencing on hippocampi from male APP/PSEN1;Iba1Tyrobp vs APP/PSEN1 mice, we identified Apoe as a potential (activation z-score: 2.44; p-value overlap: 0.224) upstream regulator (Supp Fig. 3), suggesting the possible existence of a relationship between Tyrobp upregulation and Apoe. While a TREM2-APOE pathway has been described15, it is interesting to note that these data support the possibility that the TYROBP-APOE relationship is detectable even in the absence of Trem2 upregulation (Fig. 3F and 4F).
Induction of microglial Tyrobp and Apoe is Trem2-independent in a model of cortical stab injury
To assess the interactions amongst Trem2, Tyrobp and Apoe in microglia, we used an injury paradigm by introducing a small penetrating cortical stab injury via stereotactic surgery into one hemisphere of the mouse brain in order to induce a recruitment of microglia around the injury site32. We first used injured-WT mice and combined RNA in situ hybridization and immunohistochemistry for Apoe and IBA1 respectively. In the intact hemisphere, most Apoe mRNA was not located in microglia but rather in astrocytes, the source of most APOE in brain. However, Apoe mRNA was dramatically increased in microglia recruited on the lesioned side (Fig. 5A). Following the same procedure in Tyrobp−/− mice, Apoe mRNA was not induced in microglia on either side (Fig. 5B), but strikingly, mRNA levels of Tyrobp and Apoe were highly upregulated in the recruited microglia of injured Trem2−/− mice (Fig. 5C, Supp Fig. 4). Taken together, these data indicate that Tyrobp upregulation in recruited microglia around the traumatic lesion is Trem2-independent. Moreover, the increase of Apoe transcripts in recruited microglia in the same mouse model of injury appears to be Tyrobp-dependent and Trem2-independent.

(A) Stab-injured WT mice were sacrificed 3 days after injury and dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe mRNA (red) and anti-IBA1 (green) respectively was performed. The injured ipsilateral area (red dotted line) is shown on the top row and the uninjured contralateral area is show on the bottom row. Scale bar = 200 μm. (B-C) The same stab injury protocol was utilized in Tyrobp−/− (B) and Trem2−/− (C) mice. Anti-IBA1 and DAPI stainings are shown in green and blue, respectively. Top row: Trem2 mRNA (red); middle row: Tyrobp mRNA (red); bottom row: Apoe mRNA (red). Mice were 4 months of age and slice thickness = 10 μm. The red asterisk indicates the injured side. White and orange arrows indicate examples of non-recruited and recruited microglia, respectively.
Induction of microglial Tyrobp and Apoe around amyloid plaques is Trem2-independent, and Apoe upregulation is dramatically decreased when Tyrobp is absent
In order to investigate further these interactions among Trem2, Tyrobp and Apoe in microglia in the presence of mutant human APP, we first performed dual RNA in situ hybridization and immunohistochemistry for Tyrobp, IBA1 and 6E10 in TgCRND8 mice33 on either a WT or Trem2-null background. Despite reduced recruitment of microglia around the plaques when Trem2 is deleted24, Tyrobp mRNA was still increased in plaque-associated microglia (Fig. 6A). as was Apoe mRNA in plaque-associated microglia in the same TgCRND8;Trem2−/− mice (Fig. 6B). We then assayed APP/PSEN1 mice that were either WT or deficient for Tyrobp and, while the expression of Apoe was not completely abolished by deletion of Tyrobp, we confirmed a substantial decrease in the induction of Apoe mRNA in plaque-associated microglia when Tyrobp was absent (Fig. 6C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Tyrobp mRNA (red), anti-IBA1 (green) and human Aβ (6E10 antibody) (blue) in TgCRND8 mice on WT (top row) or Trem2−/− (bottom row) background. Scale bar = 200 or 50 μm. (B) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe mRNA (red), anti-IBA1 (green) and human amyloid (6E10 antibody) (blue) in the same mice as in (A). Scale bar = 200 or 50 μm. (C) Dual RNA fluorescent in situ hybridization and immunohistochemistry for Apoe mRNA (red), anti-IBA1 (green) and human Aβ (6E10 antibody) (blue) in APP/PSEN1 mice on a WT (top row) or Tyrobp-null (bottom row) background. Scale bar = 50 μm. Right panel: quantification of the number of plaque-associated microglia with upregulated Apoe mRNA in the same mice as in (D). N = 2-3 mice per group (A-C).
DISCUSSION
TREM2, TYROBP and APOE are three microglial genes linked in a pathway contributing to the pathogenesis of AD and in the transition to DAM10. TYROBP was identified as a key driver in late onset AD9. TREM2 binds to TYROBP, its intracellular adaptor, to initiate its signal transduction pathway, and naturally occurring loss-of-function mutations of either TYROBP or TREM2 can lead to Nasu-Hakola disease13,34. There is a general assumption among investigators in this research area that genetic deletion or overexpression of either TREM2 or TYROBP would result in identical phenotypes in disease models, but, until now, this has not been tested directly. We previously demonstrated amelioration of behavior and electrophysiologic impairments in APP/PSEN1 and MAPTP301S mice on a Tyrobp-null background, despite a concurrent absence of effect on amyloid pathology and an apparent increase in the stoichiometry of phosphorylated TAU vs total TAU23, 27, 28. Homozygous deletion of Trem2 can also lead to amelioration of both amyloidosis and tauopathy 35,36, but those effects vary according to the mouse model, and the age and level of deficiency at sacrifice 23, 37, 38. Lee et al.26 used bacterial artificial chromosome (BAC)-mediated transgenesis to overexpress the human TREM2 in the mouse genome and showed that TREM2 overexpression reduces amyloid accumulation in 5xFAD mice. Using a similar BAC system, Gratuze et al.39 assessed the impact of TREM2R47H in MAPTP301S mice but no TREM2 overexpression was reported in that study.
The possible existence of an early TREM2-independent phase in conversion of microglia to DAM was described by Keren-Shaul et al.10 but was not evident in studies by either Krasemann et al.15 or Zhou et al.40 Apoe has also been described as a participant in stage 1 of DAM with Tyrobp10 and it has been suggested that it drives the DAM transition through a TREM2-APOE pathway15. Moreover, Apoe has been reported to influence both amyloidosis and tauopathy histological phenotypes in mouse models41,42. A complete elucidation of the choreography of the regulatory interactions among these genes and their cognate proteins therefore remains an area of intense interest. We would suggest that the discrepancies across the various analyses might be explained in part by the fact that DAM microglia are located in the immediate proximity of the plaques, and that neither bulk-nor single-cell-RNA sequencing can distinguish homeostatic vs DAM phentoypes since both techniques generate an average transcriptome analysis from all microglia in a given tissue sample. This formulation played a major role in prompting us to use dual RNA in situ hybridization and immunohistochemistry in the current study in which we sought to determine 1) the effects of transgenic overexpression of Tyrobp on amyloid and tau pathologies and 2) the relationship of the induction of Tyrobp to these pathologies and to the induction of Trem2 and Apoe.
While this manuscript was in preparation, Chen et al. (2020) reported spatial transcriptomics and in situ sequencing in AppNL-G-F mice to avoid the averaging of the transcriptome in a tissue sample. These investigators proposed the formulation of a plaque-induced gene (PIG) network in microglia and astrocytes in immediate proximity to amyloid plaques with differential expression of 57 genes43. Top genes highly upregulated in the proximity of the plaques and as early as 3 months of age were Tyrobp, Apoe and several complement-related genes. Notably, Tyrobp is a key regulator of the complement subnetwork9 and C1q is down-regulated in APP/PSEN1 and MAPTP301S mice in the absence of Tyrobp23,28. The authors performed the same type of experiment in human AD brain slices and confirmed the enrichment of Tyrobp and several complement components (C1qA, C1qB, C1qC, and Clu). Of particular relevance to our data herein, Trem2 was not included in the human PIG network. Furthermore, Srinivasan et al.44 recently used RNA sequencing to profile fluorescence-activated cell sorted (FACS)-purified human microglia from frozen AD and control brains and showed that one of the top gene upregulated in human-AD microglia was Apoe, confirming potential importance of the interplay between Tyrobp and Apoe.
With the inclusion of Tyrobp as a PIG gene43 and because of our prior validation of its actions as a driver of AD9,23,27,28, we hypothesized that constitutive overexpression of microglial Tyrobp via transgenesis would alter both amyloid and tau pathologies. In APP/PSEN1 mice, Tyrobp upregulation decreased the amyloid burden, similar to what occurs with the upregulation of Trem226 and supported by a recent report showing that higher microglia activation shows protective effects on subsequent amyloid accumulation45. In MAPTP301S mice, we previously reported that deficiency of Tyrobp increased TAU phosphorylation and spread23, and were puzzled, therefore, to observe a similar increase in TAU phosphorylation in the MAPTP301S mice with overexpression of Tyrobp. MAPTP301S;Iba1Tyrobp microglia are more reactive than MAPTP301S microglia, and reactive microglia have been reported to drive TAU pathology, so these two observations are compatible46,47. This exacerbation of pathology under conditions of either down- or up-regulation of Tyrobp in MAPTP301S mice reveals the complexity of the microglial events underpinning tauopathy. Our formulation is that, for any particular microglial activation status, there exists some optimum level of Tyrobp expression, and that either elevation or deficiency of Tyrobp levels can be detrimental. These data also support a key role for microglial TYROBP in AD pathology progression, as we previously proposed in our reports on mice deficient for Tyrobp23,27,28.
Our data indicate that Tyrobp upregulation is an early marker of recruited microglia and can occur even in the brains of Trem2-deficient mice. Similarly, we observed that the increased Apoe mRNA level in microglia is Trem2-independent, whether in injury or AD-related mouse models. Finally, we observed that microglial Apoe mRNA level was greatly attenuated in plaque-associated microglia in Tyrobp-deficient mice. These data confirm the model proposed by Keren-Shaul et al.10 in which Tyrobp and Apoe transcripts are increased first, and neither transcription event requires the presence of Trem2. Moreover, Meilandt et al.48 recently reported that microglial APOE expression was not reduced, but, on the contrary, was increased in PS2APP;Trem2−/− when compared with microglial APOE expression in PS2APP mice. In that same study, they also analyzed the expression profiles of FACS-purified microglia from 5xFAD mice either in the presence or absence of Trem2 and showed a two-fold reduction in Apoe in one dataset (GSE132508)10,48 but no reduction at all in the other (GSE65067)48,49. However, Parhizkar et al.50 reported that the absence of functional TREM2 reduces plaque-associated APOE. This is in line with what Krasemann et al.15 proposed when they showed that genetic targeting of Trem2 suppresses the APOE pathway. Our observations and conclusions herein apparently differ from those of Parhizkar et al.50 to the extent that, in our hands, microglial amyloid-plaque sensing followed by upregulation of Tyrobp and Apoe are preserved despite the absence of Trem2 and, as a consequence of the Trem2 deficiency, microglia recruitment into the proximity of amyloid plaques is reduced. This relationship points to the fact that the absence of functional TREM2 will block appearance of the full DAM phenotype and therefore the associated clearance of the plaques is reduced. Nevertheless, we propose a model wherein the sensing of amyloid plaques --which takes place upstream of amyloid plaque clearance--involves Tyrobp and Apoe but not necessarily Trem2.
Considering the central role of Tyrobp described in the microglial sensome8, its upregulation even in the absence of Trem2, and the consequences of its overexpression in APP/PSEN1 and MAPTP301S mice, we propose that Tyrobp is one of the key genes upregulated in the switch from the homeostatic phenotype to the DAM phenotype. Taking all the data together, we propose a model in which microglia perceive stimuli and initiate their responses during an early and relatively brief time window, corresponding to what Keren-Shaul et al.10 described as stage 1. During this phase, Trem2 is not required but Apoe will be upregulated presumably as a downstream consequence of Tyrobp upregulation. TYROBP is a 113 amino acid polypeptide with a minimal extracellular region51,52, making it unlikely that TYROBP is the sole player in a signal transduction pathway involving both the perception of the environment and the triggering of the switch from homeostatic phenotype to DAM. However, TYROBP is the adaptor for many receptors other than TREM216, and therefore, it is plausible and perhaps likely that other TYROBP receptors could play key roles in sensing the deposition of amyloid. For example, numerous SIGLEC proteins (sialic acid-binding immunoglobulin-type lectins) carry a positively charged residue in their transmembrane domain that participates in oligomerization of the SIGLEC with TYROBP. The primary SIGLEC ligand is a sialic acid that accumulates in many pathological conditions including cerebral Aβ amyloidosis53,54. Moreover, Siglec-H interacts with TYROBP, and its expression has been reported to be elevated in 5xFAD mice vs WT26. CD33 (SIGLEC-3) is also one of the most abundant SIGLECs in human brain, and genome-wide association studies (GWAS) implicated a polymorphism near CD33 as a genetic risk factor for AD2, 4, 55. CD33 and TREM2 both interact with TYROBP, either directly (TREM2) or via common intracellular signaling factors (CD33). Griciuc et al.56 recently investigated crosstalk between CD33 and TREM2 and proposed that CD33 acts upstream of TREM2. They also showed that Cd33 and Tyrobp expression levels did not change in Trem2−/− versus WT microglia. This formulation provides evidence that CD33-TYROBP signaling could occur upstream of the recruitment and upregulation of TREM2.
Rather than the somewhat “Trem2-centric” view of DAM proposed in the existing AD microglia literature15, 40, 50, we propose that Tyrobp plays a central role in an alternative and early pathway in the microglial sensome8, even in the absence of any change in Trem2 levels. The data that we present here document the robust consequences of TYROBP overexpression in both APP/PSEN1 and MAPTP301S mice. We confirm here that upregulation in microglia of both Tyrobp and Apoe constitute interconnected events in microglia sensing of amyloid deposits, and these events take place independently of Trem2.
AUTHOR CONTRIBUTIONS
MA, JVHM, SG and MEE designed the study. MA performed the experiments and analyzed the data. JM contributed to the RNA in situ hybridization related experiments. MW and BZ contributed to the RNA sequencing analysis. JKG and PF provided the TgCRND8 mice. MA, SG and MEE wrote the manuscript.
FUNDING
The study was supported by the National Institute on Aging (U01 AG046170 and R01 AG057907 to MEE, SG and BZ), the Alzheimer’s Disease Research Division of the BrightFocus Foundation (grant A2018253F to MA and grant A2016482F to JVHM), the Mount Sinai Alzheimer’s Disease Research Center (ADRC P50 AG005138 and P30 AG066514 to Mary Sano, with internal pilot grant awarded to MA).
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
REFERENCES




