

ABSTRACT
COVID-19 causes cardiac dysfunction in up to 50% of patients, but the pathogenesis remains unclear. Infection of human iPSC-derived cardiomyocytes with SARS-CoV-2 revealed robust transcriptomic and morphological signatures of damage in cardiomyocytes. These morphological signatures include a distinct pattern of sarcomere fragmentation, with specific cleavage of thick filaments, and numerous iPSC-cardiomyocytes that lacked nuclear DNA. Human autopsy specimens from COVID-19 patients also displayed marked sarcomeric disruption and similar fragmentation, as well as prevalently enucleated cardiomyocytes. These striking transcriptomic and cytopathic changes provide a roadmap to understand the mechanisms of COVID-19 cardiac damage, search for potential treatments, and determine the basis for prolonged cardiac morbidity observed in this pandemic.
INTRODUCTION
Initial descriptions of COVID-19, the pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), characterized it as a primarily respiratory syndrome1. However, increasing clinical evidence now implicates multiple organ systems in COVID-19, including the heart, gastrointestinal tract, and kidneys2–5. Notably, multiple independent reports have found that COVID-19 patients frequently present with significant myocardial damage6–8, even without prior cardiovascular disease9 (CVD), indicating that viral infection may be directly responsible for the cardiac damage. Meta-analyses identify elevated high-sensitivity troponin-I or natriuretic peptides, biomarkers of cardiac damage or dysfunction, as the strongest predictor of mortality in hospitalized patients, eclipsing both CVD and congestive obstructive pulmonary disease8–11. Alarmingly, evidence of elevated troponin can be found even in mild cases of COVID-19, and a recent study observed that the majority of recovered patients presented with impaired cardiac function, indicating that long-term heart sequelae from COVID-19 may not be limited to ICU cases12.
The identification of therapeutic strategies to prevent or manage myocardial injury in COVID-19 patients is hindered by limited understanding of the mechanisms by which SARS-CoV-2 induces cardiac damage. Cardiac damage may be caused by systemic impacts of SARS-CoV-2, such as hypoxic stress due to pulmonary damage, microvascular thrombosis, and/or the systemic immune response to viral infection13. However, cardiomyocytes are known to express the primary receptor for viral entry, ACE214 and may be infectable by SARS-CoV-215. A recent study identified SARS-CoV-2 viral particles in the myocardium of a child with COVID-19 related multisystem inflammatory syndrome16, and recent histological studies of deceased COVID-19 patients detected viral RNA in the myocardium without inflammatory cell infiltrates17. Whether these transcripts arise from infected myocytes, cardiac stroma, or blood vessels is unknown18. However, these results support the possibility that direct myocardial infection may underlie cardiac injury in COVID-19.
The alarming clinical consequences of COVID-19 on the heart have been puzzling since the vast majority of pathological studies of autopsy patients have not shown any specific signature of COVID-19 in cardiomyocytes. Pathological studies have been hampered by biosafety issues, and the fact that most studies are limited to the hematoxylin and eosin (H&E) stain. In addition, sample availability is restricted to post-mortem specimens, which limits observations to a late stage disease endpoint.
Ex vivo studies using human cell-based models of the heart, such as cardiac tissue derived from human induced pluripotent stem cells (iPSCs), afford the most direct route for the prospective and clinically-relevant study of the effects of cardiac viral infection. Stem-cell derived models have already demonstrated the susceptibility of hepatocytes19, intestinal epithelium20,21, and lung organoids22 to SARS-CoV-2 infection. While two recent reports confirmed that human iPSC-cardiomyocytes are susceptible to SARS-CoV-2 infection23,24, specific cardiac cytopathic features yet to be identified. In addition, the viral tropism for other cardiac cell types, which may be involved in microthrombosis25 or weakening of the ventricular wall, has not been explored, nor has there been direct correlation of in vitro results to clinical pathology specimens. Here, we examined the relative susceptibility of three iPSC-derived cardiac cell types: cardiomyocytes (CMs), cardiac fibroblasts (CFs), and endothelial cells (ECs), to SARS-CoV-2 infection, and identify clear hallmarks of infection and cardiac cytopathy that predicted pathologic features found in human COVID-19 tissue specimens. By identifying phenotypic biomarkers of SARS-CoV-2 infection and cytopathy in cardiomyocytes that recapitulate features of patient tissue, this system may enable rapid development of cardioprotective therapies for COVID-19.
RESULTS
SARS-CoV-2 infects and propagates in cardiomyocytes, but not endothelial cells or cardiac fibroblasts
The relative susceptibility of different cardiac cell types to SARS-CoV-2 infection has not been characterized, leading to ambiguity over the sources of cardiac damage and relevant therapeutic targets. Analysis of previously reported single-cell RNA-sequencing and immunofluorescence staining of ECs, CFs, and CMs revealed that ACE2 was only detectable in CMs. Although the cell surface protease TMPRSS2, involved in viral cell entry, was not detected in any cell type, cathepsin-L and cathepsin-B were detected in all cells (Supplementary Information 1, Supp. Figs. 1A-D). These observations suggest potential infectivity of CMs by SARS-CoV-2, and an expectation of poor infectivity in ECs and CFs. We determined the tropism of SARS-CoV-2 for different cardiac cell types by incubating human iPSC-derived CMs, CFs, ECs, or a mix of all three that mimics native myocardial composition, with SARS-CoV-2 at a relatively low MOI (MOI = 0.006).
After 48 hours, CFs and ECs showed little to no viral nucleocapsid (N5) transcript relative to a housekeeping control (RPP30), whereas CMs expressed >104 greater levels of N5 transcript than CFs or ECs (Fig. 1A). These differences in viral expression largely correlated with ACE2 expression levels in each cell type (Supp. Fig. 1A,B). There was no significant difference in viral detection between CMs and mixed cultures, indicating that CMs are primarily responsible for viral infection in the mixed cell condition. In contrast to CMs, undifferentiated iPSCs were uninfectable (Supp. Fig. S1D). Plaque assays on the supernatants of exposed cells confirmed that CFs, ECs, and iPSCs did not support productive infection, but CMs robustly produced replication-competent virions (Supp. Fig. S1E).

In all experiments, cells were exposed to SARS-CoV-2 virus for 48 hours, unless otherwise specified, at an MOI of 0.006 before lysis or fixation. A. RT-qPCR quantification of viral RNA (Fold change of SARS-CoV-2 N gene transcript, N5, over housekeeping gene transcript, RPP30) in cell cultures exposed to SARS-CoV-2. CF: iPSC-derived cardiac fibroblasts; EC: iPSC-derived endothelial cells; CM: iPSC-derived cardiomyocytes. Error bars: SEM. **: p-val < 0.01, one-way ANOVA with Tukey’s multiple comparisons. B. Representative images of immunostaining of cardiac cells exposed to SARS-CoV-2. PECAM-1 (CD31) was used as an endothelial cell marker, and cTnT as a cardiomyocyte specific marker. Cardiac fibroblast (CF) cells expressed GFP constitutively. Viral signal was detected by staining for SARS-CoV-2 Spike protein or viral origin double stranded RNA, as noted. C. Toxicity of SARS-CoV-2 to cardiac cell types, quantified by nuclear retention. Y-axis depicts the % of nuclei counted (relative to mock). Nuclei were counted automatically at 10x magnification (10 images/condition). Light gray: Vehicle treatment (mock), Dark gray: Heat inactivated SARS-CoV-2 (MOI = 0.1), Magenta: SARS-CoV-2 (MOI = 0.006). D. Representative images of immunostaining of infected cardiomyocytes at 24h, 48h or 72h after addition of SARS-CoV-2 virus E. Transmission electron microscopy of SARS-CoV-2 viral particles in an infected CM. Left: Montage view with nucleus (dashed line), in addition to remnant ER-Golgi (light blue arrowheads), with viral particles enclosed in a membrane compartment. Middle: Close up of SARS-CoV-2 virions (red arrowheads) and surrounding membrane. Right: Magnified view of SARS-CoV-2 virions, showing the 500-750nm diameter membrane and the 60-100nm diameter viral particles within.
Immunostaining for replicating virus, as revealed by the presence of double-stranded viral RNA (dsRNA) or Spike protein, further confirmed that only infected CMs supported viral replication (Fig. 1B, white arrows). However, all three cell types exhibited significant cytopathic effects after 48 hours of viral exposure, characterized by fragmented cell bodies and dissociation from neighboring cells (Fig. 1B, red arrows) and significant cell loss, as measured by nuclei counts (Fig. 1C). Interestingly, visual cytopathic effects were most prevalent in CFs, while the greatest nuclear loss was observed in ECs, indicating they result from viral exposure rather than infection. However, inoculation with heat-inactivated SARS-CoV-2 did not cause cell death or dissociation in any of the cell types assayed (Fig. 1C), indicating the observed toxicity was due to exposure to replication-competent virus.
Replication of (+) single stranded RNA (ssRNA) viruses, including SARS-CoV and MERS-CoV, involves budding of double-membrane vesicles (DMVs) from the endoplasmic reticulum, with viral particle assembly occurring in cisternae of the ER-Golgi intermediate compartment (ERGIC)26. In CMs infected with SARS-CoV-2, dsRNA and Spike protein initially (24h post-infection) accumulated near the nucleus in small perinuclear puncta, the typical location of the ERGIC region, indicating potential active centers of replication (Fig. 1D). At 48h post infection, many cells exhibited dsRNA signal dispersed throughout their cytoplasm, which may correlate with breakdown of the ER-Golgi membrane as viral replication accelerates and the cell deteriorates, evidenced by a decrease in sarcomeric integrity and intensity. By 72h post infection, SARS-CoV-2 had spread throughout the culture and a large portion of the CMs had died, with the remaining cells displaying dispersed viral stain localization, dissociation from neighboring cells, and heavily reduced sarcomeric signal (Fig. 1D).
Using transmission electron microscopy of infected CMs, we readily identified the remnants of the ER-Golgi membranes (light blue arrowheads) and large vesicles in the proximity of the nucleus. These vesicles, approximately 500-750 nanometers in diameter, contained multiple particles 50-60 nm in diameter which were identified as SARS-CoV-2 virions (red arrowheads) (Fig. 1E), consistent with the dsRNA/Spike+ aggregates detected in infected CMs. Altogether, these results indicate that SARS-CoV-2 is able to readily infect, replicate in, and rapidly propagate through CMs.
SARS-CoV-2 infection of cardiomyocytes is dependent on an endolysosomal route
We next sought to elucidate the mechanism of SARS-CoV-2 entry into CMs. Pretreatment of cells with an ACE2 blocking antibody or with cathepsin inhibitor E64d significantly reduced the number of viral transcripts detected in infected CMs (Fig. 2A, S1F). Despite detection of FURIN in CMs (Fig. S1A), inhibition of FURIN did not reduce infection rate (Fig. S1F). Further probing revealed that cathepsin-L (CTSL) inhibition via Z-Phe-Tyr(tBu)-diazomethylketone (Z-FY-DK) decreased the level of virus in infected cells to approximately 10% of vehicle-treated controls, whereas inhibition of cathepsin-B (CTSB) with CA-074 did not have this effect (Fig. 2B). In addition, the PIKfyve inhibitor apilimod or autolysosome acidification blocker bafilomycin also successfully reduced viral infection to ∼0.1% or 1% of vehicle-treated controls, respectively. In contrast, TMPRSS2 inhibitors aprotinin or camostat mesilate did not significantly inhibit viral infection. Taken together, these results indicate that SARS-CoV-2 employs the ACE2 receptor to bind to iPSC-CMs, and utilizes a CTSL (but not CTSB)-dependent endolysosomal route to infection without TMPRSS2/serine protease-mediated activation at the cellular membrane.
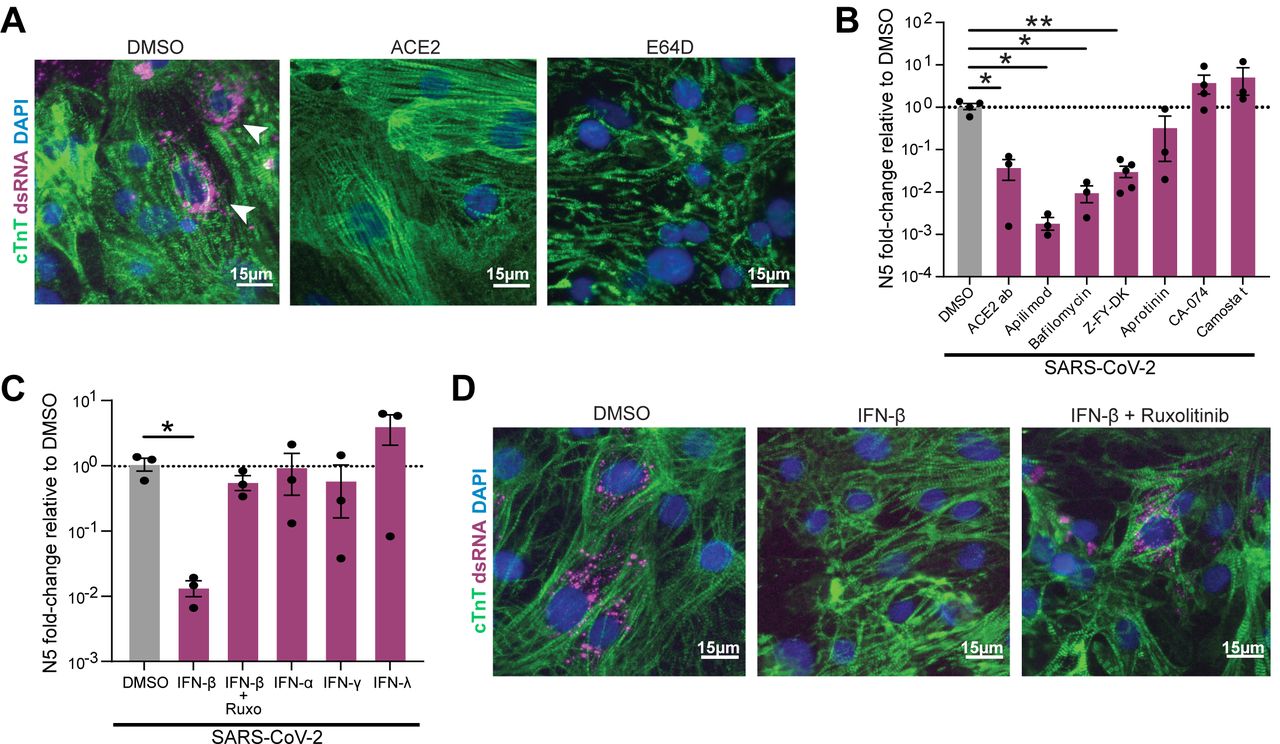
A. Representative immunofluorescence images from SARS-CoV-2 infected (MOI=0.006) CMs pretreated with either vehicle (DMSO), ACE2 blocking antibody (‘ACE2ab’) or cathepsin-B and -L inhibitor E64d for 2h before infection. Double-stranded RNA (dsRNA) staining (white arrowheads) denotes presence of replicating virus. B-C. RT-qPCR quantification of viral RNA (N5) in CM samples exposed to SARS-CoV-2 for 48h (MOI=0.006) after 2h pretreatment with the indicated reagents to block viral entry (B) or prime the cells’ innate immune response (C). Dots represent separate replicates. *: p-val < 0.05, **: p-val < 0.01. N>=3 for all conditions. One-way ANOVA with Tukey’s multiple comparisons. D. Representative immunofluorescence images from cardiomyocytes pre-treated with vehicle (DMSO), IFN-β, or IFN-β with JAK inhibitor ruxolitinib.
We next examined if priming the innate immune response or triggering inflammatory pathways could effectively combat SARS-CoV-2 infection. CMs were primed with IFN-α, IFN-β, IFN-γ, or IFN-λ, in addition to a combination of IFN-β and a JAK/Stat inhibitor (ruxolitinib) prior to infection. Only pre-exposure to IFN-β decreased infection, and this effect was reversed by co-treatment with ruxolitinib (Fig. 2C-D). Our observation that IFN-β but not IFNα pretreatment was able to reduce infection is in agreement with a previous report of differential antiviral activity of these two type-I interferon molecules in a mouse model of myocarditis27.
SARS-CoV-2 exposure induces transcriptional changes in genes involved in contractile machinery
To evaluate the transcriptional impact of exposure to SARS-CoV-2 in cardiac cells, we performed RNA-sequencing of CFs, ECs, and iPSCs exposed to an MOI of 0.006, and of CMs exposed to a range of MOIs (0.001, 0.01, and 0.1). Sequencing recovered a high proportion of SARS-CoV-2 transcripts in an MOI- and cell type-dependent fashion (Fig. 3A), with SARS-CoV-2 reaching >50% of the recovered reads in CMs at the highest MOI. Principal component analysis (PCA) of the biological conditions revealed the expected clustering primarily based on cell type, with CFs and ECs clustering near each other and CMs and iPSCs forming two separate clusters (Fig. 3B). Loading plots of the principle components supported this interpretation: the genes determining the spectrum of variation between CMs and CF/ECs were associated with CMs (MYH7, MYH6, TNNT2) at one pole (Fig. 3C) and anti-correlated with CF/EC specific genes at the other (FN1, COL1A2, TFPI2, MME) (Supplementary Fig. S2A). Notably, the distance between mock CMs and the furthest infected CMs was slightly further than the distance between CMs and CFs or ECs, indicating that viral infection altered cellular expression profiles at least as strongly as cell type. Along this axis, however, we also observed that the level of transcriptional disruption correlated poorly with MOI across all CM samples, potentially due to natural stochasticity in the kinetics of infection. Regrouping conditions by the level of transcriptional disruption allowed us to more clearly deduce transcriptional trends resulting from viral exposure (Fig. S2A-D). Infected cardiomyocytes downregulated pathways corresponding to cardiac muscle tissue organization and cellular respiration, while upregulating pathways related to innate immune response and apoptosis, although innate response to viral infection decreased, suggesting active repression (Fig. 3D). Anomalous upregulation of pathways associated with olfactory receptors and dysregulation of proteasome were also observed with increasing transcriptional disruption. (Fig. 3D, S2E).

A. Percentage of total reads that map to the SARS-CoV-2 viral genome in various cell types. iPSCs, ECs or CFs were exposed at an MOI of 0.006, and CMs at three different MOIs: 0.001 (‘Low’), 0.01 (‘Mid’) and 0.1 (‘High’). **: p-val < 0.01; ***: p-val < 0.001. B. Principal component analysis of transcriptomic samples. Dot shapes and colors represent the different cell types, whether they were exposed to SARS-CoV-2 virus and, in the case of CMs, the different MOIs used. C. Loading plot for genes, with color indicating cardiomyocyte state (orange), fibroblast/endothelial cell state (green), iPSC state (light gray), SARS-CoV-2 infection-related factors (dark gray), immune response (blue). D. Heat map depicting transcriptional expression profiles for mock-infected CMs compared to the least (Low Inf), middle (Mid Inf), and most (High Inf) transcriptionally disrupted CM samples for genes mapping to GO terms of interest (genes |log2 fold change| > 1 between high infection and mock, FDR < 0.05; GO terms containing at least 25 enriched genes, FDR < 0.01). E. Expression ratio of genes involved in sarcomeric structure and myosin contractility of the high-infection CM groups relative to the mock-infection CM group. F. Schematic drawing of a sarcomere showing localization of differentially regulated factors. Yellow outlines denote proteins which possess the putative SARS-CoV papain-like protease cleavage site LKGGK. Background colors denote log2FC of the high-infection condition over mock. Myosin heavy chains and unconventional myosins are upregulated but almost all thin filament members and myosin light chains are downregulated.
Differentially regulated genes involved in inflammation and innate immunity reflected the observed preferential infectivity of CMs, as exposed CFs and ECs had a depressed cytokine response compared to CMs at all three MOIs (Supplementary Fig. S2E). Infected CMs had enrichment of genes involved in cytokine production and T-cell activation (OAS2, MX1, IFIT1, IL1B, IL6, TNF) (Supplementary Fig. S2E). Interestingly, we noted that CMs at each MOI showed very clear dysregulation of contractile machinery, proteasomal subunits and ubiquitination. In particular, genes involved in the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex were disrupted, especially members of the nesprin family, and calmin, both critical for anchoring the nucleus to the actin cytoskeleton (Fig S4). Further, sarcomeric structural proteins, myosin light chains, and proteasome kinases and chaperones were strongly downregulated, while most myosin heavy chains were significantly upregulated (Fig. 3E, F, Supplementary Fig. 3A-G), indicating a potential effect of SARS-CoV-2 infection in the contractile and structural integrity of CMs.
SARS-CoV-2 infection disrupts multiple intracellular features of cardiomyocytes
Motivated by the discovery of expression changes for various structural and contractile genes in our transcriptomic data, we performed high-content imaging of CMs following SARS-CoV-2 infection. We immediately noticed a number of abnormal structural features in infected CMs that were not seen in parallel mock samples. Most commonly, we observed widespread myofibrillar disruption throughout the cytoplasm, which manifested as a unique pattern of very specific and periodic cleavage of myofibrils into individual sarcomeric units of identical size but without any alignment (Fig. 4A, Supplementary Fig. 4A-D). Myofibrillar fragmentation was evident as early as 24 hours after infection, and more widespread and common after 48 hours, when up to 20% of cells exposed to virus displayed myofibrillar fragmentation at the same time, suggesting that fragmentation may develop continuously over time (Fig. 4B). Curiously, myofibrillar fragmentation was present in cells independently of signs of actively replicating virus (as per dsRNA staining; Chi-square test for independence p-value = 0.81), making it more prevalent in bystander CMs, while cells positive for dsRNA rarely showed signs of myofibrillar fragmentation (Fig. 4C).

A. Representative immunofluorescence images of myofibrillar fragmentation in CMs at different timepoints after exposure to SARS-CoV-2. White arrowheads indicate fragments consisting of two bands of cTnT positive staining. B. Quantification of number of cells presenting myofibrillar fragmentation at 24h and 48h post-exposure (defined as at least one event of a cTnT doublet unaligned and dissociated from other myofibrils). Number of cells was normalized to total number of nuclei in the images counted. Each dot represents a separate infection sample. Each replicate is the additive count of 9 randomly acquired fields of view. **: p-val < 0.01. C. Representative immunostaining showing a cell staining positively for viral dsRNA, adjacent to cells with different degrees of myofibrillar fragmentation. White squares indicate zoomed-in areas, with labels corresponding to insets. White arrowheads point to examples of cTnT doublets (myofibrillar fragments). D. cTnT and ACTN2 double-staining of CMs displaying myofibrillar fragmentation. White arrowheads indicate cTnT-ACTN2-cTnT myofibrillar fragments. E. TEM images of sarcomeres in mock treated and SARS-CoV-2 infected (MOI=0.006) CM cultures. Blue arrows denote the sarcomeric z-disk; Yellow arrow indicates M-line location; dashed line delimits nucleus. Sarcomeres of mock treated cells display clear I and A-bands, but fragmented sarcomeres only possess thin filaments. Below: Representative TEM image of a healthy nucleus, and the nucleus of a cell infected with SARS-CoV-2. F. Immunofluorescence staining of SARS-CoV-2-exposed CMs displaying loss of nuclear DNA staining (48h post exposure). White arrowheads in the inset indicate locations of sarcomeric retraction and absence of nuclear material.
Co-staining of CMs exposed to SARS-CoV-2 with the thin filament marker cardiac, cardiac Troponin T, (cTnT) and the Z-disk marker, α-actinin 2, revealed that the virus-induced myofibrillar fragments consisted of two cTnT-positive bands flanking a single α-actinin 2 band, indicating cleavage at the M-line or a separation of thick and thin filaments (Fig. 4D, Supplementary Figure S5E). To examine sarcomeric fragmentation in greater detail, we employed TEM imaging of SARS-CoV-2 infected and mock-treated CMs. While intact sarcomeres were clearly identified with a classic dark Z-disk, light I-band, and dark A-band, single fragmented sarcomeres displayed an extended I-band and complete absence of the A-band (Fig. 4E), suggesting a mechanism by which thick filaments are liberated from sarcomere subunits.
Since transcriptomic profiling data indicated viral infection altered the proteasome system (Fig. 3F, Supplementary Fig. S3F), we asked whether inhibition of the proteasome would recapitulate similar structural abnormalities. Indeed, high doses of the proteasome inhibitor bortezomib induced myofibril fragmentations in CMs, although the effect was less frequent and less severe than the effect of SARS-CoV-2 infection, and was generally accompanied by diffuse cTnT staining throughout the cell cytoplasm (Supplementary Fig. S5G. Notably, the well-known cardiotoxic drug doxorubicin did not induce myofibril fragmentation (data not shown), indicating that myofibrillar fragmentation may be a specific result of proteasomal inhibition. Finally, we also observed a second structural phenotype where infected CMs often appeared to lack nuclear DNA staining. This phenomenon was observed most frequently in localized patches of nearby cells which lack dsRNA staining, and was characterized by withdrawn sarcomeres and an empty region present in completely enucleated CMs (Fig. 4F).
In vitro findings predict disruptions in myocardium of COVID-19 patients
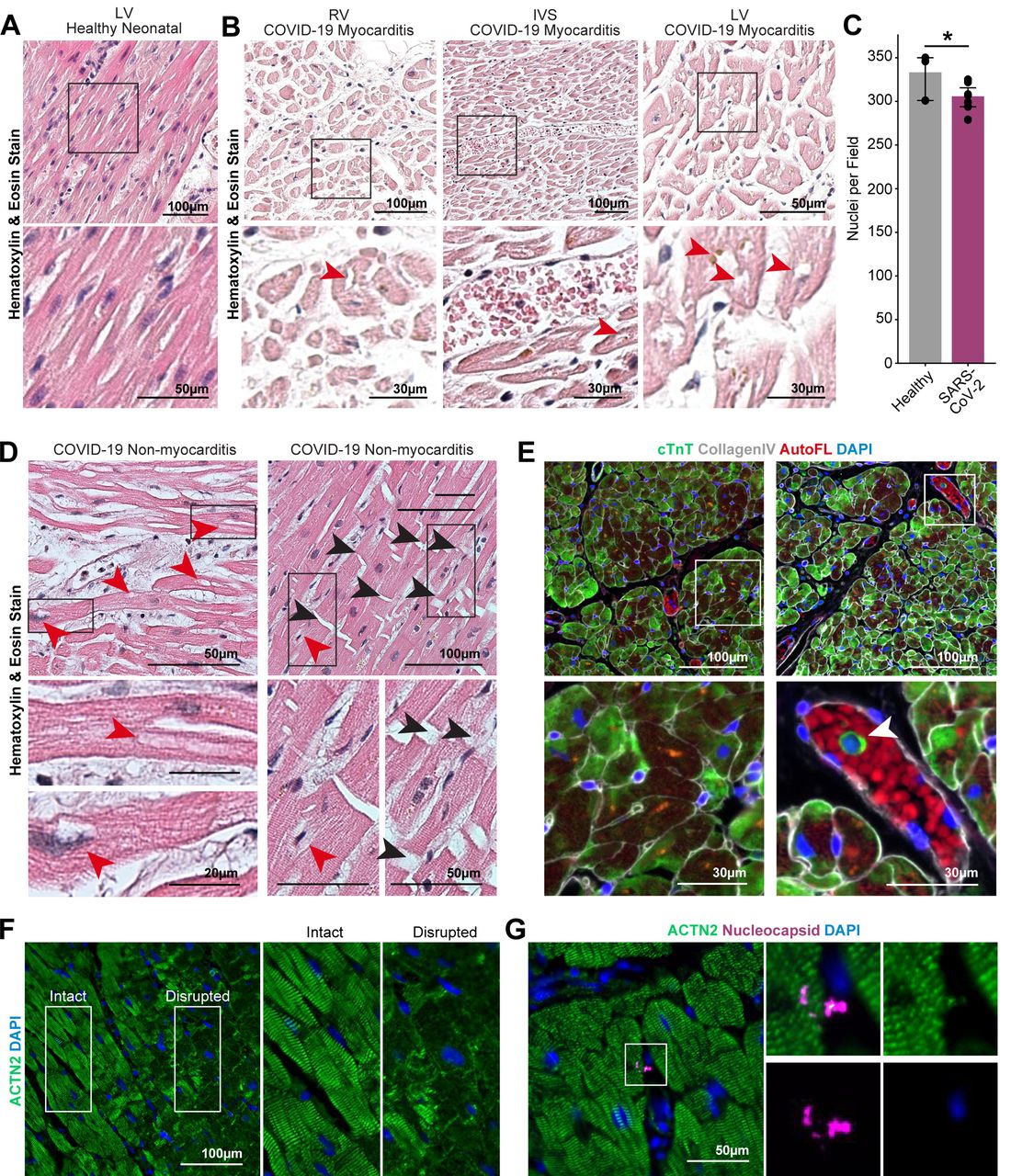
We next asked whether the phenotypes we observed in vitro could predict similar patterns of cardiac cell damage in vivo. We obtained autopsy specimens from three COVID-19 positive patients, one of whom was diagnosed with viral myocarditis. We observed significant histological alterations of the myocardium in the COVID-19 myocarditis case compared to healthy myocardial tissue (Fig. 5A), in addition to moderate levels of immune infiltration (Fig. 5B; Supplementary Fig. S5B). The tissues from COVID-19 patients with or without clinical myocarditis exhibited signs of edema with increased spacing between adjacent cardiomyocytes (Fig. 5B-D) and highly inconsistent staining for cardiac troponin-T, suggesting sarcomere disruption (Fig. 5E; Supplementary Fig. S5C). There was also evidence of troponin-T positive cells in the blood vessels, indicating the possibility of phagocytosis of compromised myocytes (Fig. 5E, right, arrowheads). Some of the cardiomyocytes lacked hematoxylin staining for nuclei, recapitulating the in vitro phenotype of DNA loss (Fig. 5B-D red arrowheads; Supplementary Fig. S5B). In a COVID-19 patient not diagnosed with myocarditis (Fig. 5D; Supplementary Fig. S5B), we observed clear evidence of loss of chromatin staining (red arrows) and abnormal nuclei (blue arrows). Strikingly, immunohistochemical labeling of contractile proteins revealed regions of extreme myofibrillar anomalies. In a sample from a patient without myocarditis, immunostaining revealed that large regions of myofibrils (ACTN2+) within cardiomyocytes were entirely missing or collapsed (Fig. 5F, disease region, right; Supplementary Fig. S5D) and many cells appeared to lack nuclear DNA. We attempted to identify features of viral presence in the tissues using antibodies against Spike, dsRNA, and two epitopes of the nucleocapsid, but were not able to identify clear features of viral presence (Fig. 5G). Overall, these results indicate similar cytopathic features in the myocardium of COVID-19 patients as predicted by in vitro exposure of SARS-CoV-2 to iPSC-CMs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Hematoxylin and Eosin (H&E) staining of healthy neonatal left ventricle tissue. B. H&E staining images of myocardial tissue from a COVID-19 patient with diagnosed myocarditis. Black boxes indicate zoomed in areas of the bottom figures. Red arrowheads indicate cardiomyocytes lacking chromatin stain. C. Quantification of nuclei per field of view of intact myocardium and disrupted myocardium from SARS-CoV-2 patients. Statistical significance was determined by fitting to a Poisson generalized linear model. *: p-val < 0.02. D. Representative H&E staining images of myocardial tissue from COVID-19 patients without diagnosed myocarditis. Red arrowheads denote putative nuclear locations lacking chromatin stain. Black arrowheads indicate breakage at the intercalated disks between cardiomyocytes. E. Representative immunofluorescence staining of the myocardial tissue from a COVID-19 myocarditis patient. Cardiomyocytes show diffuse and disorganized cardiac troponin T (cTnT) staining with occasional cells in the blood vessel staining positively for cTnT (green). White boxes indicate zoomed in areas of the bottom figures. White arrowhead indicates cardiac troponin T material in the cytoplasm of a mononuclear cell within a blood vessel. F. Region of COVID-19 patient heart spanning the transition from healthy to sick myocardium. White boxes indicate zoomed in areas of the figures on the right. Sick myocardium region is characterized by extensive breaks in α-actinin 2 (ACTN2) staining. G. Immunohistochemical staining for viral nucleocapsid protein (magenta) and α-actinin 2 (green) yielded no recognizable signal aside from occasional, unidentified puncta.
DISCUSSION
In this work, we carried out a comprehensive analysis of the cytopathic effects of SARS-CoV-2 on human iPSC-derived cardiac cell types, and identified unique structural consequences of SARS-CoV-2 infection in cardiomyocytes in vitro that predict similar pathological features found in myocardial tissue from COVID-19 patients. The most striking of these phenotypes is a pattern of highly specific, distinctive myofibrillar fragmentation into individual sarcomeres, in addition to loss of nuclear DNA from intact cell bodies. Surprisingly, these effects appear to occur independently of the presence of actively replicating SARS-CoV-2 virus, suggesting a larger landscape of cytopathological impacts than initially assumed. Guided by these results, our examination of myocardium specimens from COVID-19 patients revealed striking similarities to our in vitro findings, including localized regions of severely disrupted sarcomeric structure and numerous cells lacking nuclear DNA. Together, these results indicate new opportunities for the development of cardioprotective interventions against COVID-19, and also raise significant concerns about the prevalence and severity of cardiac involvement, even in mildly ill patients.
The distinguishing features of myofibrillar fragmentation in CMs due to SARS-CoV-2 exposure are extremely precise, ordered disruptions to the sarcomeric structure and complete dissolution of the cardiac contractile machinery. Reduced contractility and elevated circulating cardiac troponin I are the main cardiac indications of COVID-19 afflicting the heart12, thus understanding the mechanisms responsible for specific damage to the contractile machinery is critically important.. The striking consistency and periodicity of this fragmentation suggests that it is the product of cleavage by a specific protease. Our immunofluorescence and TEM imaging suggests a detachment of the sarcomeric thick filaments from the thin filament. Coincidentally, our transcriptomic analyses showed very strong upregulation of myosin heavy chain-coding genes upon SARS-CoV-2 infection, which could be due to a compensatory production of this protein in response to degradation (Fig 3D). We also observed a significant depression of the ubiquitin-proteasome system upon infection, and that myofibrillar fragmentation could be recapitulated by the addition of the proteasome inhibitor bortezomib (Sup Fig S3; Sup. Fig S5G). Taking these results altogether, we speculate SARS-CoV-2 infection triggers cleavage of myofibrils at the A-band, more specifically at myosin, resulting in the observed myofibrillar fragmentation phenotype. Interestingly, the SARS-CoV-2 papain-like protease (PLpro/Nsp3) has been described to preferentially cut the LKGG↓K motif,28,29 which is found primarily in multiple members of the family of myosin heavy chain proteins and Myomesin-1, which are well-known targets of papain proteases30. However, further studies are needed to determine whether the observed myofibrillar fragmentation is a direct consequence of viral protease cleavage since the observed pattern is most frequently detected in dsRNA-negative cells.
The majority of histological examinations of myocardium from COVID-19 patients have revealed unremarkable features aside from edema and occasional mononuclear infiltrate17. Although 50% or more of COVID-19 patients manifest clinical signs of cardiac dysfunction12, a detailed description of cardiac pathogenesis has remained elusive. Our own cursory examinations of hematoxylin and eosin staining of COVID-19 myocardial samples revealed only mild disruption and generally unremarkable tissue anatomy. However, informed by our in vitro data, we identified clear features of myocardial damage that only became apparent after staining tissues for sarcomeric proteins, revealing frequent focal regions of myofibrillar disruption similar to our in vitro findings. This correlation strongly hints at the underlying etiology of SARS-CoV-2’s impact on cardiac function and demonstrates that human iPS-derived models of myocardium are able to predict previously unrecognized features of cardiac pathogenesis in COVID-19 patients.
In addition to myofibril disruption, we also identified changes in the nuclei of CMs after SARS-CoV-2 exposure, with many cells exhibiting a complete lack of nuclear DNA. In addition, our transcriptomic data indicated disruption of the nuclear LINC complex, through which the cytoskeleton supports the shape and structure of the nucleus31,32. Similarly, autopsy specimens revealed myocytes devoid of chromatin staining in regions of disrupted sarcomeric tissue. Physically intact cardiomyocytes lacking nuclear DNA would normally reflect a terminal event33, but, based on our in vitro data, may instead represent a weakened nuclear envelope resulting from a generalized stress response or apoptosis. Irrespective of the cause(s), compromised CM nuclear integrity appears to be another feature of COVID-19 in the heart.
Aside from myocytes in the heart, non-myocyte elements of the cardiac stroma may also be involved in mediating some of the outcomes observed in COVID-19, such as cardiac hypertrophy and vascular dysfunction25. In our system, CFs and ECs were not expected to be susceptible to infection due to their low levels of expression of ACE2, and qPCR of viral genes matched these predictions. However, we did observe a potent cytopathic effect in both ECs and CFs, irregardless of productive viral infection. Interestingly, heat inactivated virus failed to recapitulate this effect, suggesting that cytotoxicity is due to exposure to live virus. Further inquiry into the mechanism by which CFs and ECs sense and respond to SARS-CoV-2 may be required in order to determine how these cells may contribute to SARS-CoV-2-induced cardiac dysfunction.
The ability of iPS-CMs to accurately model and predict cytopathic consequences of infectious pathogens, such as SARS-CoV-2, in the heart opens a wide array of potential avenues for discovery and validation of candidate cardioprotective therapies for COVID-19 and other diseases. Although cell-based drug screens exist for many pathogens, including SARS-CoV-234, the unique cytoarchitecture of cardiomyocytes and the specific impacts of SARS-CoV-2 on myocytes offer novel screening possibilities. For example, the observed myocyte-specific myofibrillar fragmentation enables cell-based drug screens with an exceptionally sensitive reporter of CM infection, since as few as 1 virion per 150 cells was sufficient to induce cytopathic myofibrilar fragmentation. The fact that myofibrillar fragmentation is independent of active viral replication suggests an asynchronous driving mechanism. For example, fragmentation and replication may have different kinetics, or fragmentation may occur when a virus infects but fails to replicate, or fragmentation may even be some form of non-cell-autonomous response. Identification of efficacious cardioprotective therapies may require both preventing viral replication and maintaining the sarcomeric integrity of the cell to achieve optimal therapeutic benefit.
Aside from serving directly as a valuable platform for cardioprotective therapies, CMs also provide a highly vulnerable substrate to interrogate basic viral biology principles that are critical to determine the efficacy of candidate therapies. In this study, we utilize a drug treatment approach to conclude that SARS-CoV-2 infects CMs independent of TMPRSS2-mediated protease activity via an endolysosomal route. While inhibition of TMPRSS2 is currently targeted in clinical trials to prevent COVID-19 (CamoCO-1935 and RACONA36 studies), our results suggest that effective cardioprotection may require orthogonal targeting of endosomal proteases37. More generally, our results indicate that carefully dissecting the route of viral entry in different human cell types may be critical to develop clinical therapies that protect different vulnerable organ systems.
Although our results indicate that direct infection of CMs may not be required for toxic effects in the cardiac muscle, the question of whether cardiomyocytes get directly infected by SARS-CoV-2 in COVID-19 patients remains important to the clinical interpretation of our report. Previous studies have shown that myocardium of post-mortem COVID-19 patients is positive for viral dsRNA,17 but failed to detect staining of viral particles in cardiomyocytes. Importantly, a recent report described for the first time detection of SARS-CoV-2 particles inside the cardiomyocytes of a COVID-19 patient presenting acute multisystem inflammation who died rapidly after admittance16. This study provides initial evidence that direct myocardial infection can occur in COVID-19. We hypothesize that if virus is present in myocytes during an early to progressive stage of the disease, terminal assays from deceased patient tissue may be unable to observe its presence if viral detection is transitory, but that the rapid decline and death of this patient may have enabled observation of the peak of infection. Similarly, our in vitro system of CM infection allows for a controlled study of the progression of the myocardial damage caused by SARS-CoV-2, modeling even the earlier stages of infection.
Recent reports have indicated that aside from fatal cases of COVID-19, a majority of patients who recover from COVID-19 display significant levels of cardiac damage (∼80%)17,38,39, and nearly half of all mildly ill COVID-19 patients have echo abnormalities17,40–42. Our infection studies are more analogous to mild cases of COVID-19, due to the low levels of initial viral exposure, yet similarly demonstrate striking features of viral cytopathy that are readily observed in patient samples without any clinical diagnosis of cardiac involvement. Due to the heart’s innate lack of regenerative capacity43,44, these results suggest that a majority of patients could suffer long-term consequences of COVID-19 on cardiac function, and that even the mildest cases could cause permanent heart damage.
Currently, the long-term effects of COVID-19 cardiomyopathy are unknown, and our findings raise several important concerns. We do not know if the sarcomeric fragmentation is a terminal event for cardiomyocytes, but the analogous sarcomere disruption observed in autopsy specimens strongly suggests that permanent damage has occurred in a non-regenerative organ. Similarly, detailed longitudinal studies will be needed to determine if viral-induced nuclear DNA loss or envelope weakening occurs as a precursor to cardiac cell death. While the damage inflicted by SARS-CoV-2 is severe, it is possible that post-infection treatments could aid in recovery. By identifying clear features of COVID-19 cardiomyopathy, our findings contribute to a better understanding of the pathological sequelae of this disease and can accelerate the development of efficacious cardioprotective and anti-viral therapies.
METHODS
hiPSC Maintenance; iPS-Cardiomyocyte differentiation and purification
Human iPSCs (WTC11 line1) were maintained in mTESR or mTESR+ (STEMCELL Technologies) on Matrigel (8 μg/ml, BD Biosciences)-coated cell culture plates at 37°C, 5% CO2. Cells were passaged every 3 days using Relesr (STEMCELL Technologies) and supplemented with Rock Inhibitor Y-27632 (SelleckChem) for 24 hours after each passaging. hiPSCs were differentiated into cardiomyocytes following a modified Wnt pathway modulation-based GiWi protocol2. Briefly, hiPSC cultures were harvested using Accutase (STEMCELL Technologies) and seeded onto Matrigel-coated 12-well plates. Three days later, cells were exposed to 12 uM CHIR99021 (Tocris) in RPMI1640 (Gibco, 11875093) supplemented with B27 without insulin (Gibco, A1895601) (R/B-) for 24 hours. After an additional 48 hours, media was changed to R/B-supplemented with 5 uM IWP2 (Tocris) for 48 hours. On day 7, media was changed to RPMI1640 medium supplemented with B27 with insulin (Gibco, 17504044) (R/B+) and refreshed every 3 days thereafter. Beating was generally observed around day 8-11. At day 15, cells were cryopreserved using CryoStor CS10 (STEMCELL Technologies). After thawing, cell cultures were enriched for iPS-cardiomyocytes following metabolic switch purification3. Briefly, cells were washed once with saline buffer and incubated in DMEM (without glucose, without sodium pyruvate; Gibco, 11966025) supplemented with GlutaMax (Gibco, 35050061), MEM Non-Essential Amino Acids (Gibco, 11140050) and sodium L-lactate (4mM, Sigma-Aldrich). Lactate media was refreshed every other day for a total of 6 days. Four to six days later (day 28-30), iPS-CMs were replated into assay plates for infection using 0.25% Trypsin (Gibco, 15050065) at a density of approximately 60,000 cells/cm2.
scRNAseq analysis of SARS-CoV-2 entry factors
A historic single cell RNA sequencing data set consisting of iPSC-derived cardiomyocytes, primary fetal cardiac fibroblasts, and iPSC-derived endothelial cells was re-analyzed to compare relative expression levels of SARS-CoV-2 relevant receptors and proteases (GSE155226)4. Briefly, day 30 lactate-purified cardiomyocytes were force aggregated either alone or with a single supporting cell type and cultured in suspension culture. Aggregates were dissociated and libraries prepared using the Chromium 3’ v2 library preparation platform (10X Genomics). Libraries were sequenced on a NextSeq 550 sequencer (Illumina) to a depth of at least 30 million reads per sample. Samples were demultiplexed and aligned to GRCh38 with CellRanger v3.0.2. Samples were normalized and clustered with Seurat v3.2.05, yielding four primary clusters corresponding to each cell type, which were used to profile cell-type specific expression of SARS-CoV-2 relevant factors.
Cardiac Fibroblast Differentiation
Second heart field-derived cardiac fibroblasts (SHF-CFs) were differentiated following the GiFGF protocol, as previously published6. Briefly, hiPSCs were seeded at 15,000 cells/cm2 in mTeSR1 medium. Once they reached 100% confluency, they were treated with R/B-supplemented with 12μM CHIR99021 (day 0), and refreshed with R/B-24 hours later (day 1). From days 2-20, cells were fed every 2 days with cardiac fibroblast basal media (CFBM) (Lonza, CC-3131) supplemented with 75ng/mL bFGF. On day 20, CFs were singularized with Accutase for 10 minutes and replated at 7,000 cells/cm2 onto tissue culture plastic 10cm dishes in FibroGRO medium (Millipore Sigma, SCMF001). FibroGRO media was changed every two days until the CFs reached approximately 80-90% confluency, at which point they were passaged with Accutase. SHF-CFs were validated to be >80% double-positive for TE-7 and vimentin by flow cytometry.
Endothelial Cell Differentiation
WTC11 iPSCs were directed towards an endothelial cell (EC) lineage by the addition of E8 media supplemented with BMP4 (5 ng/ml) and Activin A (25 ng/ml) for 48 hours followed by E7BVi media, consisting of E6 medium supplemented with bFGF (50ng/ml), VEGF-A (50 ng/ml), BMP4 (50 ng/ml) and a TGFβ inhibitor, SB431542, (5 μM) for 72 hours. After 5 days of successive media changes, ECs were split and plated at high density in EGM media (Lonza, CC-3162) on tissue culture flasks coated with fibronectin (1:100, Sigma Aldrich F0895). On day 8, all cells were cryo-preserved and a fraction of ECs were assayed for >95% purity by flow cytometry using antibodies against mature EC markers CD31 and CDH5.
Mixed Cultures of CMs, CFs, and ECs
Mixed cultures of iPS-CMs, iPS-ECs, and iPS-CFs were created by combining single cell suspensions of each cell types in a ratio of 60:30:10 CM:EC:CF at a density of 200,000 cells/mL. The mixed suspension was replated onto Matrigel-coated tissue culture plates 48 hours prior to infection at a density of 62,500 cells/cm2.
SARS-CoV-2 Infection
The WA-1 strain (BEI resources) of SARS-CoV-2 was used for all experiments. All live virus experiments were performed in a Biosafety Level 3 lab. SARS-CoV-2 stocks were passaged in Vero cells (ATCC) and titer was determined via plaque assay on Vero cells as previously described7. Briefly, virus was diluted 1:102-1:106 and incubated for 1 hour on Vero cells before an overlay of Avicel and complete DMEM (Sigma Aldrich, SLM-241) was added. After incubation at 37°C for 72 hours, the overlay was removed and cells were fixed with 10% formalin, stained with crystal violet, and counted for plaque formation. SARS-CoV-2 infections of iPSCiPS-derived cardiac cells were done at a multiplicity of infection of 0.006 for 48 hours unless otherwise specified. For heat inactivation, SARS-CoV-2 stocks were incubated at 85°C for 5 min.
Immunocytochemistry
Infected and mock-treated cell cultures in coverslips were washed with Phosphate Buffered Solution (PBS) and fixed in 4% paraformaldehyde (PFA) overnight, followed by blocking and permeabilization with 0.1% Triton-X 100 (T8787, Sigma) and 5% BSA (A4503, Sigma) for one hour at RT. Antibody dilution buffer (Ab buffer) was comprised of PBS supplemented with 0.1% Triton-X 100 and 1% BSA. Samples were incubated with primary antibodies overnight at 4°C (Table 2), followed by 3 washes with PBS and incubation with fluorescent-conjugated secondary antibodies at 1:250 in Ab buffer for 1 hour at RT (Table 2). Coverslips were mounted onto SuperFrost Slides (FisherBrand, 12-550-15) with ProLong Antifade mounting solution with DAPI (Invitrogen, P36931. Images were acquired with a Zeiss Axio Observer Z.1 Spinning Disk Confocal (Carl Zeiss) or with an ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices) and processed using ZenBlue and ImageJ.
List of reagents.
List of dyes, primary and secondary antibodies used for immunocytochemistry and paraffin sections.
List of primers.
Histology
Paraffin section of healthy and COVID-19 patient hearts were deparaffinized using xylene, re-hydrated through a series of decreasing ethanol solutions (100%, 100%, 95%, 80%, 70%) and rinsed in PBS. Hematoxylin and eosin staining was performed according manufacturer instructions and the slides were mounted with Cytoseal 60 (Richard-Allan Scientific) and glass coverslips. For immunofluorescence staining, epitopes were retrieved by immersing slides through 35 min incubation at 95°C in citrate buffer (Vector Laboratories, pH 6) or Tris-EDTA buffer (Cellgro, pH 9). Slides were cooled for 20min at RT and washed with PBS. Samples were permeabilized in 0.2% Triton X-100 (Sigma) in PBS by slide immersion and washed in PBS. Blocking was performed in 1.5% normal donkey serum (NDS; Jackson ImmunoResearch) and PBS solution for 1h at RT. Primary and secondary antibody cocktails were diluted in blocking solution (Table 2). PBS washes were performed after primary (overnight, 4°C) and secondary antibody (1h, RT) incubations. Nuclei were stained with Hoechst and coverslips were mounted on slides using ProLong™ Gold Antifade Mountant. Samples were imaged on the Zeiss Axio Observer Z1.
RT-qPCR
Cultured cells were lysed with Qiagen buffer RLT (Qiagen, 79216) supplemented with 1% β-mercaptoethanol (Bio-Rad, 1610710) and RNA was isolated using the RNeasy Mini Kit (Qiagen 74104) or Quick-RNA MicroPrep (ThermoFisher, 50444593) and quantified using the NanoDrop 2000c (ThermoFisher). Viral load was measured by detection of the viral Nucleocapsid (N5) transcript through one-step quantitative real-time PCR, performed using PrimeTime Gene Expression Master Mix (Integrated DNA Technologies, 1055772) with primers and probes specific to N5 and RPP30 as in internal reference. RT-qPCR reactions were performed on a CFX384 (BioRad) and delta cycle threshold (ΔCt) was determined relative to RPP30 levels. Viral detection levels in pharmacologically treated samples were normalized to DMSO-treated controls.
RNA-Seq
To generategenerating libraries for RNA-sequencing, RNA isolate quality was assessed with an Agilent Bioanalyzer 2100 on using the RNA Pico Kit (Agilent, 5067-1513). 10ng of each RNA isolate was then prepared using the Takara SMARTer Stranded Total RNA-Seq Kit v2 – Pico Input Mammalian (Takara, 634412). Transcripts were fragmented for 3.5 minutes and amplified for 12 cycles. Library concentrations were quantified with the Qubit dsDNA HS Assay Kit (Thermo Fisher, Q32851) and pooled for sequencing. Sequencing was performed on an Illumina NextSeq 550 system, using the NextSeq 500/550 High Output Kit v2.5 (150 Cycles) (Illumina, 20024907) to a depth of at least 10 million reads per sample. Raw data is available at GEO under the accession number GSE156754.
Bioinformatic analyses of transcriptomic data
Samples were demultiplexed using bcl2fastq v2.20.0 and aligned to both GRCh38 and the SARS-CoV-2 reference sequence (NC_045512) using hisat2 v2.1.08. Aligned reads were converted to counts using featureCounts v1.6.29. Cell-type clustering, gene loadings, and technical replication were assessed using the PCA and MDS projections implemented in scikit-learn v0.2310. Differential expression analysis was performed using limma v3.44.3 with voom normalization11 and GO term enrichment analysis was performed using clusterProfiler v3.16.012. Unbiased GO term selection was performed by non-negative matrix factorization using scikit-learn. Correlation of gene expression with distance in PCA space from mock CMs was assessed using Pearson’s R test. Pathways for sarcomere organization and the LINC complex were adapted from WikiPathways (WP383 and WP4535 respectively) using Cytoscape v2.8.013.
TEM/CLEM
Cells grown on gridded 35mm MatTek glass-bottom dishes (MatTek Corp., Ashland, MA, USA) were fixed in 2.5% glutaraldehyde and 2.5% paraformaldehyde in 0.1M sodium cacodylate buffer, pH 7.4 (EMS, Hatfield, PA, USA) following fluorescence imaging. Samples were rinsed 3 x 5 min at RT in 0.1M sodium cacodylate buffer, pH 7.2, and immersed in 1% osmium tetroxide with 1.6% potassium ferricyanide in 0.1M sodium cacodylate buffer for 30 minutes. Samples were rinsed (3 ⨯ 5 min, RT) in buffer and briefly washed with distilled water (1 x 1 min, RT) before sample were then subjected to an ascending ethanol gradient (7 min; 35%, 50%, 70%, 80%, 90%) followed by pure ethanol. Samples were progressively infiltrated (using ethanol as the solvent) with Epon resin (EMS, Hatfield, PA, USA) and polymerized at 60°C for 24-48 hours. Care was taken to ensure only a thin amount of resin remained within the glass bottom dishes to enable the best possible chance for separation of the glass coverslip. Following polymerization, the glass coverslips were removed using ultra-thin Personna razor blades (EMS, Hatfield, PA, USA) and liquid nitrogen exposure as needed. The regions of interest, identified by the gridded alpha-numerical labeling, were carefully removed and mounted with cyanoacrylate glue for sectioning on a blank block. Serial thin sections (100 nm) were cut using a Leica UC 6 ultramicrotome (Leica, Wetzlar, Germany) from the surface of the block until approximately 4-5 microns in to ensure complete capture of the cell volumes. Section-ribbons were then collected sequentially onto formvar-coated 50 mesh copper grids. The grids were post-stained with 2% uranyl acetate followed by Reynold’s lead citrate, for 5 min each. The sections were imaged using a Tecnai 12 120kV TEM (FEI, Hillsboro, OR, USA), data recorded using an UltraScan 1000 with Digital Micrograph 3 software (Gatan Inc., Pleasanton, CA, USA), and montaged datasets were collected with SerialEM (bio3d.colorado.edu/SerialEM) and reconstructed using IMOD eTOMO (bio3d.colorado.edu/imod). Reconstructed images were denoised using total variational denoising, followed by gamma correction and unsharp masking (https://peerj.com/articles/453/).
Analysis of immunofluorescence images
Immunostained images of cells were coded and manually counted by four blinded individuals, with a 20% overlap for concordance. Features counted were dsRNA positive cells (defined as a cell with clear dsRNA staining in the cytoplasm over background), cells presenting myofibrillar fragmentation (defined as presenting at least one instance of a cTnT positive doublet obviously separated from other myofibrils), and cells positive for both dsRNA and myofibrillar fragmentation. Each datapoint represents the normalized sum of counts for nine randomly acquired fields of view in a separate well using high magnification (40x). Counts were normalized to total nuclei counts per field of view. For viability estimations, each data point is the sum of nine randomly acquired fields of view in a separate well, using low magnification (10x). Nuclei counts were performed automatically using the EBImage package 14 on R15.
Statistical Analyses
Statistical testing for qPCR experiments was performed using GraphPad Prism 8 software, using 1-way ANOVA with post-hoc Tukey’s multiple comparisons test. Statistical analysis for the immunofluorescence cell counts was performed using R (REF ABOVE) using Student’s t-test with Bonferroni correction for multiple testing. Statistical differences in expression between bioinformatic samples were performed on corrected, log transformed counts using Welch’s t-test with Benjamini Hochberg false discovery rate correction with a threshold of 0.05, except where described above.
Funding
S.K was supposed by AHA 20POST35211143. G.N.R. was supported by the NSF Graduate Research Fellowship Program. B.C. was supported by R01-HL130533, R01-HL13535801, P01-HL146366. T.C. was supported by ERC 1648035. Both B.C and T.C were supported by U01 ES032673-03. M.O. was supported by NIH 5DP1DA038043. H.L. and K.N. were supported by NIH 1R01AG065428. All three corresponding authors acknowledge support through a gift from the Roddenberry Foundation and Pauline and Thomas Tusher.
Conflict of interest
B.R.C. is a founder of Tenaya Therapeutics (https://www.tenayatherapeutics.com/), a company focused on finding treatments for heart failure, including genetic cardiomyopathies. B.R.C. and T.C.M. hold equity in Tenaya.
Author Contributions
J.P.B., S.K., S.J.R., B.R.C. and T.C.M. designed and supervised the study. J.P.B., S.K., S.J.R., G.N.R., performed the cell culture and differentiation. C.R.S. prepared SARS-CoV-2 virus, performed the infections and all BSL3 sample collection. J.P.B., S.K., S.J.R. performed the immunostaining and imaging. J.P.B., S.K., S.J.R., G.N.R. performed the qPCR analyses. J.P.B., S.K., S.J.R., D.A.J, performed the RNA library preparation and sequencing. D.A.J. and W.R.F. performed the bioinformatic analyses. J.P.B., S.K., H.L. performed the electron microscopy studies. J.D.W. provided patient samples. A.C.S. performed the histological processing and tissue staining. All authors contributed to data analyses, writing the manuscript and figure preparation.
ACKNOWLEDGEMENTS
The following reagents were obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281, deposited by the Centers for Disease Control; and Monoclonal Anti-SARS-CoV S Protein (Similar to 240C), NR-616.
We thank the Gladstone Light Microscopy and Histology Core, the Gladstone Assay Development and Drug Discovery Core, and the Gladstone Stem Cell Core for their support and experimental expertise. We also would like to thank Danielle Jorgens at the University of California Berkeley Electron Microscope for electron microscopy sample preparation and data collection. We gratefully thank the Zuckerberg-San Francisco General anatomic pathology services, including Dr. Stephen Nishimura, Mark Weinstein, and Andrew Lewis, for processing and donation of patient samples.
Endothelial cells were a kind gift from Dr. Sanjeev Ranade at the Gladstone Institutes. Patient samples were very generously contributed by Dr. Timothy Kamp at the University of Wisconsin, Madison. We thank Dr. Anita Sil, Dr. Bastian Joehnk, Dr. Lauren Rodriguez and Keith Walcott for BSL-3 laboratory support.
References for methods section
References




