

Abstract
The sub-cellular positioning of endolysosomal compartments is crucial for regulating their downstream function. In particular, the positioning of endolysosomes at two sub-cellular locations: the cell periphery versus the cell peri-nuclear region; impacts autophagy, mTOR (mechanistic target of rapamycin) signaling and other cellular processes. Yet, the mechanisms that regulate the trafficking and positioning of endolysosomes at these two sub-cellular locations are poorly understood. Here, using super-resolution microscopy, we show that the retrograde motor dynein forms nano-clusters on endolysosomal membranes. Surprisingly, dynein nano-clusters are larger on peripherally positioned endolysosomes that contain higher cholesterol levels compared to perinuclearly positioned ones. By perturbing endolysosomal membrane cholesterol levels, we show that dynein clustering directly depends on the amount of cholesterol on the endolysosomal membrane. Finally, we show that the dynein adapter protein ORP1L (Oxysterol Binding Protein Homologue) regulates dynein clustering and endolysosomal positioning through its cholesterol binding domain in response to membrane cholesterol levels.
Introduction
Late endosomes, lysosomes and autolysosomes constitute a broad class of sub-cellular compartments1,2 that we will refer to here as endolysosomal compartments or endolysosomes for simplicity. These compartments play key cellular roles including transport of cellular proteins destined for degradation, metabolic sensing, membrane repair and signaling3. The maturation level, fusion capacity with other sub-cellular compartments and downstream function of endolysosomes are regulated by their intracellular trafficking and sub-cellular positioning. For example, dispersal of lysosomes from the cell periphery to the peri-nuclear region impacts their association with mTOR (mechanistic target of rapamycin) and leads to downregulation of autophagy4-7. The trafficking and subcellular positioning of endolysosomes are in turn regulated by a myriad of mechanism8 including motor activation9-11, motor tug-of-war mechanisms12-14 and association of motors with microtubule tracks having distinct post-translational modifications15-17.
Peripheral trafficking of endolysosomes is mediated by kinesin motors belonging to different kinesin families (Kif5, Kif1, Kif3)16,18-21, whereas dynein is responsible for their retrograde trafficking22,23. Recent discovery of dynein activating adapters have revolutionized our understanding of how dynein mediates efficient retrograde transport11,23-26. Dynein assembles into an autoinhibitory, weakly processive conformation, while dynein activating adaptors are crucial for dynein’s assembly with dynactin and the processive motility of the dynein-dynactin complex on microtubules24,26-28. Recent Cryo-EM studies showed that certain early endosomal dynein activating adapters including BICD2 and Hook3 recruit two dynein dimers27. In vitro assays further showed that these heteromeric dynein complexes move faster and navigate obstacles better compared to single dynein27,29,30. Hence, in addition to mediating dynein’s interaction with dynactin and bringing dynein out of its auto-inhibitory confirmation, these activators further improve the efficiency of dynein mediated motility by allowing two copies of dynein to assemble together into complexes. Yet, to date, the stoichiometry of adapter-dynein-dynactin complexes on sub-cellular compartments in vivo and how these complexes are spatially organized on the membrane of sub-cellular compartments are not known. Dynein is recruited to endolysosomal compartments via the cholesterol-sensing tripartite complex Rab7-RILP-ORP1L31-33. Whether this tripartite complex can recruit multiple copies of dynein dimers to improve the efficiency of retrograde transport of endolysosomal compartments in response to cellular cues is unknown.
Previously, using super-resolution microscopy, we showed that dynein forms nano-clusters on microtubules consisting of 1-3 dynein dimers34,35. However, whether these nano-clusters are formed on the membrane of endolysosomal compartments, the mechanisms of nano-cluster formation and whether formation of larger nano-clusters containing more dynein motors lead to more efficient retrograde transport are not known. Here, using super-resolution microscopy, we show that dynein forms nano-clusters on endolysosomal compartments and these nano-clusters are larger on peripheral endolysosomes having higher membrane cholesterol levels compared to peri-nuclear endolysosomes having lower cholesterol levels. By manipulating the endolysosomal membrane cholesterol content through treatment with pharmacological drugs, we show that cholesterol levels regulate the level of dynein clustering on endolysosomal membranes; with higher cholesterol leading to larger dynein nano-clusters. Increased dynein clustering in turn leads to more efficient retrograde trafficking. The cholesterol sensing adaptor protein ORP1L is similarly clustered on the membrane of endolysosomal compartments in cells treated with pharmacological drugs to increase membrane cholesterol levels. Further, increased ORP1L clustering under high cholesterol is directly dependent on the cholesterol sensing domain of ORP1L. Finally, we show that increased dynein clustering under high cholesterol is also mediated by the cholesterol sensing domain of ORP1L. Overall, we present a new mechanism by which dynein recruitment and clustering on endolysosomal compartments and retrograde trafficking of endolysosomal compartments are regulated by the membrane cholesterol levels in a manner dependent on ORP1L’s cholesterol sensing domain.
Results
Dynein forms nano-clusters on endolysosomal membranes with increased clustering for peripheral endolysosomes having higher membrane cholesterol levels
To visualize the spatial organization of dynein on endolysosomal membranes we expressed mCherry-ORP1L in HeLa cells that are a knock-out for ORP1L36. There was a large overlap between the mCherry-ORP1L signal and CD63, an endolysosomal marker37-39, indicating that ORP1L marks endolysosomal compartments (Figure S1A). We carried out super-resolution imaging of dynein labeled with an antibody against the dynein intermediate chain (IC74). These images revealed nano-clusters within the cell cytoplasm similar to what we have previously demonstrated35 (Figure 1A-B). Our previous quantitative analysis of these dynein nano-clusters had shown that they consist of 1-3 full dynein motors35. We used the mCherry signal to specifically segment dynein nano-clusters that overlapped with an endolysosomal compartment (Figure 1A-B, Methods). Dynein super-resolution images were further segmented into individual nano-clusters using a previously developed Voronoi tessellation approach (Figure S1B)40. We then quantified the number of localizations per dynein nano-cluster for peripherally and peri-nuclearly positioned endolysosomes (Figure 1C). The peripheral and peri-nuclear endolysosomes were separated manually based on their proximity to the cell nucleus (Figure S1C). The number of localizations per nano-cluster is proportional to the number of dyneins within nano-clusters34,35. Surprisingly, we found that the dynein nano-clusters associated to peripheral endolysosomes contained significantly higher number of localizations compared to peri-nuclear endolysosomes (Figure 1C). We asked if the membrane cholesterol content may be responsible for increased dynein clustering of peripheral endolysosomes. To start addressing this question, we measured the cholesterol levels of peripheral and peri-nuclear endolysosomes by labeling mCherry-ORP1L expressing HeLa cells with filipin, a toxin that binds cholesterol. We measured the filipin intensity on mCherry-ORP1L positive endolysosomes and found that membrane cholesterol levels of peripheral endolysosomes were indeed higher compared to peri-nculear endolysosomes (Figure 1D). These results indicate a positive correlation between endolysosomal membrane cholesterol levels and dynein clustering.

(A) Cropped wide-field image of mCherry-ORP1L (yellow) overlaid with super-resolution image of dynein (blue). Cell edge and nucleus are highlighted in yellow dashed lines. Scale bar is 10 µm. A zoom of the white rectangle is shown. Scale bar is 200 nm.
(B) An overlay of wide-field image of ORP1L (yellow) and super-resolution image of dynein (image is color coded according to localization density with higher density corresponding to white and lower density corresponding to cyan) for an endolysosome positioned at the cell periphery (PP) and peri-nuclear region (PN). Scale bar is 200 nm.
(C) Violin plot showing the number of localizations per dynein nano-cluster for peripherally positioned endolysosomes (PP) (n=195 endolysosomes from n=6 cells, n=2experiments) versus peri-nuclearly positioned endolysosomes (PN) (n=210 endolysosomes from n=6 cells, n=2 experiments). Statistical significance was assessed using a Kolmogorov-Smirnov-test with a p-value of 0.012.
(D) Box plot showing the intensity of filipin, which binds cholesterol, on peripherally positioned endolysosomes (PP) (n=50 endolysosomes from n=4 cells, n=2 experiments) versus peri-nuclearly positioned endolysosomes (PN) (n=80 endolysosomes from n=5 cells, n=2 experiments). Statistical significance was assessed using an unpaired t-test with a p-value of 0.011
Membrane cholesterol levels determine the level of dynein clustering on endolysosomes
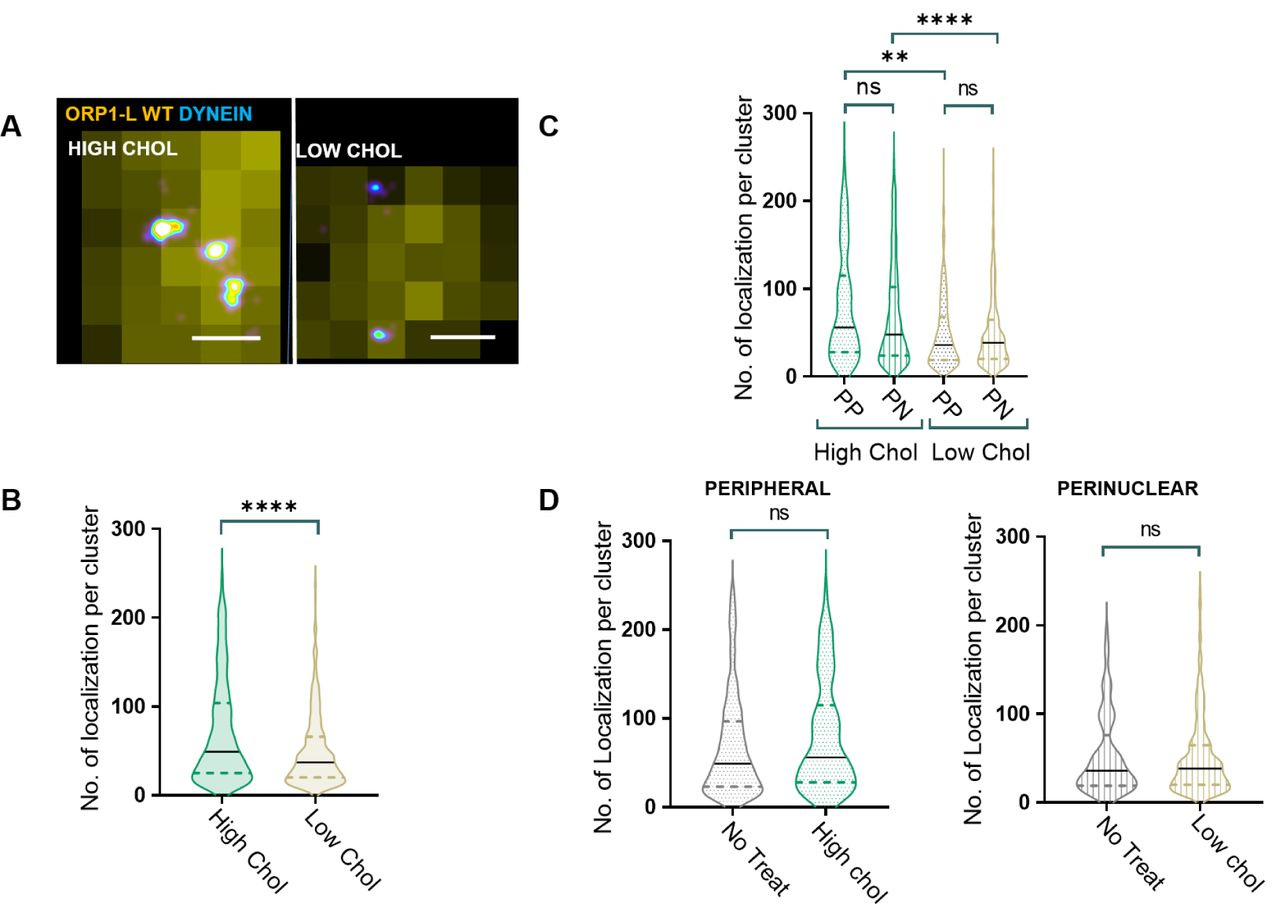
It is known that positioning of endolysosomal compartments in the cell affects their membrane composition, maturation and signaling41-43. To better understand the mechanisms behind dynein clustering and causally relate dynein clustering to the endolysosomal membrane cholesterol levels, we manipulated cholesterol levels with two commonly used drugs: U-1866A to increase endolysosomal membrane cholesterol and Lovastatin to decrease cellular and endolysosomal membrane cholesterol levels31,44. mCherry-ORP1L-positive endolysosomes in HeLa cells treated with U-1886A indeed had higher membrane cholesterol levels (by 4.5-fold) compared to those in HeLa cells treated with lovastatin as measured by filipin intensity (Figure S2A). We will refer to the endolysosomal membrane cholesterol content of the U18666A and Lovastatin treated cells as ‘high cholesterol’ and ‘low cholesterol’ condition, respectively. Super-resolution imaging revealed that dynein nano-clusters on endolysosomal compartments contained a significantly higher number of localizations under high cholesterol compared to low cholesterol conditions (Figure 2A-B). These differences were not due to changes in dynein expression level upon cholesterol drug treatments, as western blot analysis showed that dynein expression was similar in both cases (Figure S2B). Further, in contrast to physiological conditions, peripheral and peri-nuclear endolysosomes had similar cholesterol levels (Figure S2C) and similar dynein clustering (Figure 2C) under both high and low cholesterol conditions. Interestingly, the dynein clustering level of peri-nuclear endolysosomes under physiological conditions was similar to those under low cholesterol conditions and the dynein clustering level of peripheral endolysosomes under physiological conditions was similar to those under high cholesterol conditions (Figure 2D). These results indicate that cholesterol levels and not endolysosome positioning determine the level of dynein clustering on endolysosomal membranes. Low cholesterol levels also led to an increase in the percentage of endolysosomes that completely lacked dynein (Figure S2D). Finally, the positioning of endolysosomes were impacted by cholesterol levels, with low cholesterol leading to more scattered and high cholesterol leading to more peri-nuclearly positioned endolysosmes (Figure S2E).

(A) An overlay of cropped wide-field image of ORP1L (yellow) and super-resolution image of dynein (image is color coded according to localization density with higher density corresponding to white and lower density corresponding to cyan) for an endolysosome in cells treated with U-1866A (High Chol) or Lovastatin (Low Chol). Scale bar is 200 nm.
(B) Violin plot showing the number of localizations per dynein nano-cluster for endolysosomes in U-1866A treated cells (High Chol) (n=224 endolysosomes from n=6 cells, n=2 experiments) versus Lovastatin treated cells (Low Chol) (n=217 endolysosomes from n=6 cells, n=2 experiments). Statistical significance was assessed using a Kolmogorov-Smirnov-test with a p-value <0.0001.
(C) Violin plot showing the number of localizations per dynein nano-cluster for peripherally positioned (n=70 endolysosomes from n=6 cells, n=2 experiments) or perinuclearly positioned (n=154 endolysosomes from n=6 cells, n=2 experiments) endolysosomes in U-1866A treated cells (High Chol PP and High Chol PN, respectively) or Lovastatin treated cells (Low Chol PP; n=110endolysosomes from n=6 cells, n=2 experiments and Low Chol PN; n=107 endolysosomes from n=6 cells, n=2 experiments, respectively). Statistical significance was assessed using a Kolmogorov-Smirnov-test with a p-value of n.s.: 0.27, n.s.: 0.93, **: 0.001, ****: <0.0001.
(D) Violin plot showing the number of localizations per dynein nano-cluster for for peripherally positioned endolysosomes in untreated cells (No Treat) (n=195 endolysosomes from n=6 cells, n=2 experiments) and U-1886A treated cells (High Chol) (n=70 endolysosomes from n=6 cells, n=2 experiments); as well as in perinuclearly positioned endolysosomes in untreated cells (No Treat) (n=210 endolysosomes from n=6 cells, n=2 experiments) and Lovastatin treated cells (Low Chol) (n=107 endolysosomes from n=6 cells, n=2 experiments). Statistical significance was assessed using a Kolmogorov-Smirnov-test with a p-value of 0.23 and 0.7
Taken together our results show that membrane cholesterol levels impact both dynein recruitment and dynein clustering on endolysosomal membranes, impacting the balance between antero-and retro-grade transport of these compartments and leading to their re-positioning with respect to the cell peri-nuclear region.
The cholesterol sensing domain of ORP1L regulates ORP1L clustering on endolysosomal membranes in a cholesterol dependent manner
Dynein does not bind directly to endolysosomal membranes but is recruited through a tripartite complex of Rab7-ORP1L-RILP31-33. ORP1L contains multiple lipid binding domains that allow it to bind to either oxysterols or phospholipids36,45,46. Hence, we asked whether the cholesterol sensing domain of ORP1L is responsible for regulating dynein clustering on endolysosomal membranes. To address this question, we first imaged ORP1L’s spatial distribution on endolysosomes in wild type or ORP1L-knock-out HeLa cells that express mCherry-ORP1L36 under low or high cholesterol treatment conditions using super-resolution microscopy (Figure 3A). ORP1L appeared uniformly distributed on the endolysosomal membrane under low cholesterol conditions and more clustered under high cholesterol conditions (Figure 3A). To quantify the level of ORP1L clustering, we once again used Voronoi tessellation to segment individual ORP1L clusters on endolysosomal membranes. This approach revealed an increased number of ORP1L clusters under high cholesterol compared to low cholesterol conditions (Figure S3A). However, we also found that the localization density of ORP1L was higher on endolysosomes under low cholesterol compared to high cholesterol conditions (Figure S3B), suggesting that more ORP1L binds to endolysosomes when their membrane cholesterol levels are lower. To ensure that the increased protein density does not confound the clustering analysis using Voronoi tessellation, which is dependent on localization density, we developed an alternative quantification method that is insensitive to differences in localization density and protein amount (Methods). To this end, we carried out Voronoi tessellation and re-scaled the distribution of Voronoi polygon areas so that the mean area was set to unity. We refer to the re-scaled quantities as reduced areas. The distribution of reduced areas provides a means to measure the clustering tendency of ORP1L from various conditions without explicitly compensating for different localization densities. For a clustered distribution, we expected to see a shift in the mode of the reduced Voronoi polygon area distribution towards a smaller value. Indeed, the mode of the distribution for endolysosomal compartments under high cholesterol conditions was shifted to smaller polygon areas compared to low cholesterol conditions or compared to a simulated random distribution, indicating that ORP1L is more clustered on endolysosomes with high membrane cholesterol levels (Figure 3B). We further used a statistical test (Kullback-Leibler Divergence or KL Divergence) to determine how much ORP1L organization differed from a random distribution under low and high cholesterol conditions. The difference from the normalized random distribution of points to the experimental distribution, which we call the clustering tendency score, was 0.75 under high cholesterol and 0.14 under low cholesterol (a 5-fold difference). These results further confirm that ORP1L’s organization on endolysosomal membranes deviated significantly more from a random distribution under high cholesterol compared to low cholesterol conditions. When endogenous ORP1L on CD63 positive endolysosomal compartments was imaged in wild type HeLa cells using an ORP1L antibody, it also was more clustered and had lower localization density under high cholesterol compared to low cholesterol conditions (Figure S3C-D), demonstrating that the results are not an artifact of ORP1L over-expression. Overall, these results show that ORP1L, like dynein, is more clustered on endolysosomes having higher membrane cholesterol levels.

(A) Super-resolution images of full length ORP1L (WT ORP1L) in cells treated with U-1886A (High Chol, upper panels) and in cells treated with Lovastatin (Low Chol, lower panels). Scale bar is 200 nm.
(B) Log plot of the Reduced Voronoi Polygon area distribution for super-resolution images of full length ORP1L in cells treated with U-1886A (dark green, High Chol) (n=110 endolysosomes from n=6 cells, n=2 experiments), Lovastatin (light green, Low Chol) (n=120 endolysosomes from n=6 cells, n=2 experiments) and for a random distribution of localizations (yellow, Random).
(C) Super-resolution images of sterol binding deficient ORP1L mutant lacking residues 560-563 (mCherry-ORP1L-SBD) in cells treated with U-1886A (High Chol, upper panels) and in cells treated with Lovastatin (Low Chol, lower panels). Scale bar is 200 nm.
(D) Log plot of the Reduced Voronoi Polygon area distribution for super-resolution images of sterol binding deficient ORP1L mutant lacking residues 560-563 (mCherry-ORP1L-SBD) in cells treated with U-1886A (dark green, High Chol) (n=120 endolysosomes from n=6 cells, n=2 experiments), Lovastatin (light green, Low Chol) (n=170 endolysosomes from n=6 cells, n=2 experiments) and for a random distribution of localizations (yellow, Random).
To determine if the differences in ORP1L’s spatial distribution on endolysosomes were due to its cholesterol binding, we expressed a sterol binding deficient ORP1L mutant lacking the residues 560–563 (mCherry-ORP1L-SBD) 36,47 fused to mCherry in HeLa ORP1L knock-out cells (Figure 3C-D and Figure S3E-F). Super-resolution images of the mCherry-ORP1L-SBD mutant (Figure 3C) and both Voronoi cluster segmentation (Figure S3E) and reduced Voronoi polygon area distribution analysis (Figure 3D) (Clustering tendency score for ORP1L-SBD: 0.1 for high and 0.09 for low cholesterol conditions) showed that the distribution of the ORP1L-SBD mutant was uniform on endolysosomal membranes independent of cholesterol levels. The membrane localization density of this mutant was overall high under both high and low cholesterol conditions (Figure S3F) and at a similar level to the full length ORP1L under low cholesterol conditions (Figure S3B). Finally, under physiological conditions (no cholesterol treatment) the full-length ORP1L was more clustered on peripherally positioned endolysosomes that have higher cholesterol levels whereas these differences in ORP1L clustering were absent for the ORP1L-SBD mutant (Figure S4A-B).
Overall, these results strongly support that the cholesterol sensing domain of ORP1L regulates its spatial distribution on endolysosomal membranes in a cholesterol dependent manner, with high cholesterol leading to more specific ORP1L recruitment (Figure S3B) and a clustered ORP1L spatial organization (Figure 3A-B).
The cholesterol sensing domain of ORP1L regulates dynein clustering and endolysosomal positioning in a cholesterol dependent manner
Having established that ORP1L’s cholesterol sensing domain regulates ORP1L’s level of clustering on endolysosomal membranes in a cholesterol dependent manner, we next asked if ORP1L’s spatial organization impacts dynein clustering. We thus imaged dynein using super-resolution microscopy in HeLa cells expressing the mCherry-ORP1L-SBD mutant fused to mCherry in an ORP1L-null background. Analysis of dynein nano-clusters on endolysosomes under high and low cholesterol conditions showed that dynein clustering was no longer sensitive to cholesterol levels in cells expressing the mCherry-ORP1L-SBD mutant (Figure 4A-B). In addition, the level of dynein clustering in cells expressing the mCherry-ORP1L-SBD mutant was similar to the level of dynein clustering under low cholesterol conditions in cells expressing the wild type mCherry-ORP1L.

(A) An overlay of cropped wide-field image of ORP1L (yellow) and super-resolution image of dynein (image is color coded according to localization density with higher density corresponding to white and lower density corresponding to cyan) for an endolysosome in cells expressing sterol binding deficient ORP1L mutant lacking residues 560-563 (mCherry-ORP1L-SBD) and treated with U-1866A (High Chol) or Lovastatin (Low Chol). Scale bars are 200 nm.
(B) Violin plot showing the number of localizations per dynein nano-cluster for endolysosomes in cells expressing the sterol binding deficient ORP1L mutant lacking residues 560-563 (mCherry-ORP1L-SBD) and treated with U-1866A (High Chol) (n=450 endolysosomes from n=6 cells, n=2 experiments) versus Lovastatin (Low Chol) (n=300 endolysosomes from n=6 cells, n=2 experiments). Statistical significance was assessed using a Kolmogorov-Smirnov-test with a p-value of 0.06.
(C) Violin plot showing the number of localizations per dynein nano-cluster for peripherally positioned endolysosomes (n=100 endolysosomes from n=5 cells, n=2 experiments) and peri-nuclearly positioned endolysosomes (n=150 endolysosomes from n=5 cells, n=2 experiments) in cells expressing the sterol binding deficient ORP1L mutant lacking residues 560-563 (mCherry-ORP1L-SBD). Statistical significance was assessed using a Kolmogorov-Smirnov-test with a p-value of 0.44.
The sub-cellular positioning of endolysosomes also became insensitive to cholesterol treatment in cells expressing the mCherry-ORP1L-SBD mutant and the endolysosomes were overall more scattered throughout the cell under high or low cholesterol conditions as well as under physiological conditions (Figure S4C). Finally, peripheral and peri-nuclear endolysosomes had similar level of dynein clustering in cells expressing the mCherry-ORP1L-SBD mutant (Figure 4C). Taken together, these results show that the increased clustering of dynein on peripherally located endolysosomes with higher cholesterol content is dependent on the cholesterol binding and clustering ability of ORP1L (Figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cartoon model showing how cholesterol levels and ORP1L regulate dynein nano-clustering and endolysosome positioning. Peripheral lysosomes with higher cholesterol content have a more clustered organization of ORP1L and dynein leading to efficient retrograde transport (blue arrow). Perinuclear lysosomes having lower cholesterol content have more uniform ORP1L organization leading to lower dynein recruitment and clustering likely facilitating endolysosome anchoring at the peri-nuclear region or anterograde trafficking back to the cell periphery.
Discussion
Here using a combination of super resolution microscopy and perturbation experiments in which we manipulated endolysosomal membrane cholesterol levels we show that the level of dynein clustering on endolysosomal membrane is increased under high cholesterol. This increased clustering is regulated by the cholesterol binding ability of ORP1L that forms part of the Rab7-ORP1L-RILP tripartite complex, which recruits dynein to endolysosomal membranes.
Cryo-EM experiments demonstrated that BICD2 and Hook3 bind two copies of dynein dimers27 but whether the endolysosomal adapter proteins Rab7-ORP1L-RILP also recruit multiple dynein dimers is unknown. Our results demonstrate that besides the stoichiometry between motor proteins and their adapter proteins, additional mechanisms inside cells are at play to increase motor protein clustering and retrograde transport efficiency. We show that clustering of adapter proteins into cholesterol enriched domains impacts the clustering of dynein motor on endolysosomal membranes, increasing the efficiency of retrograde transport.
Previous in vitro work showed that early phagosomes engulfing 2-micron sized polystyrene beads isolated from dictyostelium cells had a uniform dynein distribution on their membrane whereas late phagosomes that move unidirectionally towards the retrograde direction had a clustered dynein distribution48. These changes in dynein distribution were correlated to the membrane cholesterol content of phagosomes. In contrast, our results show that instead of uniformly covering the entire membrane, dynein forms small teams on endolysosomal membranes in vivo. Interestingly, the number of dyneins within these small teams is higher for peripherally located endolysosomes, which also had higher cholesterol levels. It is well-known that endolysosomes contact the endoplasmic reticulum as they are trafficked within the cell cytoplasm31,36,49. Such contacts lead to exchange of membrane lipids and maturation of endolysosomes 36,38. It is plausible that multiple endolysosome-ER contacts during retrograde transport play a role in lowering the cholesterol content of endolysosomes, leading to re-organization of ORP1L and decreased dynein clustering. Hence, the initial high level of dynein clustering on the peripheral endolysosomes likely leads to their efficient retrograde trafficking. Loss of membrane cholesterol through ER contacts, on the other hand, potentially leads to loss of dynein clustering facilitating stalling/positioning at the perinuclear region or anterograde trafficking back to the cell periphery via kinesin.
Previous studies showed that ORP1L, depending on its conformation, can either bind dynein or make contacts with the ER-membrane2,31,36,45. A change in ORP1L’s conformation leads to shedding of dynein and initiation of contact between ORP1L and the ER-membrane protein VAP to regulate endolysosomal positioning31. Our results are consistent with these former studies as we show that in addition to the level of dynein clustering, the amount of dynein on endolysosomal compartments is also dependent on cholesterol levels. Under low cholesterol, the percentage of endolysosomes completely lacking dynein was increased by ∼2-fold. However, here we additionally show that ORP1L’s nanoscale spatial organization on the endolysosomal membranes is also dependent on cholesterol levels and regulates dynein clustering in addition to dynein recruitment.
Overall, our results provide an in vivo mechanism dependent on cholesterol levels by which multiple dyneins can be recruited and clustered on endolysosomal membranes leading to their efficient retrograde trafficking and positioning. Increased dynein clustering in response to cholesterol levels is likely to be functionally significant as it impacts the sub-cellular positioning of endolysoosmal compartments. Metabolic disorders that lead to accumulation of lipids including cholesterol in endolysosomal compartments like Niemann Pick Disease (NPC) are typically associated with alterations in endolysosomal homeostasis and function50. In the future it would be interesting to explore if the nanoscale organization of ORP1L and dynein is altered on endolysosomal membranes in NPC and other lysosomal storage disorders leading to their mislocalization within the cell and whether restoring the proper nanoscale organization of these cytoskeletal proteins can restore endolysosomal function. It would also be interesting to determine if similar mechanisms play a role in regulating kinesin clustering or in regulating transport of other organelles including Golgi vesicles and autophagosomes. It would further be exciting to determine the precise stoichiometry of adapter-motor complexes on organelle membranes to determine how the stoichiometry can be precisely tuned to regulate organelle transport and positioning. Our work establishes the methodology needed and opens the door to carry out these future studies.
Author Contributions
ML and ST conceived of the study. ST carried out experiments, wrote software and analyzed data. P.K.R. wrote software and implemented the renormalized voronoi area distribution and KL-Divergence analysis methodology. E.M.S. maintained cell lines, carried out western blot experiments, carried out all transfections, provided reagents and prepared samples. ML wrote the manuscript, acquired funding and supervised the work. All authors provided feedback on the manuscript.
Materials and Methods
Cells and transfections
Wild type HeLa cells were obtained from the American Type Culture Collection (CCL-2, ATCC, Manassas, VA). HeLa-ORP1L-null cell lines, as well as mCherry-tagged ORP1L and ORP1L-SBD constructs were a kind gift from Prof. Neale Ridgway (Dalhousie University, Depts. of Pediatrics, and Biochemistry and Molecular Biology, Atlantic Research Centre, Halifax, Nova Scotia, Canada). HeLa cells were grown in DMEM (GIBCO Laboratories, Grand Island, NY) supplemented with 10% fetal bovine serum and antibiotics, and maintained in 5% CO2 at 37°C. Cells were transiently transfected with mCherry-tagged wild type or mutant ORP1L at 70% confluency using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol. Cells were subjected to experimental treatments 24 h after transfection.
Pharmacological treatment of cells
Lovastatin (Sigma 1370600) was converted from its inactive prodrug form to its active open acid form by dissolving Lovastatin (52gms) in ethanol (95%, 1.04 ml), followed by addition of 1N NaOH (813 µl), followed by heating for 2 hrs at 50°C and neutralized with 1N HCL (pH 7.2). The volume was made up to 13 ml by adding distilled water, giving 10mM active Lovastatin solution later aliquoted and stored at −20°C44. Mevalonic acid lactone (Sigma M4667, 1 gm) was converted to its active form by dissolving in ethanol (3.5 ml), followed by addition of 1N NaOH (4.2 ml) and heating for 2 hrs at 55°C. The solution was made up to 15.4 ml with distilled water and neutralized with 1N HCL (pH 7.2), giving 500 mM of stock solution, later aliquoted and stored at −20. U18666a (Sigma U3633) was dissolved in ethanol giving final concentration of 10mg/ml31. For cholesterol depletion treatment, cells were cultured in DMEM, 10% Lipoprotein deficient serum (Sigma S5394), 50uM Lovastatin, 230uM Mevalonate for 6 hrs before fixing for immunostaining. For high cholesterol treatment cells were cultured in DMEM, 10% FBS and 3µg/ml U18666A for 12 hrs before fixing.
Immunostating
Hela ORP1L KO cells were transiently transfected with ORP1L WT-mCherry or ORP1L SBD-mCherry and immunostained for ORP1L and dynein. For ORP1L immunostaning, cells were fixed with 4% (Vol/Vol) Paraformaldehyde in PBS for 20 mins and for dynein immunostaining, cells were fixed in prechilled 1:1 Ethanol/Methanol solution for 3 mins on ice. The cells were blocked in blocking buffer (3% BSA, 0.2% Triton X-100 in PBS) for 1 hr. Cells were incubated with primary anitbodies: Chicken anti-mCherry (Novus biotech nbp2-2515, 1:500) and mouse anti-dynein (Abcam ab23905, 1:50) in blocking buffer for 1 hr on a rocker. Cells were washed with washing buffer (0.2% blocking buffer, .05% Triton X-100 in PBS) three times. Custom made secondary antibodies were labeled with an Alexa Fluor 405–Alexa Fluor A647 activator/reporter dye pair combination at concentrations (0.1-0.15 mg/µl) and used in the ratio of 1:50 in blocking buffer for 40 mins, at RT on a rocker. Sample was then washed three times in PBS.
Filipin staining
Filipin (Sigma F4767) was lyophilized, aliquoted (250 µg per aliquot) and stored at −80°C. Filipin was resuspended in 5 µl DMSO. Cells were fixed in 4% Paraformaldehyde for 20 mins and then rinsed 3 times with PBS. Background autofluorescence was quenched with 50mM NH4CL for 10 mins. Cells were incubated for 2 hrs with 100 µg/ ml working solution of Filipin in 3% BSA. Cells were washed with 1% BSA in PBS three times before imaging. Imaging was performed immediately.
Western blot
Western blot analysis was performed using the two-color Odyssey LI-COR (Lincoln, NE) technique according to the manufacturer’s protocol. A rabbit monoclonal antibody to ORP1L (ab131165, Abcam), mouse monoclonal antibody to dynein (ab23905, Abcam), and a mouse monoclonal antibody to detect GAPDH (clone 3B1E9, GenScript A01622–40) were used at a dilution of 1:1,000 in blocking buffer. The secondary antibody IRDye800CW Donkey anti-Rabbit and IRDye680RD Donkey anti-Mouse (LI-COR) were used in 1:10000 dilution for imaging in the green 800-nm and red 700-nm channels, respectively.
STORM Imaging
Single-molecule imaging was done using imaging buffer comprising of 50 mM Tris, pH 7.5, 10 mM NaCl, 0.5 mg/mL glucose oxidase (Sigma, G2133), 40 μg/mL catalase (Roche Applied Science, 106810), 10% (w/v) glucose and 10% (v/v) Ciseamine (77mg/ml of 360mM HCL)51. Images were acquired on the Oxford Nanoimager-S microscope which has the following configuration: 405, 488, 561, and 640 nm lasers, 498–551 and 576–620 nm band-pass filters in channel 1, and 665–705 nm band-pass filters in channel 2, 100× 1.4 NA objective (Olympus), and a Hamamatsu Flash 4 V3 sCMOS camera. Localizations were acquired with 10-ms exposure over 50,000 frames with 405 nm activation and 647 nm excitation. Images were processed and localizations were obtained using the NimOS localization software (Oxford Nanoimaging).
Data Analysis
For quantitative analysis we used custom written MATLAB codes. A previously described method was adapted that segments super-resolution images based on Voronoi tessellation of the fluorophore localizations40,52. Voronoi tessellation of a STORM image assigns a Voronoi polygon to each localization, such that the polygon area is inversely proportional to the local localization density. The spatial distribution of dynein or ORP1L localizations from each ORP1L positive endolysosome is represented by a set of Voronoi polygons such that smaller polygon areas correspond to regions of higher density. The Voronoi polygons at the endolysosomal edge are extremely large and were omitted for any quantification. Dynein and ORP1L clusters were segmented by grouping adjacent Voronoi polygons with areas less than a selected threshold and imposing a minimum number of localizations. For mCherry-ORP1L, mCherry-ORP1L-SBD, dynein (ORP1L positive endolysosomes) and dynein (ORP1L-SBD positive endolysosomes), the selected area thresholds were 0.0156 px2, 0.02 px2, 0.01 px2 and, 0.01 px2, respectively and the minimum number of localizations imposed were16,10, 7 and 7, respectively. ORP1L localization density was calculated by normalizing the total number of localizations per endolysosme by endolysosome area. Each endolysosme area was calculated by summing up all of its Voronoi polygon areas. The low cholesterol treatment yielded endolysosomes with ORP1L localization densities 4.5 times higher as compared to the high cholesterol treated endoslysosome localization densities. To compare endoslysomal ORP1L distribution following cholesterol treatments, the ORP1L localization densities should be comparable. To address this we divided the Voronoi polygon areas of each endolysosome by its mean Voronoi polygon area, such that the mean localization density of each endolysosome is in reduced units of 1. All reduced Voronoi polygon areas from each endolysosme for each treatment were pulled together, and the histogram was plotted. The distribution of Voronoi areas from uniformly simulated random points is fit to an analytical distribution53. A method for calculating the KL divergence between two histograms or between a histogram and an analytical distribution54 was implemented in Matlab. The KL divergence scores between the experimental reduced voronoi areas and the theoretical random distribution were calculated to determine the clustering tendency score of each cholesterol treatment. All ORP1L and dynein analysis has been done with endolysosmes with radius < 200nm for consistency. Filipin intensity was calculated using Image J ‘plot profile’ tool. All ORP1L positive endolysosomes were analyzed by drawing a line segment across it and looking at its intensity profile using the ‘plot profile’ tool. The average of 3-4 highest intensity points were taken and the average background intensity was subtracted from that. For the analysis of dynein clusters on ORP1L positive endolysosmal compartments, a intensity plot of the conventional ORP1L image was taken and all dynein clusters falling within the Full Width Half Maxima of the profile was accepted
Acknowledgements
We thank Prof. Neale D. Ridgway, Dalhousie University for the Hela cells that are a knockout for ORP1L and for the mCherry-ORP1L and mCherry-ORP1L-SBD constructs. ML acknowledges funding from the National Institutes of Health/National Institutes for General Medical Sciences (NIH/NIGMS) under the grant numbers: RO1 GM 133842-01 and 1RM1GM136511-01.
References




