

Abstract
COVID-19 caused by the SARS-CoV-2 virus has become a global pandemic. 3CL protease is a virally encoded protein that is essential to the viral life cycle across a broad spectrum of coronaviruses with no close human analogs. The designed phosphate prodrug PF-07304814 is metabolized to PF-00835321 which is a potent inhibitor in vitro of the coronavirus family 3CL pro, with selectivity over human host protease targets. Furthermore, PF-00835231 exhibits potent in vitro antiviral activity against SARS-CoV-2 as a single agent and it is additive/synergistic in combination with remdesivir. We present the ADME, safety, and in vitro antiviral activity data to warrant clinical evaluation.
One Sentence Summary The phosphate prodrug PF-07304814 is disclosed as an investigational novel intravenous small molecule 3CL protease inhibitor for COVID-19.
Main Text
In December 2019, COVID-19 was identified as a new, potentially fatal, respiratory infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1, 2). Unlike previous coronavirus outbreaks that receded relatively quickly, the resultant COVID-19 pandemic spread across the globe. As of August 2020, over 24 million people have been infected and over 821,000 people have died globally with no approved drugs available to treat the disease (3).
The RNA-dependent RNA polymerase (RdRp) inhibitor remdesivir is currently undergoing clinical investigation for the treatment of SARS-CoV-2 and was granted emergency use authorization by the U.S. Food and Drug Administration (FDA) in May 2020 (4). To date, trials of remdesivir have shown significantly decreased recovery time for COVID-19 patients, but the drug has not had a significant effect on morbidity (5). Other classes of antivirals that exhibit single agent efficacy or that are complementary to remdesivir for use in combination regimens are essential to meet this substantial unmet need.
SARS-CoV-2 produces two large viral polyproteins, pp1a and pp1ab, which are processed by two virally encoded cysteine proteases, the main protease, also called 3C-like protease (3CL protease or 3CLpro) and the papain-like protease. Mutagenesis experiments with other coronaviruses have demonstrated that the activity of the 3CLpro is essential for viral replication (6, 7). 3CLpro proteolytically processes the virus p1a/p1ab polyproteins at more than 10 junctions to generate a series of non-structural proteins critical for virus replication and transcription, including RdRp, the helicase, and the 3CLpro itself (8). No close human analogs of the coronavirus 3CLpro are known, suggesting that selective 3CLpro inhibitors should avoid unwanted polypharmacology (9). The essential functional importance of proteases in virus replication has led to the clinical success of protease inhibitors in combating both human immunodeficiency virus (HIV) and hepatitis C virus (HCV) (10–13). This together with the opportunity for selectivity, makes 3CLpro an attractive antiviral drug target (14).
Following the severe acute respiratory syndrome (SARS) outbreak in 2002-2003 we identified a potential small molecule protease inhibitor (PF-00835231) for the treatment of SARS-CoV, using structure based drug design (15). Due to the SARS pandemic being brought under control in July 2003 following public health measures which incorporated patient isolation and travel restrictions, this project was discontinued. Given that the SARS-CoV and SARS-CoV-2 3CLpro sequences share 96% identity overall and 100% identity in the active site (1, 16, 17), following the emergence of SARS-CoV-2, PF-00835231 was identified as a potential SARS-CoV-2 3CLpro inhibitor for the treatment of COVID-19 disease.
Herein we describe the 3CLpro inhibitor, PF-00835231, and its novel phosphate prodrug, PF-07304814, and present broad-spectrum antiviral activity along with absorption, distribution, metabolism, excretion (ADME) and safety data highlighting its potential for the intravenous (IV) treatment of COVID-19 disease.
Results and Discussion
PF-00835231 exhibits tight and specific binding to SARS-CoV-2 3CL in vitro
Previously, it was shown that PF-00835231 potently inhibited SARS-CoV-2 3CLpro with a Ki value of 0.27±0.1nM (15). An X-ray co-crystal structure of PF-00835231 bound to SARS-CoV-2 3CLpro is consistent with PF-00835231 binding to the 3CLpro enzyme with a covalent and reversible interaction at the catalytic cysteine residue of the active site, thus inhibiting the activity of the 3CLpro (PDB-6XHM) (15). A thermal-shift assay was used to evaluate the direct binding between PF-00835231 and SARS-CoV-2 3CLpro. The melting temperature of SARS-CoV-2 3CLpro was shifted by 14.6°C upon binding of PF-00835231, from 55.9±0.11°C (n=16) to 70.5±0.12°C (n=8). These data support tight and specific binding of PF-00835231 to SARS-CoV-2 3CLpro (Fig. 1) and, thereby, provide further evidence for the molecular inhibitory mechanism of PF-00835231.

X-ray structures of SARS CoV-2 3CLpro apoenzyme (left) and SARS CoV-2 3CLpro in complex with PF-00835231 (right).
PF-00835231 has potent and broad-spectrum inhibitory activity against a panel of coronavirus 3CLpros
Since the active sites of 3CLpro are fairly well conserved across different coronaviruses, it was hypothesized that PF-00835231 could be active against other coronaviruses besides SARS-CoV (15, 18). Consistent with this hypothesis, it was shown that PF-00835231 was also active against 3CLpros from SARS-CoV-2 and HCoV-229E (15). To further explore the notion that PF-00835231 could have pan-coronavirus activity, PF-00835231 was evaluated against 3CLpro from a variety of other coronaviruses representing alpha, beta and gamma groups of Coronaviridae, using biochemical Förster Resonance Energy Transfer (FRET) protease activity assays. PF-00835231 demonstrated potent inhibitory activity against all tested coronavirus 3CLpro including members of alpha-coronaviruses (NL63-CoV, PEDV, FIPV), beta-coronaviruses (HKU4-CoV, HKU5-CoV, HKU9-CoV, MHV-CoV, OC43-CoV, HKU1-CoV), and gamma-coronavirus (IBV-CoV), with Ki values, ranging from 30 pM to 4 nM (Table 1). The demonstrated activity is consistent with a potential therapeutic use against emerging coronaviruses. This inhibitory activity is restricted to coronavirus 3CLpros as PF-00835231 was inactive against a panel of human proteases and HIV protease (Table S1). PF-00835231 showed detectable activity against human cathepsin B but 1000-fold weaker affinity compared to 3CLpro (Table S1). Thereby, these data collectively support PF-00835231 is a selective in vitro protease inhibitor with broad coronavirus activity.
Activity of PF-00835231 against 3CLpro of coronaviruses
In vitro cellular antiviral activity of PF-00835231 against SARS-CoV-2
The antiviral activity of PF-00835231 against SARS-CoV-2 in cell culture was evaluated with a cytopathic effect (CPE) assay using either VeroE6 cells enriched for ACE2 (VeroE6-enACE2) receptor or VeroE6 cells constitutively expressing EGFP (VeroE6-EGFP). These cell lines were infected with the SARS-CoV-2 Washington strain 1 (WA1 - EPI_ISL_404895) or the BetaCoV GHB-03021/2020 strain (GHB-03021 - EPI_ISL_407976) (19), respectively, which have identical 3CLpro amino acid sequences. PF-00835231 exhibited viral CPE EC50 values of 39.7µM and 88.9µM, respectively (EC50, Fig. 2). However, Vero cells express high levels of the efflux transporter P-glycoprotein (P-gp) (also known as MDR1 or ABCB1) (20), of which PF-00835231 is a known substrate (15) suggesting that the intracellular concentration of PF-00835231 was lower than the concentration added. Therefore, the assays were repeated in the presence of a P-gp efflux inhibitor, CP-100356 (21). PF-00835231 exhibited a 117-to 173-fold increase in activity in the presence of 2 µM P-gp inhibitor, with EC50 values of 0.23µM in VeroE6–enACE2 cells and 0.76µM in the VeroE6-EGFP cells (Fig. 2). The P-gp inhibitor alone had no antiviral or cytotoxic activity at these concentrations and did not cause cytotoxicity in the presence the protease inhibitor. Consistent with many viral protease inhibitors (22), there was a steep response to increasing doses of PF-00835231, with a ∼2-3 fold difference between EC50 and EC90 in both cell types (EC90 = 0.48µM in VeroE6-enACE2 cells and EC90= 1.6µM in VeroE6-EGFP cells in the presence of the P-gp inhibitor). When lung cell lines were tested for antiviral potency in the presence and absence of P-gp inhibitor (A549-ACE2 (23) and MRC5), no statistical difference in antiviral potency was observed (Fig. 2A). Additionally, antiviral activities in both VeroE6 cell lines with 2µM P-gp inhibitor are similar to those observed in more physiologically relevant human cell culture systems, including A549-ACE2 and polarized human airway epithelial cells (23), where P-gp expression is lower. These data support the potential for single agent antiviral activity.

(A) In vitro antiviral activity, and cytotoxicity for PF-00835231 and PF-07304814 with and without the P-gp efflux inhibitor, CP-100356. (B) EC50 values with PF-00835231 with increasing P-gp inhibitor in human lung and monkey kidney cell lines. A549-ACE2 human lung carcinoma data (red) as reported in (23).
Potential for antiviral combination benefit of PF-00835231 in combination with remdesivir
Combinations of antiviral agents, especially those targeting different steps in the virus replication cycle, are a frequently employed therapeutic strategy in treating viral diseases (24). As PF-00835231 and remdesivir, a nucleoside RNA-dependent RNA polymerase inhibitor, target different steps in the virus replication cycle, the antiviral activity of the two compounds was evaluated alone and in combination using HeLa-ACE2 cells (25). Viral proteins were detected in this assay using convalescent human polyclonal sera from two different COVID-19 patients. PF-00835231 alone inhibited SARS-CoV-2 replication with an average EC50 of 0.14µM and EC90 of 0.40µM; whereas remdesivir had an average EC50 of 0.074µM and EC90 of 0.17µM (Fig. 3A). Combination studies were performed using a drug testing matrix, and the data for the drug combination were analyzed using reference models (Loewe, Bliss, HSA) to classify the effects of the drug combination as either additive, synergistic or antagonistic (isobologram, synergy scores, and combination indices).

(A) (Top) Representative antiviral dose response curves of PF-00835231 in combination with remdesivir against SARS-CoV-2. Serial dilutions of PF-00835231 with a range of fixed concentration of remdesivir. (Bottom) In vitro absolute antiviral activity shift in 50% and 90% antiviral activity with fixed concentrations of remdesivir. (B) (Top) A representative 3-dimensional drug interaction landscape plotting synergy scores analyzed using Synergyfinder (median scores of 3 replicates). (Bottom) Average in vitro combination synergy scores from the 3 experiments using 2 different patients’ sera (shown separately).
As summarized in Fig. 3B, the combination of PF-00835231 and remdesivir exhibited synergy from patient #1 sera in 2 independent experiments and additivity in a single experiment with sera from patient #2 (Fig. 3B). The different classification is most likely due to the different convalescent serum used as detection reagents. These same antiviral data were also analysed using Synergyfinder, which also indicated that the 2 drugs were additive to synergistic, with a representative graph shown in Fig. 3B. Antagonism was not demonstrated for the combination of PF-00835231 and remdesivir in these studies. The observed additivity/synergy was not due to cytotoxicity, as there was no noticeable cytotoxicity in virus infected host cells for all the combinations tested. This additivity/synergy is similar to other protease inhibitors used for the treatment of HCV which has led to substantial clinical benefit (26).
Favorable preclinical ADME and pharmacokinetic profile of PF-00835231
The metabolic stability of PF-00835231 was evaluated in vitro using pooled human liver microsomes (HLM). PF-00835231 was shown to be metabolized by cytochrome P450 enzymes exhibiting an unbound CLint 14µl/min/mg. Using recombinant heterologously expressed enzymes and HLM with the CYP3A selective inhibitor ketoconazole, CYP3A4 was identified as the major CYP involved in the metabolism of PF-00835231. It was also noted that the polymorphically expressed CYP3A5 can also metabolize PF-00835231 and that clearance may be slightly greater in CYP3A5 expressers. The potential for PF-00835231 to reversibly inhibit human cytochrome P450 enzymes (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A) was evaluated using probe substrates in pooled HLM and provided IC50 values >200 µM and a weak signal for time dependent inhibition (TDI) of CYP3A4/5. These data indicate PF-00835231 provides a low risk of causing drug-drug interactions (DDIs) on coadministration with other drugs. The potential for PF-00835231 to inhibit a range of transporters (BCRP, P-gp, OATP1B1/1B3, OCT1/2, OAT1/3 and MATE1/2K) was evaluated using in vitro systems. The IC50 values were >20µM and indicate a low risk of causing DDIs due to transporter inhibition at the projected clinical exposure. The plasma protein binding of PF-00835231 was measured across species using equilibrium dialysis and demonstrated moderate binding to plasma proteins with plasma free fractions of 0.33 to 0.45 across species.
PF-00835231 was administered IV to rats, dogs and monkeys (1 or 2mg/kg) and exhibited moderate plasma clearances (50-80% liver blood flow), low volumes of distribution (<1.5 L/kg) and short half-lives (<2 hours) across species in keeping with its lipophilic (LogD7.4=1.7) and neutral physicochemistry. Following oral administration to rats (2 mg/kg) and monkeys (5 mg/kg) PF-00835231 exhibited low oral bioavailability (<2%), likely due to a combination of low absorption because of its low permeability (apparent MDCK-LE permeability of 1.3×10− 6cm/sec(27, 28), low solubility, potential for active efflux in the gut by P-gp and BCRP, and the potential for amide hydrolysis by digestive enzymes in the gastrointestinal tract. In rat, dog and monkey approximately 10% or less of PF-00835231 was eliminated unchanged in the urine indicating renal elimination may also play a minor role in the clearance of PF-00835231 in humans. Additional data presented in the supplementary data (Tables S1-S7).
Human pharmacokinetic predictions suitable for IV administration
Taking into account the human in vitro metabolism data and in vivo pharmacokinetic (PK) data in rats, dogs and monkeys, PF-00835231 was predicted to exhibit a plasma clearance (CLp) of ∼6mL/min/kg (major CYP, minor renal pathways), steady state volume of distribution (Vdss) of 1L/kg and half-life of approximately 2 hours in humans. Due to the limited oral bioavailability, short elimination half-life, and the likely need to maintain free systemic concentrations over time to achieve antiviral activity, a continuous IV infusion was proposed as the optimal dosing route and regimen.
Efficacious target concentration and feasible human dose projection
The projected minimally efficacious concentration (Ceff) was chosen to match the in vitro EC90 (please see supplemental methods for rationale), consistent with the preclinical to clinical translation of approved protease inhibitors (29). Since PF-00835231 was proposed to be administered by continuous infusion, the projected steady-state exposure is equal to the Cmin maintained over the dosing interval. The dose response assay performed in the most physiologically relevant cell type, human lung carcinoma, resulted in an average EC90 value of 0.44µM (23). This is consistent with additional antiviral data in Hela-ACE2 cells (EC90=0.4µM) and Vero cell lines (EC90= 0.48-1.6µM) when a P-gp inhibitor was added to better reflect the lack of substantial P-gp transporter in the lung (Fig. 2B). Furthermore, the antiviral inhibition is supported by the antiviral time course experiment performed in a primary human airway epithelial model (preliminary data indicates an unbound EC90 <0.5 µM) (23), indicating a consistent intrinsic anti-SARS-CoV-2 activity of PF-00835231 across different cell types. Therefore, the proposed minimal Ceff is ∼0.5µM.
Due to the rapid blood perfusion through the lungs and the continuous steady state intravenous infusion regimen, the free plasma and free lung concentrations are assumed to be in equilibrium and, therefore, the free plasma concentration provides a reasonable surrogate for the concentration at the main site of action of the disease. Based on the human PK predictions, the minimally efficacious dose of PF-00835231 necessary to achieve this exposure is ∼320 mg/day administered as an intravenous continuous infusion. The required duration of dosing for efficacy remains uncertain and will need to be evaluated in humans. Based on clinical results from remdesivir a duration of up to 10 days of dosing may be required to provide improved patient outcomes (5).
Formulation and solubility profile of PF-00835231 to enable IV administration
PF-00835231 is a moderately lipophilic (LogD7.4 = 1.7), neutral molecule with no ionizable centers throughout the physiologically relevant pH range. Consequently, PF-00835231 exhibits a pH independent solubility with an intrinsic aqueous solubility of less than 0.1mg/mL and limited opportunities for solubility-enabling formulation approaches. Preliminary work using standard solubilizing excipients indicated that achieving a solubility >0.5mg/mL would likely be challenging.
Based on a maximum desired intravenous infusion volume of ∼1L per day a solubility of 0.5mg/mL would be sufficient to deliver the minimal efficacious dose estimate of ∼320 mg/day to maintain a ∼0.5µM steady state unbound concentration (Fig. 4B). Due to the nascent understanding of the virus and the current lack of in vivo data to aid clinical translation, the required target levels of inhibition for clinical benefit remain uncertain and the ability to evaluate exposures up to ∼10x Ceff in early clinical development is desirable. As a potential option to increase exposures, and/or decrease the required infusion volume, the use of a strong CYP3A inhibitor (itraconazole 200mg QD for 15 days) was considered but preliminary, physiologically-based pharmacokinetic (PBPK) modeling predicted only a ∼2-fold increase in PF-00835231 exposure at steady state (supplemental Table S10).

(A) Chemical structure of conversion of prodrug PF-07304814 to the active moiety and PF-00835231 by alkaline phosphatase. (B) Dose feasibility matrix illustrating the ability to achieve higher target exposures with increasing solubility and the limitations of dosing PF-00835231. with dosing either aqueous PF-00835231, clinically formulated PF-00835231, or aqueous PF-07304814 (prodrug). The infeasible limit (red) is assumed to be 1 L per day with a 2x potential benefit with a Cyp inhibitor (orange). Any dose under that is considered feasible (green).
The ability to achieve higher doses could also potentially mitigate a higher than predicted clearance, or variations in patient body weight. Therefore, a medicinal chemistry strategy to significantly enhance the aqueous solubility of PF-00835231, by designing a phosphate prodrug was pursued.
Considering an intravenous phosphate prodrug approach to improve solubility
IV phosphate prodrugs have precedence with several commercially available drugs such as fosfluconazole and fosphenytoin which are rapidly cleaved by human alkaline phosphatase to provide high systemic exposures of their respective active moieties following IV administration (Fig. 4) (30, 31). Alkaline phosphatase is ubiquitously expressed across tissues with high levels expressed in the liver, lung, and kidney (ALPL tissue data from v19.proteinatlas.org (32)). High levels of conversion from prodrug to active moiety for fosfluconazole and fosphenytoin have also been observed in rats and dogs supporting cross species translation to human for the conversion of prodrug to active moiety (31, 33). Overall, the use of a phosphate prodrug is an established approach for IV administration to provide rapid conversion to its active moiety and was considered for PF-00835231.
Synthetic route to provide phosphate prodrug
The synthesis of PF-00835231 has been described previously (15). The subsequent synthesis of the phosphate prodrug of PF-00835231 was achieved via two steps (Fig. S1). Briefly, treatment of 1 (PF-00835231) with di-tert-butyl N,N-dipropan-2-ylphosphoramidite and tetrazole in tetrahydrofuran followed by oxidation with aqueous hydrogen peroxide delivered intermediate 2. The phosphate t-butyl groups were subsequently hydrolyzed using trifluoroacetic acid in dichloromethane to deliver phosphate prodrug 3 (PF-07304814) as a solid.
Enhanced formulation and solubility profile of PF-07304814 to provide clinical flexibility
PF-07304814 rapidly undergoes in vivo conversion to the active metabolite PF-00835231 (Fig. 3A). The phosphate prodrug is weakly acidic, with pKas of 1 and 6.4, and a predicted LogD7.4 of −3.7. At pHs above the compound’s first pKa, the phosphate functional group is de-protonated and negatively charged, which enables a significant improvement in aqueous solubility to greater than 200mg/mL over a pH range compatible with intravenous infusion. The higher intrinsic solubility of PF-07304814 eliminates the need for solubility-enabling formulations and enables the use of standard IV compatible excipients. Furthermore, the improved solubility enables higher doses to be explored in the clinic and gives clinicians greater flexibility in terms of dose volume to account for patient-specific co-administration requirements.
PF-07304814 (prodrug) preclinical in vitro and in vivo ADME profile
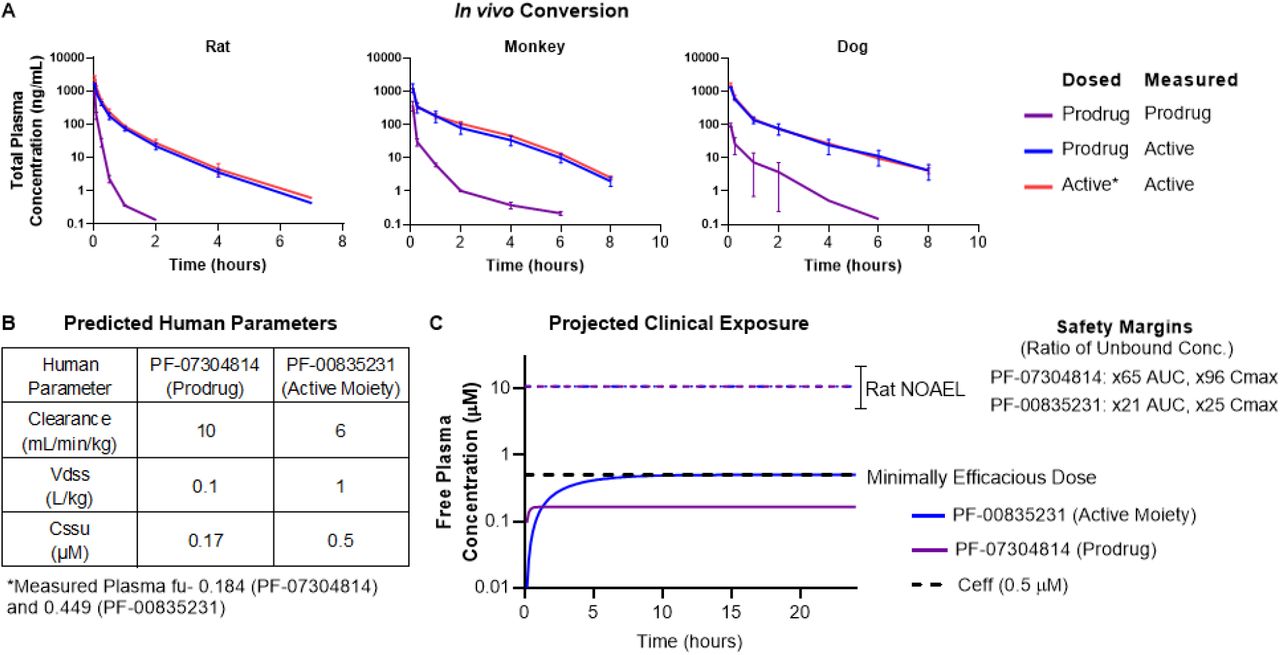
To understand the metabolic stability and conversion of PF-07304814 to its active moiety (PF-00835231), PF-07304814 enzyme kinetics were evaluated in vitro using liver S9 fractions and was shown to exhibit rapid conversion to PF-00835231 with unbound CLint values of 51, 84, 168 and 428µl/min/mg in rat, dog, monkey and human respectively. In these in vitro systems, PF-00835231 was the only metabolite formed from PF-07304814. Conversion was rapid in phosphate-free incubations but abolished in the presence of phosphate buffer supporting the role of alkaline phosphatase in this conversion (Table S8, Fig. S2). To evaluate the in vivo conversion and systemic availability of the active moiety PF-00835231, PF-07304814 was administered intravenously to rats, dogs and monkeys. PF-07304814 exhibited high systemic clearance and short half-life across species forming 68, 81, 76% PF-00835231 in rats, dogs and monkey respectively in comparison to the systemic exposure achieved with IV administration of PF-00835231 (Fig. 5A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Rat, dog and monkey PK following administration of PF-07304814 or PF-00835231 demonstrating high levels of PF-00835231 formed following administration of PF-07304814. (B) Predicted human PK parameters and measured protein binding for PF-07304814 and PF-00835231 used for human dose prediction. (C) Projected human systemic exposure profiles at the minimally efficacious dose of 500mg/day of PF-07304814 delivered as a continuous IV infusion. The predicted unbound steady state concentrations for the prodrug PF-07304814 (purple) and the active moiety PF-00835231 (blue) are 0.17µM and 0.5µM respectively.
PF-07304814 was also evaluated for the potential to cause reversible and time dependent inhibition of human cytochrome P450 enzymes using pooled HLM and probe substrates for a range of CYP enzymes (CYP1A2, 2C8, 2C9, 2C19, 2D6, and 3A4/5) and showed low risk with IC50 values >100µM and no evidence of TDI. The potential for PF-07304814 to inhibit a range of transporters (BCRP, P-gp, OATP1B1/1B3, OCT1/2, OAT1/3 and MATE1/2K) was evaluated using in vitro systems providing IC50 values >130µM indicating a low risk of causing DDIs due to transporter inhibition at the projected Ceff. The plasma protein binding of PF-07304814 was measured across species using equilibrium dialysis showing moderate binding to plasma proteins with plasma free fractions of 0.18 to 0.38 across species. Additional data presented in the supplementary data (Tables S3, S5-S7).
Encouraging human PK predictions for PF-00835231 formation
The predicted human plasma clearance of PF-07304814 is ∼10mL/min/kg based on scaling in vitro human liver S9 CLint data (using equations 9 and 10, see Methods) and represents a conservative prediction of total Clp as it only accounts for conversion of prodrug to active moiety in the liver. The human Vdss for PF-07304814 is predicted to be ∼0.1L/kg based on its acidic physiochemistry and observed human Vdss values of other phosphate prodrugs (30, 31). Based on the predicted Clp, Vdss, and a ∼75% conversion to PF-00835231 based on the mean conversion in animals, PF-07304814 is anticipated to exhibit a short half-life of ∼0.1hour with high conversion to the active moiety (Fig. 5B).
PF-07304814 unlikely to contribute to antiviral activity in vivo
In a direct comparison, using the same SARS-Cov-2 3CLpro assay method as described in (15), the prodrug PF-07304814 binds and inhibits SARS-CoV-2 3CLpro activity with a Ki of 174nM providing a >600-fold higher Ki in comparison to the active moiety PF-00835231 (0.27nM) (15). However, PF-07304814 shows similar antiviral activity to PF-00835231 (1-12-fold, Fig. 2A) across cellular in vitro assays, most likely due to the partial conversion of PF-07304814 to PF-00835231 in the cellular assays by alkaline phosphatase. This was consistent with PF-00835231 concentrations measured at approximately 50% of the PF-07304814 starting concentration at the end of the 3-day incubation in the VeroE6 cell assay.
PF-07304814 dose projection provides clinical flexibility to achieve target Ceff
The antiviral activity for PF-00835231 was used to derive the minimal Ceff and dose estimates. Based on the predicted human PK and 75% conversion of the prodrug, a free plasma concentration of the active moiety PF-00835231 of 0.5µM (Ceff) can be achieved with a 500 mg continuous IV infusion of the prodrug over 24 hours (Fig. 5C). The estimated time to achieve 90% steady state exposure of PF-00835231 is approximately 6 hours. Due to the improved solubility (>200mg/mL), the dose of PF-0730814 can be delivered in a volume of less than 0.25L. In addition the dose can be increased if the observed human plasma Cl exceeds 6mL/min/kg, if the percent converted from prodrug to active is less than predicted, or if exposures in excess of the minimal Ceff (0.5µM) are required to maximize clinical activity (Fig. 4B). Overall the improved solubility would theoretically enable >100-fold the proposed minimal Ceff dose in a 0.25L dose volume.
Preclinical safety profile supports progression to clinical evaluation
A toxicology assessment consisting of an in vitro battery of genetic toxicity, secondary and safety pharmacology studies, in conjunction with a single species (rat) in vivo Good Laboratory Practice (GLP) study has been completed. Data from repeat dose toxicology studies in rodent and non-rodent are planned or currently underway.
The safety profiles of PF-07304814 and PF-00835231 were assessed individually in a range of in vitro and in vivo safety studies in rats. In the in vitro studies, PF-07304814 and PF-00835231 were negative in the bacterial reverse mutation assay and did not induce micronuclei formation. Both compounds had minimal potential for secondary (off-target) pharmacology at clinically relevant exposures. Neither PF-07304814 nor PF-00835231 inhibited hERG current amplitude at up to 300µM (1,770- and 600-fold, respectively, in reference to the projected unbound human Cmax of 0.17 and 0.50µM, respectively, at the projected human efficacious dose), indicating a favorable cardiovascular safety profile. In human blood hemocompatibility assays, both compounds had no effect on hemolysis or flocculation/turbidity parameters, indicating compatibility with human blood and supporting intravenous administration.
PF-07304814 was administered to rats via continuous IV infusion for 24 hours in a GLP study. There were no test article related findings and no target organ toxicity was identified. PF-07304814 had no effects on neurological safety pharmacology parameters as assessed by functional observation battery in the 24-hour continuous IV infusion rat study. The no observed adverse effect level (NOAEL) was 1000mg/kg. PF-00835231 was also administered to male rats via continuous IV infusion for 4 days in a non-GLP exploratory toxicity study and was tolerated at 246mg/kg/day, the highest feasible dose tested. PF-00835231-related findings in this study were limited to minimal, non-adverse effects on clinical chemistry parameters including higher mean triglycerides (1.9-3.6x vs controls), cholesterol (1.3x), and phosphorus (1.1x) without any microscopic correlates or associated functional changes. No test article related adverse effects were seen in any study.
At the NOAEL from the 24-hour, GLP continuous IV infusion study with PF-07304814 in rats, the anticipated exposure margins for unbound Cmax and AUC24 are 97 and 65-fold for PF-07304814 and 25 and 21-fold for PF-00835231, at the projected minimum human efficacious dose of 500mg/day. This indicates the potential to safely evaluate multiples over EC90 in humans during clinical testing to understand the exposure response relationship and to achieve high levels of inhibition, if required. Furthermore, no overlapping or additive toxicity with medications currently being used in standard of care COVID-19 treatment is expected with administration of PF-07304814 in humans, making PF-07304814 an attractive combination partner. Based on results from the set of safety studies conducted, PF-07304814 exhibited an encouraging nonclinical safety profile and supported progression into Phase 1 clinical studies.
Conclusions
PF-07304814 is a phosphate prodrug that is rapidly converted in vivo to the active moiety, PF-00835231, which exhibits high selectivity over human proteases and acts as a broad-spectrum coronavirus 3CL protease inhibitor. Robust antiviral activity was demonstrated in a range of cellular in vitro assays in keeping with SARS-COV-2 human airway epithelial data (23) suggesting a Ceff value of ∼0.5µM. The predicted human pharmacokinetics of PF-07304814 provide the ability to achieve systemic unbound concentrations of 0.5µM (EC90) of PF-00835231 by delivering 500mg as a continuous infusion over 24 hours with infusion volumes of less than 0.25L. In addition, higher doses (up to and beyond 10x Ceff) also remain feasible if needed due to the high solubility of PF-07304814.
Overall, PF-07304814 exhibits an encouraging preclinical profile that has the SARS-CoV-2 antiviral activity, ADME, and safety profile that supports progression to the clinic as a potential novel COVID-19 single agent antiviral treatment, with potential for further additional benefit in combination with antivirals that target other critical stages of the coronavirus life cycle. The favorable profile of PF-07304814 warrants clinical evaluation.
Funding
A.D.M acknowledges partial support for this project from federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201700060C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The study performed in Dr. Jun Wang’s laboratory is partially supported by NIH grant (AI147325) and the Young Investigator Award grant from the Arizona Biomedical Research Centre (ADHS18-198859). Scripps work was supported by a grant from the Bill & Melinda Gates Foundation #OPP1107194, and the Scripps Family Impact Fund of the Miramar Charitable Foundation (MCF)
Author contributions
A.D.M, E.L and B.A contributed to conceptualization, investigation, analysis, visualization and data curation of inhibition of protease activity against a panel of coronavirus 3CLpro experiments. A.D.M contributed to funding acquisition, supervision, project administration and resources of these experiments. J.W and C.M contributed to conceptualization, investigation, analysis, visualization and data curation of the thermal shift experiments. JW contributed to funding acquisition, supervision, project administration and resources of these experiments. M.A.B, T.F.G supervised, designed and carried out antiviral synergy infection experiments. N.B, M.G.K and E.C contributed to the generation of in vitro antiviral synergy data J.B, J.H, Y.Z, L.M.A, L.L,S.N, R.O, C.S, R.K, R.H and B.B contributed to the analysis, interpretation of protease and antiviral data from collaborators and internal data. E.K designed and interpreted the ADME transporter experiments, R.S.O, H.E, R.M.J, E.P.K contributed to the metabolism, pharmacokinetics and bioanalysis of PF-07304814 and PF-00835231. J.R.L, S.A.L, M.N.O., and M.T. designed and interpreted formulation experiments and characterization of API properties. M.R.R, M.P, K.O, R.H and D.O designed and synthesized PF-07304814. R.M.J, B.B, R.S.O and E.K contributed to the conceptualization, analysis and calculations for the prediction of human PK and dose estimate for PF-07304814. J.G.S, L.W.U, R.M.J contributed to the design, supervision and interpretation of in vitro and in vivo safety study data. C.A, A.A, M.I.R contributed to the scientific discussion, experimental design, data interpretation in addition to manuscript review and editing. All authors contributed to writing drafts of the manuscript.
Competing interests
A.D.M has a sponsored program contract with Pfizer to test compounds for inhibition of coronavirus proteases. JW has a sponsored research agreement with Pfizer to test compounds for inhibition of coronavirus proteases.
Data and materials availability
All data are available in the main text or the supplementary materials.
Acknowledgments
The authors would like to thank Sarah Lazzaro, Sumathy Mathialagan, Sangwoo Ryu, Mark West and Emi Yamaguchi (Pfizer) for the transporter inhibition studies. Angela Doran, Chad Limanni, Amanda Plante and Jocelyn Rosado for their in vivo and PK study support (Pfizer). Marcus Ewing (Pfizer) and Gail Johnson (Pfizer) for preformulation studies. Li Hao (Pfizer) for sequence analysis support. Shinji Yamazaki (Pfizer) for PBPK modeling simulations. Daniel Lettiere, Michael Homiski, Michelle Kenyon, Asser Bassyouni, Declan Flynn, William Reagan, Victoria Markiewicz and Stephen Jenkinson for overseeing safety studies, and William Reagan, Shirai Norimitsu for expert clinical pathology and pathology support for the toxicology studies. Stephen Mason (Pfizer) for critical review and editorial input. Deli Huang for supplying the HeLa-ACE2 stably transfected cell line.
Footnotes
One small typo in the abstract. The compound ID number should be PF-00835231 and in the abstract the number is PF-00835321 in one place. This has been corrected in the abstract and in the manuscript.
References and Notes




