

Abstract
Mechanical stimuli such as stretch and resistance training are essential to regulate growth and function of skeletal muscle. However, the molecular mechanisms involved in sensing mechanical stress remain unclear. Here, the purpose of this study was to investigate the role of the mechanosensitive ion channel Piezo1 during myogenic progression. Muscle satellite cell-derived myoblasts and myotubes were modified with stretch, siRNA knockdown and agonist-induced activation of Piezo1. Direct manipulation of Piezo1 modulates terminal myogenic progression. Piezo1 knockdown suppressed myoblast fusion during myotube formation and maturation. This was accompanied by downregulation of the fusogenic protein Myomaker. Piezo1 knockdown also lowered Ca2+ influx in response to stretch. Conversely Piezo1 activation stimulated fusion and increased Ca2+ influx in response to stretch. These evidences indicate that Piezo1 is essential for myotube formation and maturation, which may have implications for msucular dystrophy prevention through its role as a mechanosensitive Ca2+ channel.
Introduction
Skeletal muscle is a highly specialised tissue composed of multi-nucleated, post-mitotic muscle fibres. Since myonuclei within a muscle fibre do not divide after development, the production of new myonuclei is entrusted to satellite cells (SCs), the skeletal muscle’s resident stem cell. SCs are found on the surface/periphery of postnatal skeletal muscle fibres [1–3] and in response to muscle damage, rapidly activate to generate a myoblast progeny that proliferate, undergo myogenic differentiation, and fuse to repair damaged myofibres, resulting in regeneration of a functional muscle [4]. Although much is known about the myogenic progression program, some of the underlying mechanisms remain to be determined. Among these are the molecular mechanisms involved in sensing mechanical stress (mechanosensation) and its effect on myocyte fusion at the cellular and molecular levels.
Mechanosensitive (MS) ion channels are pore-forming membrane proteins which gate in response to mechanical stimuli applied on the cell membrane [5–7]. MS ion channels have been linked to many physiological processes associated with mechanosensory transduction; including osmoregulation, proprioception, hearing, touch, blood flow regulation to name but a few examples [8–10]. Piezo1 and Piezo2 were first identified by Coste et al, (2010) as the long-sought principal types of molecular force sensors (mechanosensors) in mammalian cells [5]. Piezo1 (and Piezo2) is a very large protein containing ∼2500 amino acids with each subunit (a total of three subunits per channel) containing an estimated 24-40 transmembrane (TM) segments [6,8,11,12]. Characterisation of ionic selectivity revealed that Piezo1 was nonselective, permeating Na+, K+, Ca2+ and Mg2+ with a preference for Ca2+ [5, 13]. With such a crucial role Ca2+ regulation plays in skeletal muscle maintenance and repair, understanding Piezo1’s function may prove vital when looking at strategies for therapeutic interventions of muscular dystrophies [14, 15].
Piezo1 is widely expressed in a range of tissues, including skeletal muscle, the bladder, colon, lung, skin and stomach [5, 16]. Piezo2 shows a similar pattern with the exception of being highly expressed in the dorsal root ganglion (DRG) while being less represented in skeletal muscle [5, 16]. Another key difference between Piezo1 and Piezo2 is their activation and inactivation kinetics. Overexpression analyses revealed that the kinetics of inactivation of Piezo2-dependent mechano-activated currents was faster than Piezo1 both for inward and outward currents, thus conferring distinct channel properties [5,13,11]. Although Tsuchiya et al published the first report on Piezo1 in skeletal muscle cells [17], looking at the role of transmembrane localisation of phosphatidylserine and Piezo1 activation, the physiological function(s) of Piezo1 throughout the myogenic program remain unelucidated.
The current study aimed to analyse the role of Piezo1 in skeletal muscle proliferation, differentiation and its role in stretch-induced Ca2+ influx of primary derived myotubes. Our findings revealed that Piezo1 is dispensable for myoblast proliferation and onset of differentiation, but its knockdown suppressed myotube formation and maturation in primary myotubes derived from slow soleus and fast extensor digitorum longus (EDL) muscles in mice. In line, Piezo1 reduction was accompanied by downregulation of the fusogenic gene Myomaker, decreased accumulation of f-actin and lowered Ca2+ influx of myotubes in response to mechanical stretch. In contrast, administration of the Piezo1-specific agonist Yoda1 increased fusion of myoblasts. Piezo1 activation also showed increased Ca2+ influx in response to stretch. Using publicly available datasets, we showed a dysregulation in the expression of Piezo1 in the skeletal muscle disease facioscapulohumeral muscular dystrophy (FSHD). In summary, we propose that Piezo1 plays a crucial role at the terminal stage of myotube fusion and maturation. Piezo1 may employ direct or indirect mechanisms that regulate fusion proteins as well as proteins involved in cytoskeletal organisation. These mechanisms may be regulated by intracellular Ca2+-signals mediated by mechanosensitive Piezo1 channels.
Materials and methods
Primary myoblast cell culture
All experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Bethesda, MD, USA) and were approved by the Animal Use Committee of Toyohashi SOZO University A2018006, A2019006). Male C57BL/6J mice (8-12 weeks of age) were used. All mice were housed in a vivarium room with 12-h-12-h light-dark cycle; with temperature and humidity maintained at ∼23°C and ∼50%, respectively. Solid food and water were provided ad libitum.
After cervical dislocation, the EDL (extensor digitorum longus) and soleus muscles were carefully dissected, and manipulated only by their tendons. Muscles were digested in 0.2% Collagenase Type 1 (Sigma, UK. Ref: SCR103) in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Thermo Fisher Scientific, Ref: 11885084) with 1% penicillin/streptomycin (Pen Strep, Gibco, Thermo Fisher Scientific, Ref: 15140-122) for 2 hours. Individual myofibres were then dissociated by trituration using heat polished glass Pasteur pipettes (Marienfeld, Germany. Ref: 3233049) with variously sized apertures (coated with 5% BSA, Sigma-Aldrich, Ref: A7906-100G) and washed as described by Collins and Zammit (2009)[18]. Isolated myofibres were plated on Matrigel (Corning. Ref: 354234) and the satellite cell-derived myoblasts were then expanded in proliferation medium, consisting of; DMEM, with 30% heat-inactivated foetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific. Ref: 26140-079), 10% horse serum (Gibco, Thermo Fisher Scientific, Ref: 16050-122), 1% chick embryo extract (Sera Laboratories. Ref: CE-650-TL), 10 ng/ml basic FGF (bFGF, Gibco, Thermo Fisher Scientific. Ref: PHG0264) and 1% penicillin. Cells were kept in a 37°C incubator (Panasonic, MCO-230AICUVH) under a humidified atmosphere with 95% air and 5% CO2. Cells designated for proliferation and differentiation conditions were seeded at different densities depending on the size of wells they were cultured in. For 96-well plate proliferation conditions required 5,000 cells per well, and for differentiation, cells were seeded at 10,000 cells per well. For 6-well plates, proliferating and differentiation cohorts consisted of 50,000 cells and 70-80,000 per well, respectively. Differentiation medium was made up of DMEM, 2% heat-inactivated horse serum, and 1% penicillin.
siRNA transfection
Small interfering RNAs (siRNAs) were purchased from (Qiagen, Hiden, Germany) (Table 1) and diluted to 20 or 10 μM in double-distilled water (ddH2O) and stored at −20°C. To investigate the effects Piezo1 knockdown on proliferation, early entry into differentiation and myotube formation, Piezo1-targeting or control scrambled siRNA (siScrambled; Qiagen, Hiden, Germany) was transfected in proliferation medium. Cells were plated on 6-well plates at 50,000 cells per well in proliferation medium. Following a 24 h incubation period, the medium was replaced with 1.75 ml fresh proliferation medium and the transfection mixture was prepared: A solution 150 μl Optimem (Gibco, Thermo Fisher Scientific. Ref: 31985-070) medium with 9 μl of lipofectamine (lipofectamine RNAiMAX Thermo Fisher Scientific. Ref: 13778030) was made for each well. Separately, siRNA was diluted in 150 μl Optimem. The two solutions were then mixed and incubated for 5 min. 250 μl of the siRNA/lipofectamine mixture was added to corresponding wells dropwise. The final siRNA concentration was set at 10 nM. Following overnight incubation in the transfection medium, cells were trypsinised for RT-qPCR analysis and seeded in 96-well plates for proliferation and differentiation cohorts (day 1 and day 3 differentiation) in proliferation and differentiation medium, respectively. After a 24-hour incubation, proliferating cells were subjected to a 2-hour 5-ethynyl-2’-deoxyuridine (EdU) pulse and fixed as below. Day 1 and day 3 differentiating cohorts were also fixed.
To determine Piezo1’s role in myotube formation, siRNA transfection was performed in early differentiated myotubes. Cells were seeded at confluency in differentiation medium. Following 24-hour incubation in differentiation medium, siRNA transfection was performed. Cohorts were designated for RT-qPCR analyses and immunolabelling.
RNA Extraction and Reverse Transcription
RNA was extracted from cells using the RNeasy mini kit as per manufacturer’s requirements (Qiagen. Ref: 217004). Reverse transcription was carried out using PrimeScript RT Master Mix (Takara Bio, Otsu, Japan. Ref: RR036A). Optical density analysis using a Nanodrop ND-1000 spectrophotometer (Labtech, UK) quantified RNA concentration. Samples were then loaded to a PCR thermal cycler (Takara, Dicemini). The resulting cDNA was then diluted 1:9 to obtain a working dilution for RT-qPCR analysis.
Real-time Quantitative PCR (RT-qPCR)
Primers were designed using the Takara Bio Perfect Real Time Support System (Takara Bio, Table 2). Primers were diluted to 50 μM in ddH20 and stored at −20°C. Real-time RT-qPCR was performed on the cDNA (Thermal Cycler Dice Real Time System IIMRQ, Takara Bio) using Takara SYBR Premix Ex Taq II (Takara Bio. Ref: RR802A). 12.5 μl of SYBR Premix Ex were added to each RT-qPCR well. 8.5 μl of ddH20 and 2 μl of the corresponding primers were then added (a final concentration of 2 μM per primer). 2 μl of the respective cDNA was then added to the appropriate wells, bringing the total volume to 25μl per well. The RT-qPCR cycle consisted of 95°C for 30 s (for enzyme activation), followed by 40 cycles at 95°C for 5 s and a qPCR amplification period of 30 s at 60°C. The relative fold change of expression was calculated by the comparative threshold cycle (CT) method using Takara Thermal Cycler Dice Real Time System Software Ver. 4.00 (Takara Bio). To normalise for the amount of total RNA present in each reaction, Gapdh was used as an internal standard.
Piezo1 activation
In order to induce Piezo1 activation, early forming myotubes were subjected to the Piezo1 specific agonist Yoda1 (Cayman Chemical Company. Ref: 21904) diluted in dimethyl sulfoxide (DMSO, Sigma. Ref: D2650-5×5ML). Yoda1 binds the agonist transduction motif (ATM), located at the pore domain of the Piezo1 channel [19]. With each subunit displaying such motif, Yoda1 has potentially three binding sites. This phase of the investigation consisted of two main variables 1) drug concentration and 2) duration of time cells were incubated with the drug. Five concentrations were chosen in order to cover an increasing spectrum of Yoda1 final concentration, these were: 5, 10, 30 and 100 μM diluted in differentiation medium. Preliminary findings from the group found that a 24-hour incubation with any of the concentrations chosen, led to complete abolishment of myotube maturation (data not show). Thus, time-points thought to have potential to maximise myotube fusion/maturation were tested. The incubation time-points were set for 1 and 30 min, 1 and 4 hours. Control cohorts containing only DMSO were incubated at the allocated times to allow comparisons to be made within each condition. Following the incubation of Yoda1 or DMSO, cells were cultured in the differentiation medium for a further 2 days (i.e. myotubes were analysed 3 days post initial induction of differentiation).
Immunolabelling
Throughout the protocol, all washes were performed with Dulbecco’s phosphate-buffered saline (DPBS, Gibco, Dulbecco’s Phosphate Buffered Saline, Thermo Fisher Scientific, Ref: 14190-144). Cells were fixed with 4% paraformaldehyde for 15 min. Samples were then washed three times with PBS (5 minutes each wash) and permeabilised for 15 min using 0.5% triton-X100/PBS (Sigma-Aldrich. Ref: T9284-500ml). Cells were blocked for 1 hour in 5% bovine serum albumin (BSA, Sigma-Aldrich Ref: A7906-100G). Primary antibodies (diluted to the working concentration in PBS) (Table 3) were added to the samples and incubated overnight at 4⁰C. Primary antibodies were decanted, the samples were washed three times and appropriate secondary antibodies diluted to the working concentration in PBS added (Table 3) were added to the samples. The Samples were covered with aluminium foil to avoid light exposure and left to stand at room temperature for 1 hour. Cells were washed again (three times). To visualise nuclei, the cells were incubated for 10 min at room temperature with 1 μg/ml 4’, 6-diamidino-2-phenylindole (DAPI) (Sigma. Ref: D9542-10MG) diluted 1:1000 in PBS. After a final wash with PBS (5 min), cells were replenished with PBS and stored at 4 ⁰C until image analysis.
EdU incorporation
For the evaluation of cell proliferation, cells were incubated with 5-ehtynyl-2’-deoxyuridine (EdU: Invitrogen, Thermo Fisher Scientific) at 10 μM, added in fresh proliferation medium for 2 hours at 37 °C. EdU, the alkyne-containing thymidine analog, is incorporated into DNA during active DNA synthesis. The click-iT EdU Alexa Flour kit (Invitrogen, Thermo Fisher Scientific, Click-iT, EdU Alexa Fluor. Ref: 594 C10339) was used as per manufacturer’s instructions with either the 488 (green) or 594 (red) azide to detect incorporated EdU.
Phalloidin labelling
To evaluate the cytoskeleton, cells were treated with phalloidin (Invitrogen, Thermo Fisher Scientific, Alexa-Fluor. Ref: 488 A12379) diluted 1:40 in PBS. Phalloidin binds to f-actin, a major cytoskeleton protein in skeletal muscle fibres. Cells were incubated with phalloidin solution for 30 mins at room temperature. Cells were then washed with PBS twice for 5 min.
Image quantification
Images were taken using a fluorescence microscope (BZ-X710, KEYENCE, Osaka, Japan). Four to five images per each well (3 wells per repeat) consistent of a total of 12-15 images per repeat were analysed. One repeat refers to one mouse. For EdU incorporation, the total number of DAPI-counterstained nuclei and total number of EdU-incorporated cells were quantified. The proportion of EdU-incorporated cells relative the total number of nuclei was subsequently expressed as percentages. The relative proportion of cells expressing myogenin was also quantified in this manner.
The fusion index was calculated by quantifying the total number of nuclei within MyHC positive myotubes and expressing this value as a proportion of the total number of nuclei in each field of view. As a criterion, more than two nuclei must be within a MyHC positive myotube to be quantified for the fusion index.
To measure myotube width, the “measure” tool on ImageJ imaging software was used. This allows measurements of a chosen distance to be made. Before measurement, a scale was applied to all images. On the “set scale” option pixels are converted into μm. Taking the fluorescence microscope and magnification into account the program determines 100 μm to be 133.00 pixels or 0.75 μm/pixel. Three independent images were chosen per condition. The criterion for this analysis was to choose the widest possible distance between myotube edges without the presence of any branching points. This was carried out in three independent points within the field of view. The values were then averaged.
Stretch experiments and intracellular Ca2+ level imaging
Stretch experiments were performed at the National Institute for Physiological Sciences (NIPS). Primary derived myoblasts were seeded (30,000 cells/chamber) on modified elastic silicone chambers (Strexcell, Ooyodonaka, Ref: STB-CH-0.02). After 24-hour incubation, cells were transfected with either control (siScrambled) or Piezo1-specific siRNA in proliferation medium. After overnight incubation, cells were switched to differentiation medium and cultured for a further 3 days. Stretch experiments were conducted on the third day. For Piezo1 activation by Yoda1 administration, cells were seeded as above. Following the initial overnight incubation, cells were switched to differentiate and the resulting myotubes were analysed 3 days post differentiation induction.
For Ca2+ imaging, Fura 2-AM (Invitrogen) with 10% Pluronic® F-127 (Molecular Probes, USA) diluted in double distilled water (ddH2O), was administered to EDL and soleus-derived myotubes followed by a 30-min incubation time. Chambers were attached to an extension device (modified version of STB-150, Strex) on the microscope stage. Stretch stimulation was applied using a pre-set stretch speed and distance. After an initial 1 min rest period (0% stretch), stretch was applied at 3% (0.3 mm), 6% (0.6 mm) and 9% (0.9 mm) for 1 min followed by a 1-min resting period in between. During the initial 0% stretch timepoint, Yoda1 cohorts were administered with 30 μM of the agonist before being subjected to stretch. Ionomycin (Sigma-Aldrich) at 5 μM was applied at the final step in each experiment for normalisation and to check cell viability.
Changes in intracellular calcium [Ca2+]i were measured by ratiometric imaging with Fura 2-AM at 340 and 380 nm, and the emitted light signal was read at 510 nm. Images were then analysed on ImageJ imaging software. Three independent myotubes from each condition were selected and analysed. The changes in ratios were calculated by subtracting basal values from peak values. The values were then normalised to ionomycin data.
Statistical analysis
Data is presented as mean ± SEM from at least three experiments (at least three mice). Significance was assessed by either paired Student’s t-test or one-way ANOVA followed by followed by the Tukey-Kramer post-hoc; wherein p-values of < 0.05 were considered to be statistically significant. A paired t-test was adopted when comparing effects within the same group e.g. analysing the effects of siRNA mediated down-regulation of Piezo1 versus siRNA controls in murine derived myoblasts. A one-way ANOVA was implemented when two or more independent groups were analysed, for example; comparing the effects of varying agonist concentrations across different timepoints.
Results
Expression of Piezo1 in primary myoblasts and myotubes
We investigated the expression level of Piezo1 during myogenic progression in murine fast EDL and slow soleus muscles-derived primary myoblasts (Figure 1a). The expression of EDL-derived cells (Figure 1b) showed a significant increase in mRNA expression of Piezo1 in myotubes cultured at 3 days in differentiation medium, compared to the expression level in proliferating myoblasts (p<0.05). Soleus-derived cells (Figure 1c) showed a significant increase in Piezo1 mRNA expression after 24 and 72 hours in differentiation medium, compared to proliferation cohorts (p<0.05). The observed increase in Piezo1 in soleus-derived cells after 1 day of differentiation may be explained by the fact that soleus-derived myoblasts tend to differentiate slightly quicker than EDL, i.e. myotubes could be seen sooner in soleus-derived cells compared to EDL. Moreover, we found that Piezo1 is expressed at a higher level in day 1 and day 3 differentiated soleus cells compared to EDL counterparts (Figure S1).

Relative fold changes in expression of Piezo1 in myoblasts from EDL (green bars) and Soleus muscle (blue bars), during proliferation and through differentiation; 24 hours (Day 1) and 72 hours (Day 3) in differentiation medium. Values were normalised to Gapdh and then expressed as fold change compared to levels of proliferation in EDL samples. Data is presented as mean ± SEM from three experiments (n = 3 mice). Asterisks (*) denote significance at p < 0.05 compared to control proliferation conditions. ¶: Significant difference at p < 0.05 significant compared to either day 1 or day 3 differentiation cohorts using one-way ANOVA followed by the Tukey-Kramer post-hoc.

a) Schematic representation of muscle isolation and satellite cell expansion procedure. b) and c) Relative fold changes in expression of Piezo1 in murine EDL and soleus muscle-derived myoblasts, during proliferation and through differentiation; 24 hours (Day 1) and 72 hours (Day 3) in differentiation medium. d) and e) EDL and soleus-derived myoblasts were transfected with 10 nM of either control-siRNA (siScrambled) or targeting siRNA against Piezo1 (Piezo1-siRNA. After overnight incubation, cells were incubated for a further 24 hours and expression of Piezo1 was measured. f-i) Relative fold changes in expression of Piezo1 and Piezo2 in EDL and soleus muscle-derived myotubes. Following an initial differentiation period (24 hours at high confluency), cells were transfected with 10 nM of either control-siRNA (siScrambled) or Piezo1- siRNA. After overnight incubation, cells were incubated for a further 24 hours. Values were normalised to Gapdh and then expressed as fold change compared to either level of proliferation (b and c) or to siScrambled conditions (f-i). Data is presented as mean ± SEM from three experiments (n = 3 mice). Asterisks (*) denote significance at p < 0.05 compared to control conditions (either proliferating myoblasts or siScrambled cohorts), using one-way ANOVA followed by the Tukey-Kramer post-hoc (a and b) or a 2-tailed paired student t-test (f-i).
siRNA-mediated downregulation of Piezo1
We next set out to evaluate the effects of manipulating Piezo1 on proliferation and myogenic differentiation. This was achieved by siRNA-mediated knockdown (Figure 1d and e). Cells were transfected with either control (siScrambled) siRNA (control-siRNA) or targeting siRNA specific for Piezo1 (Piezo1-siRNA). Following a 24-hours incubation period, the medium was changed and cells were incubated for a further 24 hours. In EDL-derived cells (Figure 1d), the treatment of Piezo1-siRNA showed a significant reduction (∼48%) in mRNA expression level of Piezo1, compared to control-siRNA (p<0.05). Analysis of soleus-derived cells (Figure 1e) also revealed a significant reduction (∼34%) in expression of Piezo1 post siRNA-mediated transfection, compared to control conditions (p < 0.05).
Effects of Piezo1 downregulation on Piezo2
Using RT-qPCR analysis, the expression of Piezo2 in Piezo1-downregulated conditions was measured in both EDL and soleus primary cells (Figures 1f-i). Furthermore as Piezo1 seemed to exert most of its expression during differentiation, we evaluated Piezo2 expression in early differentiated myotubes. This encompassed a 24-hour incubation in differentiation medium at high confluence, followed by siRNA transfection. After overnight incubation with siRNA, the medium was exchanged with fresh differentiation medium and incubated for a further 24 hours before sampling. We again confirmed the reliability in our method of siRNA-mediated knockdown by showing significant reduction of Piezo1 expression in both EDL- and soleus-derived cells (Figures 1h and i). Data from the EDL (Figure 1f) showed that in Piezo1-downregulated conditions, the expression of Piezo2 is relatively stable. Soleus-derived cells subjected to Piezo1-siRNA (Figure 1g) showed a slight trending increase in Piezo2 expression, compared to control-siRNA; however this did not reach statistical significance, indicating that Piezo2 did not provide a compensatory role in the down-regulation of Piezo1. Though, we must acknowledge that perhaps if the degree of Piezo1 downregulation was more “potent”, Piezo2 may alter its expression in response. Furthermore a prolonged downregulation of Piezo1 may also affect the expression level of Piezo2.
Piezo1 is dispensable for muscle cell proliferation
We next turned our attention to the effects of Piezo1 downregulation during proliferation. Although we did not see a significant difference in the expression dynamics of Piezo1 early in the myogenic program that is not to say that the knockdown of this channel would not affect proliferating cells. Thus in order to address this question we used EdU incorporation assays (Figures 2a-d). Analyses of the proliferation rate in myoblasts derived from EDL (Figures 2a and b) showed that the downregulation of Piezo1 had no significant effect on EdU incorporation, compared to siRNA control conditions. Similarly, the downregulation of Piezo1 in soleus-derived myoblasts (Figures 2c and d) showed no statistical change in the proliferation rate between Piezo1-siRNA-treated cells and control conditions. From these results, we can summarise that on plated cell conditions, downregulation of Piezo1 does not seem to affect the proliferation capacity of EDL- and soleus-derived myoblasts.

a) and c) Representative images of EDL and soleus primary-derived myoblasts, transfected with 10nM of siRNAcontrol (siScrambled) or siRNA-Piezo1. Following overnight incubation, the medium was changed with fresh proliferation medium and cells were incubated for a further 24 hours, and then subjected to a 2-hour pulse with EdU (red panels). DAPI counterstained nuclei shown in black and white panels. Scale bar is 100 μm. b) and d) Proportion of EdU-incorporated cells relative to total (DAPI) cell count. e) and g) Representative images of EDL and soleus muscle-derived myoblasts transfected with 10nM of control-siRNA (siScrambled) or Piezo1-siRNA. Following overnight incubation, cells were incubated for a further 24 hours, immunolabelled for Myogenin (red panels) and counterstained with DAPI (black and white panels). Scale bar is 100 μm. f) and h) Percentage proportion of Myogenin positive cells relative to total nuclei. Data is represented as percentages. Data is mean ± SEM from three experiments (n = 3 mice). *: significant at p < 0.05 compared to siScrambled conditions using a 2-tailed paired student t-test.
Suppression of Piezo1 does not inhibit the onset of the myogenic program
Our next aim was to investigate whether knockdown of Piezo1 would alter the capacity of cells to enter into the differentiation phase of the myogenic program. siRNA-transfected cohorts were cultured in differentiation medium for 24 hours. This early differentiation phase was analysed by immunolabelling of the myogenic regulatory factor (MRF) Myogenin. Quantification was performed by calculating the proportion of Myogenin-positive myoblasts relative to the number of DAPI counterstained nuclei (Figures 2e-h). Analysis of EDL-derived myoblasts (Figures 2e and 2f) showed no significant difference in the relative proportion of myogenin-positive cells between control-siRNA and Piezo1-siRNA treated conditions Similarly, soleus-derived cells (Figures 2g and 2h) did not show a significant difference in the relative proportion of myogenin-positive cells in samples treated with Piezo1-siRNA, compared to control-siRNA. From these results, we may suggest that the downregulation of Piezo1 does not seem to have any effects on the ability for myoblasts to enter the differentiation phase of the myogenic program. However, the question remained whether the continuous knockdown of this receptor would affect the fusion and maturation of myotubes. To answer this, we turn to myotube analyses.
Piezo1 regulates myocytes fusion
Piezo1-siRNA-treated cells were cultured in differentiation medium for 3 days post transfection. By this timepoint myoblasts fuse to neighbouring differentiating myoblasts and begin to upregulate the sarcomeric motor protein MyHC, which is expressed in the forming myotubes. We can visualise MyHC containing myotubes through immunolabelling. Not only does this allow a visual representation of myotube formation, but it can also be used to quantify the level of fusion that has occurred - the fusion index.
The data derived from the EDL-derived cells (Figure 3a), showed a clear difference between control-siRNA and Piezo1-siRNA conditions, with the latter displaying much fewer myotubes. Indeed, we observed a significant (p<0.05) reduction of fusion index in Piezo1-siRNA conditions compared to control conditions (Figure 3b). These findings were also translated to soleus-derived cells. From the myotube images (Figure 3c), we observed smaller myotubes when Piezo1 was downregulated. This was confirmed by the significant reduction in myocytes fusion (Figure 3d) in Piezo1-siRNA conditions compared to control conditions (p<0.05).

a) and c) Representative images of primary derived myotubes from EDL and soleus muscle, transfected with 10nM of control-siRNA (siScrambled) or Piezo1-siRNA. Following overnight incubation, cells were incubated for a further 72 hours. Cells were immunolabelled for Myosin heavy chain (MyHC) (red panels) and counterstained with DAPI (black and white panels). b) and d) The fusion index was calculated by counting the total number of nuclei within each myofibre and representing this as a percentage relative to the total number nuclei in the image taken. e) and g) Representative images of EDL and soleus muscle-derived. Early forming myotubes were transfected with 10nM of control-siRNA (siScrambled) or Piezo1-siRNA. Following overnight incubation, cells were incubated for a further 72 hours. immunolabelled for Myosin heavy chain (MyHC) (red panels) and counterstained with DAPI (black and white panels). Scale bar is 100 μm. f) and h) Bar graphs display the fusion index. Data is mean ± SEM from three experiments for myotube formation data (n = 3 mice) and four experiments for myotube maturation data (n = 4 mice). *: Significant at p < 0.05 compared to siScrambled conditions using a 2-tailed paired student t-test.
A potential concern of the data in Figures 3a-d, is the fact that cells were first transfected in proliferating conditions (i.e. in proliferation medium) before being switched to differentiate; and although the initial analysis of EdU- and myogenin-positive cells did not reveal a change post siRNA-mediated knockdown of Piezo1, that is not to say that the initial knockdown in proliferating conditions did not alter the behaviour of the channel and the subsequent formation of myotubes. Thus, in order to address this, we carried out the transfection of Piezo1-siRNA in differentiating conditions. This encompassed a 24-hour incubation period in differentiation medium. By this point primary derived myoblasts seeded at high confluency, already begin to show myotube formation in the relatively small 96-well plates. These early formed myotubes were then subjected to Piezo1-siRNA transfection in differentiation medium. Following overnight incubation, the transfection mixture was exchanged with fresh differentiation medium. Myotubes were subsequently analysed 3 days post the initial Piezo1-siRNA transfection (Figures 3e-h).
Downregulation of Piezo1 in EDL-derived myotubes (Figures 3e and 3f), showed a significant reduction (p<0.05) in the fusion index, compared to control-siRNA conditions. The soleus muscle (Figures 3g and 3h) also showed a reduction in the ability of myoblasts to fuse in Piezo1-siRNA-treated conditions, compared to control-siRNA cohorts (p<0.05). In summary, the downregulation of the Piezo1 channel both in forming and maturing myotubes significantly reduces the ability of cells to fuse into new or existing myotubes. This response seems to be unique to this phase of the myogenic program as downregulation of Piezo1 does not affect proliferation or early entry into differentiation.
These thought-provoking results demand further exploration of; 1) the mechanisms of Piezo1-mediated fusion, 2) other fusion regulators, and 3) the downstream signalling pathways of the Piezo1 channel which enables correct myogenic fusion.
Piezo1 and actin remodelling
Previous research places Piezo1 regulation as a key modulator of this cytoskeletal protein [20, 21]. In skeletal muscle actin plays a pivotal role not only in myoblast fusion but myotube alignment to attachment sites [22]. Therefore, a putative mechanism(s) of Piezo1 is the regulation of cytoskeletal structures, including filamentous actin (f-actin). As part of our analyses, we examined f-actin intensity using fluorescently labelled phalloidin as a proxy to evaluate its accumulation, and thus the extent of cytoskeletal reorganisation during myogenic differentiation in EDL or Soleus derived myoblasts. Piezo1-siRNA-treated cells showed a significant decrease in the accumulation of f-actin compared to control-siRNA (Figure S2). However, activation of Piezo1 using Yoda1 did not reveal a statistically significant increase in f-actin intensity during any of the incubation timepoints or concentrations (Figure S3). Further, high concentration and relatively longer incubation time of Yoda1, suggestive of an over-activation of Piezo1, showed a significant decrease in f-actin in EDL (100 μM; 4-hour incubation) and soleus (100 μM; 30-min, 1- and 4-hour incubation) derived myotubes. These findings suggest that cytoskeletal organisation may be affected by Piezo1 activity; however our methods of measuring such changes lacked resolution. For example, f-actin stress fibre formation as well as filopodia extension in response to Piezo1 manipulation should be quantified under higher magnification.

a) and b) Representative images of EDL and soleus muscle--derived myotubes. Early forming myotubes were transfected with 10nM of control-siRNA (siScrambled) or Piezo1-siRNA. Following overnight incubation, cells incubated for a further 72 hours. The cytoskeleton protein f-actin was visualised using fluorescently labelled phalloidin (green panels). Nuclei were counterstained with DAPI (black and white panels). Scale bar is 100 μm. c) and d) Overall fluorescence intensity measured by pixel/area in each field of view (six images per conditions). e) and f) is data from c) and d) expressed as percentages relative to the maximum pixel value. Data is mean ± SEM from three experiments (n = 3 mice). *: Significant at p < 0.05 compared to siScrambled conditions using a 2-tailed paired student t-test.

Early forming myotubes were administered with either DMSO (control, white bars) or Yoda1 at the following concentration: 5 μM (red bars), 10 μM (blue bars), 30 μM (green bars) and 100 μM (grey bars). Myotubes were incubated for 1 min, 10 min, 30 min, 1 hour and 4 hours. Following the incubation period, the medium was exchanged with fresh reduced medium (without agonist) and myotubes were incubated for a further 2 days. b) and d) Representative images at relevant timepoints and concentrations treated with fluorescently labelled phalloidin (green panels and counterstained with DAPI (black and white panels). Images taken at x20 magnification. Scale bar is 100 μm. Bar graphs display proportion of f-actin intensity in a) EDL and b) soleus derived myotubes from each time point and concentration variables. Values are mean ± SEM. * indicates significance at p < 0.05 using one-way ANOVA tests followed by the Tukey-Kramer post-hoc. n = 3 mice.
Lack of Piezo1 affects the expression of fusogenic genes
An ever-increasing topic of interest within the skeletal muscle field is the role fusion proteins play. The recently discovered fusion protein Myomaker and its associated micropeptide Myomixer/Myomerger/Minion (hereafter referred to as Myomixer) are at the centre of myogenic fusion research [23]. Thus, we were curious to examine the effects Piezo1 may have on these fusion proteins. Piezo1 was first downregulated in EDL- and soleusderived myoblasts. Cells were then collected and RT-qPCR against Myomaker and Myomixer was carried out and compared to control-siRNA conditions (Figure 4a-d). EDL-derived cells (Figure 4a) showed that the expression of Myomaker is significantly reduced in Piezo1-siRNA treated cells (∼43%), compared to control-siRNA conditions (p<0.05). In Piezo1-downregulated conditions, Myomixer (Figure 4b) showed a trend to decrease, compared to control-siRNA, (p=0.0505). In soleus-derived cells (Figure 4c), downregulation of Piezo1 significantly lowered the expression of Myomaker (∼30%), compared to control-siRNA (p<0.05). Similar to the EDL, we observed a trend for Myomixer to decrease post Piezo1 knockdown (Figure 4d, p>0.05). Taken together, these results show a potential interplay between the stretch induced Piezo1 channel and the fusion protein gene Myomaker (and perhaps Myomixer). The question remains as to whether the downregulation of Piezo1 actively employs mechanisms which lower Myomaker expression which in turn reduce myogenic fusion or whether this is an indirect effect of another Piezo1 governed event. Answering these queries may have implication in skeletal muscle regeneration and muscular dystrophies.

Relative fold changes in expression of EDL-derived myoblasts (a and b) and soleus-derived myoblasts (c and d). Cells were transfected with 10 nM of either control-siRNA (siScrambled) or Piezo1-siRNA. After overnight incubation, cells incubated for a further 24 hours. The expression of the fusogenic protein genes Myomaker and Myomixer were then analysed. Values were normalised to Gapdh and then expressed as fold change compared to siScrambled conditions. Data is mean ± SEM from three experiments (n = 3 mice). *: Significant at p < 0.05 compared to siScrambled conditions using a 2-tailed paired student t-test.
Ca2+ influx in Piezo1 downregulated myotubes
The Piezo1 channel permeates Ca2+ influx at a greater preference than other cations (Na+, K+ and Mg2+) [5]. Ca2+ is itself a crucial regulatory and signalling molecule of the myogenic program as well as skeletal muscle contraction and various intracellular events. Thus, a logical query to pursue was to see whether selective downregulation of Piezo1 hinders or blunts the influx of Ca2+ in cultured myotubes exposed to stretch. To achieve this we used customised stretch silicon bio-chambers [16]. Myoblasts from both EDL and soleus muscles were transfected with either Piezo1-siRNA or control-siRNA (siScrambled). The transfection mixture was then removed, and samples were cultured in differentiation medium for 3 days. The resulting myotubes were then subjected to incremental bouts of stretch. Throughout the experiment, intracellular Ca2+ level [Ca2+]i was measured using Fura-2AM, a membrane-permeant fluorescent Ca2+ indicator (Figure 5).

a) and b) Representative images of intracellular Ca2+ imaging in EDL and soleus-derived myotubes. Myoblasts were transfected with 10 nM of either control-siRNA (siScrambled) (top panels) or Piezo1-siRNA (bottom panels). After overnight incubation, cells were incubated for a further 72 hours. Fura 2-AM was administered to myotubes followed by a 30-min incubation time. Stretch was then applied at 3% (0.3 mm), 6% (0.6 mm) and 9% (0.9 mm) for 1 min followed by a 1-min resting period in between. Ionomycin at 5 μM was then applied. Side vertical bar shows Fura 2-AM ratio emittance from low to high. c) and d) Average changes in the intracellular Ca2+ level ([Ca2+]i) calculated by difference between base and peak pixel value) normalised to ionomycin and control-siRNA at 0% stretch. Values are mean ± SEM from three experiments (n = 3 mice). *: Significant difference at p < 0.05 compared to control-siRNA at each condition. ¶: Significant difference at p < 0.05 compared to 0% stretch counterparts. †: Significant difference at p < 0.05 compared to 3% stretch counterparts. Ŧ: Significant difference at p < 0.05 compared to 9% stretch in Piezo1-siRNA conditions using one-way ANOVA followed by the Tukey-Kramer post-hoc.
Focusing first on control-siRNA, we found that [Ca2+]i was increased at certain stretch conditions. In EDL-derived myotubes for example (Figures 5a and 5c), a 6% stretch significantly increased [Ca2+]i influx compared to 0% stretch controls (p<0.05). An increase in [Ca2+]i was also observed in EDL-derived myotubes at 9% stretch conditions (p<0.05). A 9% stretch bout also showed significantly higher [Ca2+]i, compared to 3% stretch conditions (p<0.05). Soleus-derived myotubes (Figures 5b and 5d) at stretch conditions of 6% and 9%, showed a significant increase in [Ca2+]i, compared to 0% stretch condition (p<0.05). Soleus-derived myotubes in 9% stretch conditions also demonstrated higher [Ca2+]i, compared to 3% stretch bouts (p<0.05). Neither EDL- nor soleus-derived myotubes at the relatively lower stretch bout of 3%, showed a significant increase in [Ca2+]i, compared to 0% stretch controls. This implies that a certain magnitude of physical stretch is necessary to elicit measurable changes in [Ca2+]i in cultured myotubes.
Turning our attention to the effects of Piezo1 downregulation, we witnessed a striking suppression in [Ca2+]i in response to stretch compared to control-siRNA (Figures 5a and 5b). First of all, the progressive increase in [Ca2+]i was completely abolished in EDL-derived myotubes (Figure 5c). Indeed neither a 6%, nor a 9% stretch could elicit a significant increase in [Ca2+]i. In soleus-derived cells (Figure 5d), only Piezo1-downregulated myotubes at the 9% stretch condition, were able to show increased [Ca2+]i, compared to 3% stretch counterparts (p < 0.05).
Comparison of [Ca2+]i between control-siRNA and Piezo1-siRNA cohorts at each stretch condition further cemented the remarkable reduction of [Ca2+]i in Piezo1 downregulated conditions in response to stretch (Figure 5c). EDL-derived myotubes at 3% stretch in Piezo1-siRNA conditions, show a suppression in [Ca2+]i compared to control-siRNA counterparts (p<0.05). This is also true for 6% cohorts, with Piezo1-siRNA transfected EDL myotubes displaying a lower [Ca2+]i, compared to control-siRNA (p<0.05). At 9% stretch we continued to see a significant suppression of increase in [Ca2+]i in Piezo1-siRNA treated conditions, compared to control-siRNA at the same stretch bout (p<0.05). In the soleus (Figures 5b and 5d), the suppression of [Ca2+]i following Piezo1 downregulation persisted. Indeed, a 3% stretch showed a lower [Ca2+]i in Piezo1-downregulated conditions, compared to 3% stretch in control-siRNA. At 6% stretch, increase in [Ca2+]i is again blunted in Piezo1-downregulated cohorts, compared to siRNA controls (p<0.05). This is further demonstrated at 9% stretch bouts, with Piezo1-downregulated myotubes showing a reduced increase in [Ca2+]i, compared to control-siRNA (p< 0.05). The suppression of increase in [Ca2+]i following Piezo1 downregulation may be a key factor in the reduction of fusion observed in Figure 3.
Piezo1 activation and myotube fusion
The next phase of the investigation demanded we look at the effects of Piezo1 activation in myotube fusion and Ca2+ influx. This was achieved using the Piezo1 specific agonist Yoda1. Our preliminary findings showed that a 24-hour incubation of myotubes with all the chosen Yoda1 concentrations significantly inhibited myotube maturation (data not shown). Thus the subsequent experiments set out to test a series timepoints with our chosen Yoda1 concentrations, in order to uncover a potential “sweet-spot” between agonist concentration, incubation time and our hypothesised increase in myogenic fusion. Early forming myotubes (cells incubated in differentiation medium for 24 hours at high cell confluency in 96-well plates) were subjected to varying timepoints and concentrations of Yoda1. After each allocated timepoint, the agonist containing medium was removed and replenished with fresh differentiation medium and incubated for a further 2 days (Figure 6).

Early forming myotubes were administered with either DMSO (control, white bars) or the Piezo1-agonist Yoda1 at the following concentration: 5 μM (red bars), 10 μM (blue bars), 30 μM (green bars) and 100 μM (grey bars). Myotubes were incubated for 1 min, 10 min, 30 min, 1 hour and 4 hours. Following the incubation period, the medium was exchanged with fresh reduced medium (without agonist) and myotubes were incubated for a further 2 days. a) and b) Representative images of cohorts at relevant timepoints and concentrations, immunostained for Myosin heavy (MyHC) (red panels) and counterstained with DAPI (black and white panels). Micrographs taken at x20 magnification. Scale bar is 100 μm. Bar graphs display fusion index in c) EDL and d) soleus-derived myotubes from each time point and concentration variables. Values are mean ± SEM. * indicates significance at p < 0.05 using one-way ANOVA tests followed by the Tukey-Kramer post-hoc. n = 3 mice.
EDL-derived myotubes (Figures 6a and c) at the 1-minute timepoint showed that a 30 μM and 100 μM incubation with Yoda1 significantly increased the fusion index compared to DMSO controls. Similarly, soleus-derived myotubes (Figures 6b and d) had a significant increase in fusion at 30 μM and 100 μM, compared to DMSO controls (p<0.05). At the 30 min timepoint, we found that EDL-derived myotubes at Yoda1 concentrations of 30 μM had a significantly higher fusion index, compared to DMSO counterparts (p<0.05). Soleus samples incubated for 30 min with Yoda1 at 10 μM and 30 μM had a significantly higher fusion index than DMSO controls (p<0.05). Unlike the 1 min timepoint, a 100 μM Yoda1 incubation revealed a reduced fusion index when incubated for 30 minutes. Indeed, both soleus and EDL samples incubated at 100 μM beyond the 1 min mark, showed a significant decrease in fusion, compared to DMSO controls.
At the 1-hour incubation timepoint, EDL-derived myotubes exhibited increased fusion index at 5 μM, 10μM and 30 μM, compared to DMSO controls (p<0.05). Similarly soleus-derived samples incubated with Yoda1 for 1 hour showed an increase in fusion at 5 μM, 10 μM and 30 μM, compared to DMSO controls. Four hours of consistent Piezo1 activation showed a significant decrease in fusion of EDL-derived myotubes at a Yoda1 concentration of 30 μM compared to DMSO control (p<0.05). Taken together, we showed that Piezo1 activation results in enhanced fusion of myotubes. This is dependent on the duration and potency in which Piezo1 is activated, as too much of either inhibits myotube fusion
Myotube width and Piezo1 activation
We noticed that Yoda1 treated samples that exhibited increased fusion, also appeared to have thinner myotubes. In order to address this, we compared the myotube width of these samples to DMSO controls (Figure 7). Interestingly, both EDL (Figure 7a and b) and soleus derived myotubes (Figure 7c and d) showed reduced myotube width compared to controls. We further subdivided myotube width into bins of 5 μm increments and compared the distribution of Yoda1 treated myotubes at the 1 min incubation with 100 μM concentration to DMSO controls. Both EDL (Figure 7e) and soleus derived myotubes (Figure 7f), showed that Yoda1 treated cells, on average, have a greater proportion of smaller myotubes. In contrast DMSO controls have a higher distribution of lager myotubes. A similar pattern was observed in the rest of the Yoda1 treated cohorts that displayed increased fusion (Figure S5). Taken together it appears that increased fusion post Yoda1 administration comes at the cost of myotube syncytial maturation.
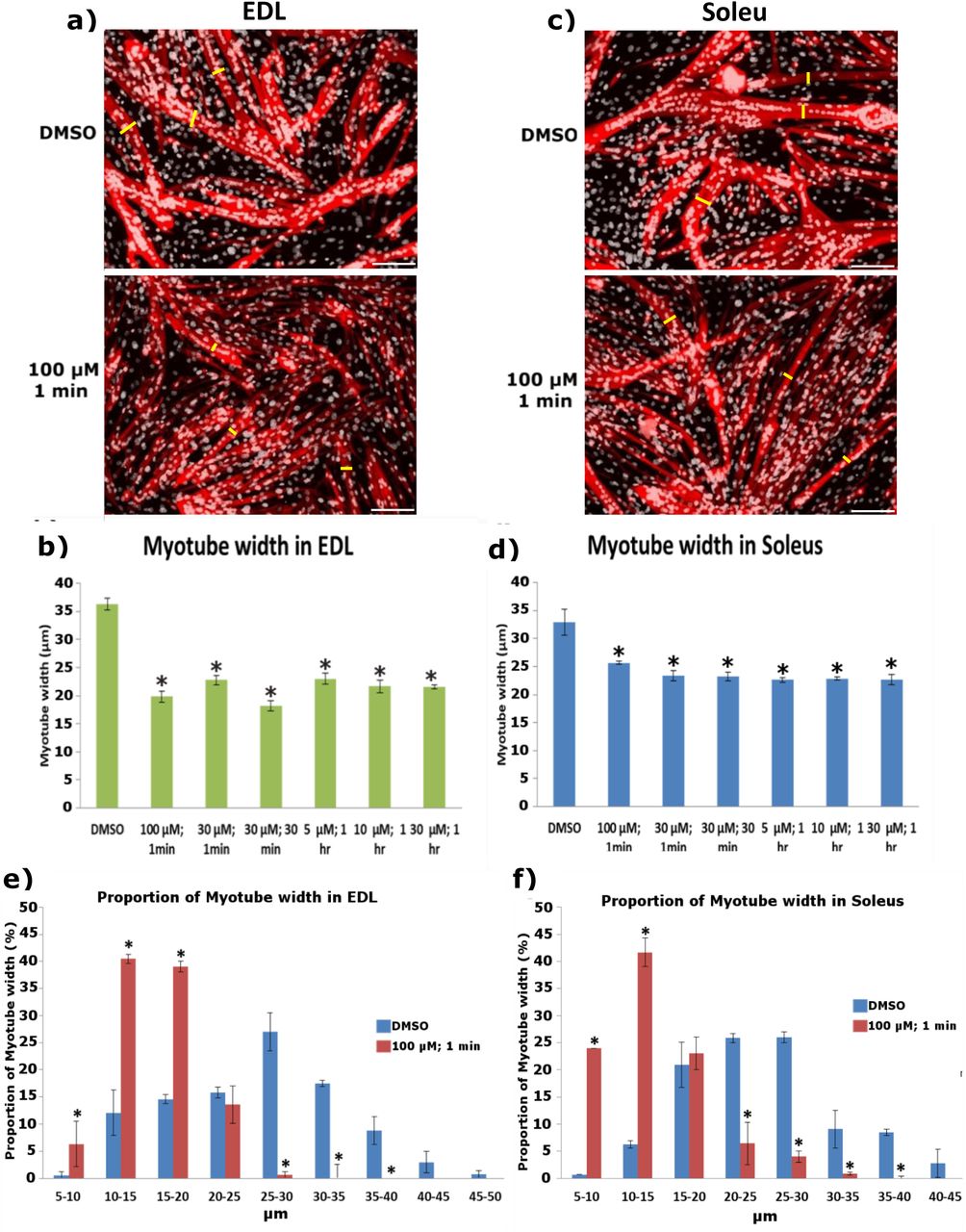
Representative images of a) EDL and c) soleus derived myotubes immunolabelled for MyHC. We measured the width of the myotubes to quantify potential differences between Piezo1-activated cohorts (lower panels) and DMSO controls (upper panels). This was achieved by taking three independent points within a field of view (yellow bars) and measuring the distance from one side of the myotube to the other. Example pictures from DMSO controls and 100 μM of Yoda1 incubated for 1 min are displayed. Scale bar is 100 μm. b) and d) Average myotube width in DMSO and Piezo1 activated samples. e) and f) Proportion of myotube width in EDL and soleus-derived myotubes. Myotube width was divided into incremental bins of 5 μm and represented as percentages relative to the total number of myotubes counted. Data is mean ± SEM from three experiments data (n = 3 mice). *: Significant at p < 0.05 compared to DMSO controls conditions using one-way ANOVA followed by the Tukey-Kramer post-hoc. Please note that only cohorts which showed increased myogenic fusion in both EDL and soleus myotubes from Figure 6 are displayed.
Ca2+ influx and Piezo1 activation
The next aim of was to determine whether Piezo1 activation leads to increased influx of Ca2+ in cultured myotubes. Using the customised stretch silicon bio-chambers [16], we cultured myotubes derived from both EDL and soleus muscle. We then divided the samples into two groups; those given the Piezo1 agonist Yoda1 (at 30 μM) and those without. The chambers were then subjected to incremental bouts of stretch, with a minute rest in between each stretch. Throughout the experiment we measured [Ca2+]i (Figure 8).
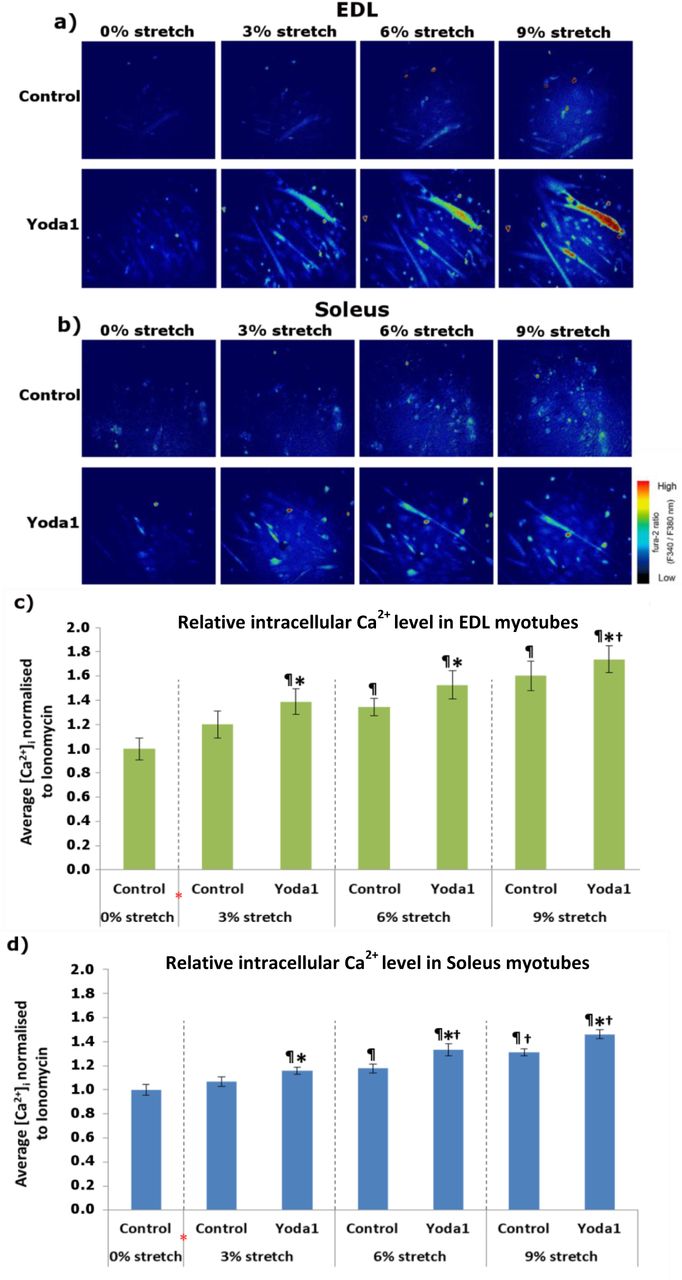
a) and b) Representative images of Ca2+ imaging in EDL and soleus-derived myotubes. Fura 2-AM was administered to myotubes followed by a 30-min incubation time. Stretch was then applied at 3% (0.3 mm), 6% (0.6 mm) and 9% (0.9 mm) for 1 min followed by a 1-min resting period in between. During the initial 0% stretch timepoint, Yoda1 cohorts were administered with 30 μM of the agonist (red *) before being subjected to stretch. Ionomycin at 5 μM was then applied. Side vertical bar shows Fura 2-AM ratio emittance from low to high. c) and d) Average changes in the intracellular Ca2+ level ([Ca2+]i,) difference between base and peak pixel value) normalised to ionomycin. Data is mean ± SEM from three experiments (n = 3 mice). *: Significant difference at p < 0.05 compared to control at each condition. ¶: Significant difference at p < 0.05 compared to 0% stretch counterparts. †: Significant difference at p < 0.05 compared to 3% stretch counterparts using one-way ANOVA followed by the Tukey-Kramer post-hoc.
First of all our results confirmed that increasing stretch amplitude results in higher [Ca2+]i, which means a greater influx of Ca2+, in both EDL- and soleus-derived myotubes (Figures 8c and 8d). Indeed under control conditions (without Yoda1), EDL-derived myotubes subjected to 6% stretch and 9% stretch show a higher [Ca2+]i, compared to 0% stretch controls (p<0.05). Similarly, soleus-derived myotubes at a stretch of 6% and 9% exhibited a higher [Ca2+]i, compared to 0% stretch counterparts (p< 0.05). Notably EDL- and soleus-derived myotubes under 3% of stretch showed no significant difference in [Ca2+]i, compared to 0% stretch counterparts. In contrast when Yoda1 was administered prior to stretch, we find that the apparent “threshold” for Ca2+ influx is decreased. Indeed at 3% stretch bouts (Figures 8c and 8d), we found that both EDL- and soleus-derived myotubes showed a significantly higher [Ca2+]i compared to their respective 0% stretch control conditions (p<0.05). Soleus-derived myotubes (Figure 8d) also exhibited a greater increase in [Ca2+]i post Yoda1 administration in both 6% and 9% stretch conditions, compared to 0% stretch controls (p<0.05). In a similar pattern (Figure 8c), EDL-derived myotubes at 6% and 9% stretch bouts in Yoda1 administered groups, showed a significant increase in [Ca2+]i compared to 0% control (p<0.05).
Comparing the differences between agonist treated groups and those without, at each stretch condition, we are able to further demonstrate the increased [Ca2+]i in response to Piezo1 activation. For example at bouts of 3% stretch, EDL-derived myotubes treated with Yoda1 (Figure 8c) showed a significantly higher [Ca2+]i, compared to untreated 3% stretch counterparts. At bouts of 6% stretch, EDL-derived myotubes given Yoda1 showed a significantly higher [Ca2+]i, compared to untreated myotubes (p<0.05). At 9% stretch, EDL-derived myotubes treated with Yoda1, once again showed a significantly higher [Ca2+]i, compared to cohorts without the agonist (p<0.05). Soleus-derived myotubes at 3% stretch (Figure 8d), showed that cohorts given Yoda1 prior, exhibited a higher [Ca2+]i, compared to untreated counterpart (p<0.05). At 6% stretch the significant difference in [Ca2+]i is also evident, with Yoda1 treated myotubes showing a higher [Ca2+]i, compared to untreated samples (p<0.05). In line with these findings we showed that soleus-derived myotubes at bouts of 9% stretch had a higher [Ca2+]i when given Yoda1, compared to myotubes without the Piezo1 agonist (p<0.05). Taken together, this series of experiments showed that Piezo1 is an essential player in the regulation and function of myotubes. This process seems to involve active modulation of Ca2+ influx. How this leads to increased myotube fusion is an area of great interest and one which demands further research.
Discussion
The current study showed that the mechanosensitive ion channel Piezo1 is required for the fusion and formation of myotubes. Indeed reduced expression of this channel hindered the fusion of myocytes to forming or maturating myotubes – a phenotype that may be partly associated with interacting fusogenic proteins like Myomaker or Myomixer. We further showcased how agonist mediated activation of Piezo1 can enhance myogenic fusion at the expense of myotube width. Additionally, our findings revealed that over-activation of Piezo1 results in the loss of myotube integrity. Using live imaging assays, we discovered that Piezo1 activation enhances Ca2+ permeation in cultured myotubes. This led to an increased influx of Ca2+ in response to stretch. Conversely, we showed how the downregulation of Piezo1 hinders Ca2+ entry into myotubes of both EDL and soleus muscle.
Piezo1 in myogenesis
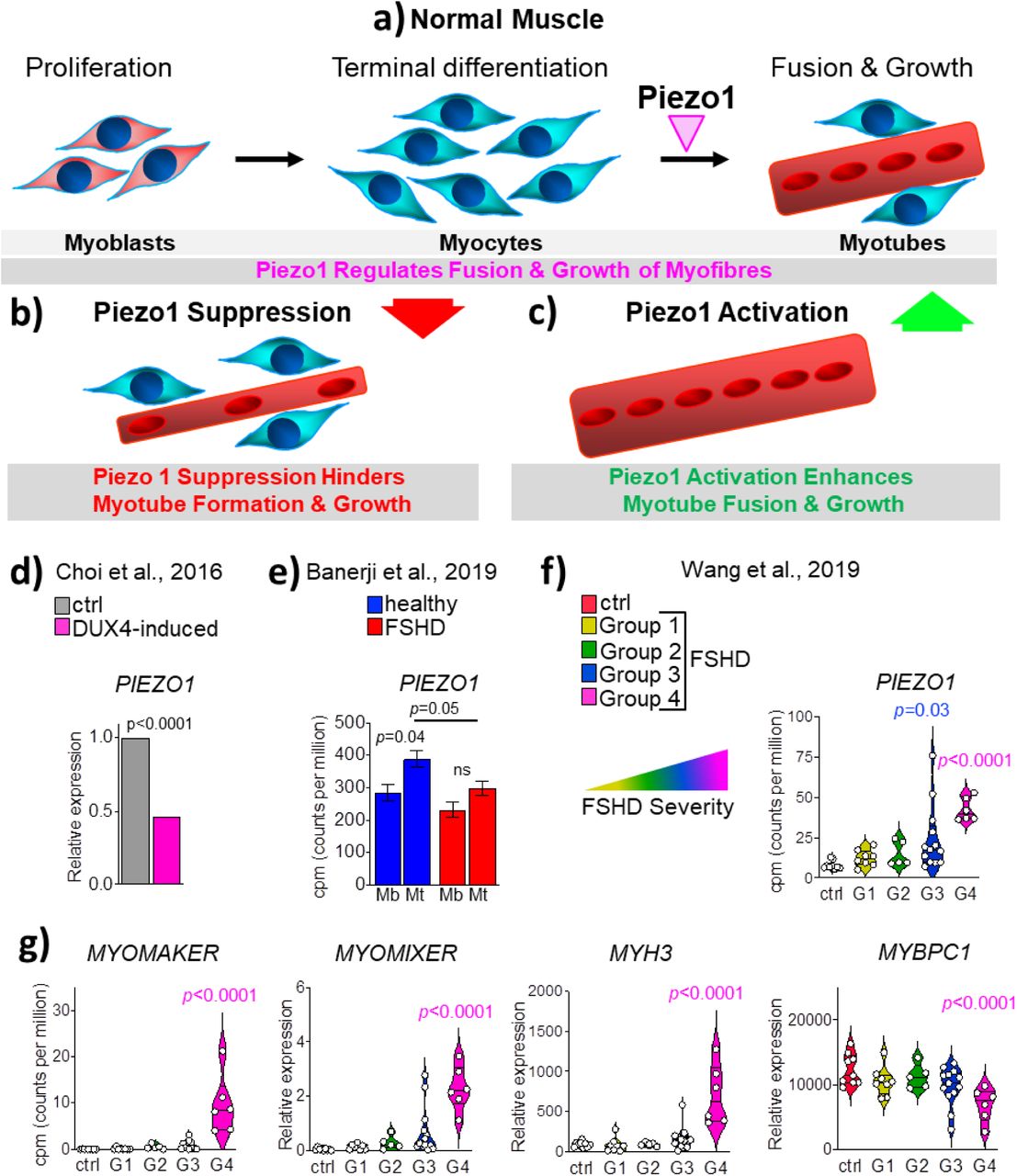
The current study investigated the effects of Piezo1 regulation throughout the myogenic program. Piezo1 is expressed at a higher proportion in terminally differentiated myotubes, compared to proliferating myoblasts. Moreover, we found that, differentiating primary muscle cells derived from the mainly slow-type muscle soleus displayed higher expression of Piezo1 compared to the fast EDL muscle (Figure S1). Understanding the potential differences in muscle/fibre types and Piezo1 regulation is an intriguing area for future research and could reflect differences in the dynamics of myogenic progression. We also confirmed that the expression of Piezo2 is not altered by the down-regulation of Piezo1. This is perhaps not surprising given the fact that Piezo2 is not as abundant in skeletal muscle compared to Piezo1 [5, 16]. Nevertheless it was important to see any potential compensatory effects Piezo2 may impose. Specific downregulation of Piezo1 by siRNA-mediated transfection showed no significant change in the proliferation rate of either EDL- or soleus-derived primary myoblasts. However, our data do not exclude the possibility that Piezo1 is not involved in earlier myogenic events, perhaps in balancing quiescence and activation of satellite cells. In proliferating myoblasts, lack of Piezo1 function does not alter onset of myogenic differentiation, evaluated by the proportion of myogenin-positive cells. In contrast, where a significant phenotype was observed was in terminally differentiated myotubes. Indeed, our data found that knockdown of Piezo1 significantly reduced fusion of myocytes and prevented myotube formation and maturation. In contrast, activation of this Ca2+ permeable channel resulted in enhanced myogenic fusion. The main findings are summarised in Figure 9a-c.

a) Under normal conditions, membrane mechanosensors, such as Piezo1 (magenta arrowhead), regulate myocyte fusion thereby ensuring efficient muscle fibre formation and maturation. b) Suppression of Piezo1 expression blunts myocyte fusion and Ca2+ homeostasis, thus hampering the formation of proper myotubes. c) In contrast, selective chemical activation of Piezo1 function enhances myogenic fusion thus significantly increasing myotube formation and maturation. d) Statistical analysis derived from Choi et al, [31], shows that DUX4 expression induced by Doxycycline, which mimics human FSHD pathogenesis, results in significant suppression of Piezo1 level compared to control non-induced cells (Unpaired two-tailed t-test). e) Time-course transcriptomics analysis, derived from Banerji et al, [33], on differentiating isogenic (bar D4Z4 repeats) myoblasts derived from a male mosaic FSHD1 patient show that while unaffected (blue) myoblasts upregulated significantly PIEZO1 expression when differentiating into myotubes, in FSHD PIEZO1 level remains unchanged from myoblasts to myotubes (red bar) (Unpaired two-tailed t-test). f) Statistical analysis of transcriptomic data on human FSHD biopsies derived from Wang et al [34]. Patients have been grouped according to FSHD severity, which increases from Group1 to Group 4. Control (ctrl) represents non-affected individual. Count of gene reads (cpm, count per million) PIEZO1 is significantly upregulated in FSHD Group 4. g) The same dataset as (f) shows concomitant upregulation of MYOMAKER, MYOMIXER and MYH3 (myosin 3) while reduced MYBPC1 (myosin binding protein C1) level in line with regeneration process ongoing and a severe dystrophic phenotype respectively. One-way ANOVA with Tukey’s post-hoc test.
Piezo1 and muscular dystrophies
Piezo1 activation showed a significant increase in the fusion index of both EDL- and soleus-derived myotubes. Although this phenotype could be viewed as beneficial in terms of muscular dystrophy prevention, we must be aware of the potential dangers of an overactive Piezo1 channel. In fact, we showed that even a 30 min incubation of myotubes with a high agonist concentration (100 μM of Yoda1 treatment) led to decreased fusion in both EDL- and soleus-derived myotubes (Figure 6). This adverse phenotype is most likely the result of a dangerously high Ca2+ influx. The publicly available data obtained from MRI-guided human biopsies from facioscapulohumeral muscular dystrophy (FSHD) affected individuals (Figure 9f and g) further highlights the necessity to examine mechanosensor channels like Piezo1 in muscular dystrophies. FSHD is associated with mutations in the distal end of chromosome 4 (4q35), resulting in epigenetic deregulation of the D4Z4 macrosatellite repeat array, causing uncontrolled expression of the transcription factor Double homeobox 4 (DUX4) [24–29]. In turn, DUX4 accumulation hampers myogenic progression promoting cytotoxicity and is considered the most possible root cause of FSHD pathogenesis [30]. We took advantage of a transcriptomic study on murine DUX4-inducible myoblasts model elucidating DUX4 role in FSHD, to assess the level of Piezo1 in such context and found that it is significantly downregulated upon accumulation of DUX4 [31] (Figure 9d), thus suggesting that Piezo1 dysregulation may contribute to FSHD pathology.
In line with the muscular atrophy usually found in patients, human primary myoblasts derived from FSHD biopsies display reduced differentiation in vitro generating hypotrophic myotubes [32, 33], thus resembling the effect of Piezo1 knockdown in our experimental setup. Indeed, analysis of transcriptomic dataset on differentiating isogenic FSHD affected or unaffected myoblasts previously performed by Banerji and colleagues [33], revealed that while during unaffected (healthy) myogenic differentiation, Piezo1 is upregulated from myoblasts to myotubes, its expression fails to increase in FSHD myogenic progression (Figure 9e). Moreover, our analyses of transcriptomic dataset from Wang and colleagues [34] found that PIEZO1 mRNA is significantly upregulated in MRI-guided muscle biopsy from FSHD patients that show the worst pathological phenotype (Group 4) (Figure 9f). Interestingly, it has recently been demonstrated that FSHD muscles display traces of ongoing regeneration, as indicated by elevated levels of embryonic Myosin, a canonical marker of ongoing muscle fibre regeneration [24]. Congruently with the accumulation of PIEZO1 mRNA, additional transcriptomic analysis also revealed a concomitant upregulation of MYOMAKER and MYOMIXER in the most affected group of muscle biopsies (Figure 9g) indicating an ongoing regeneration process. Such regeneration is further suggested by significant accumulation of embryonic MYH3 (myosin 3) mRNA (Figure 9g). In contrast, the reduced level of MYBPC1 (myosin binding protein C1), a myosin-associated protein involved in the formation and maintenance of sarcomere structure confirmed the dystrophic status of FSHD muscle biopsies (Figure 9g). Therefore, from these pieces of evidence we conclude that Piezo1 dysregulation may contribute to the pathomechanism underlying FSHD. However, the precise definition of PIEZO1 dynamics in FSHD progression would require specific investigation.
The skeletal muscle has typically a slow turnover, thus putative fusion proteins like Myomaker and Myomixer regulated by Piezo1 may be present at a relatively lower level (as shown by control patients, Figure 9d) and called upon in response to muscle damage (injury, resistive exercise etc.). However in FSHD, the disorganised and possibly inefficient activation of the regeneration program may dysregulate Piezo1 expression. From a pathophysiological angle and taking into consideration our over-activation data using Yoda1 (Figure 6), we suggest that an over-abundance of Piezo1 expression in FSHD may result in uncontrolled increase in Ca2+ influx, thus hindering myotube integrity. On the other hand, improper Piezo1 expression may also lead to defective myogenic progression and muscle formation, as indicated by our knockdown analysis. In line with this, transcriptomic analysis of human myoblasts induced to express a high level of DUX4, which is considered to mimic some aspects of FSHD pathogenesis, revealed significant suppression of PIEZO1 expression (Figure 9d), thus suggesting that reduced PIEZO1 function may contribute to FSHD pathogenesis.
Piezo1 and the myocyte fusion machinery
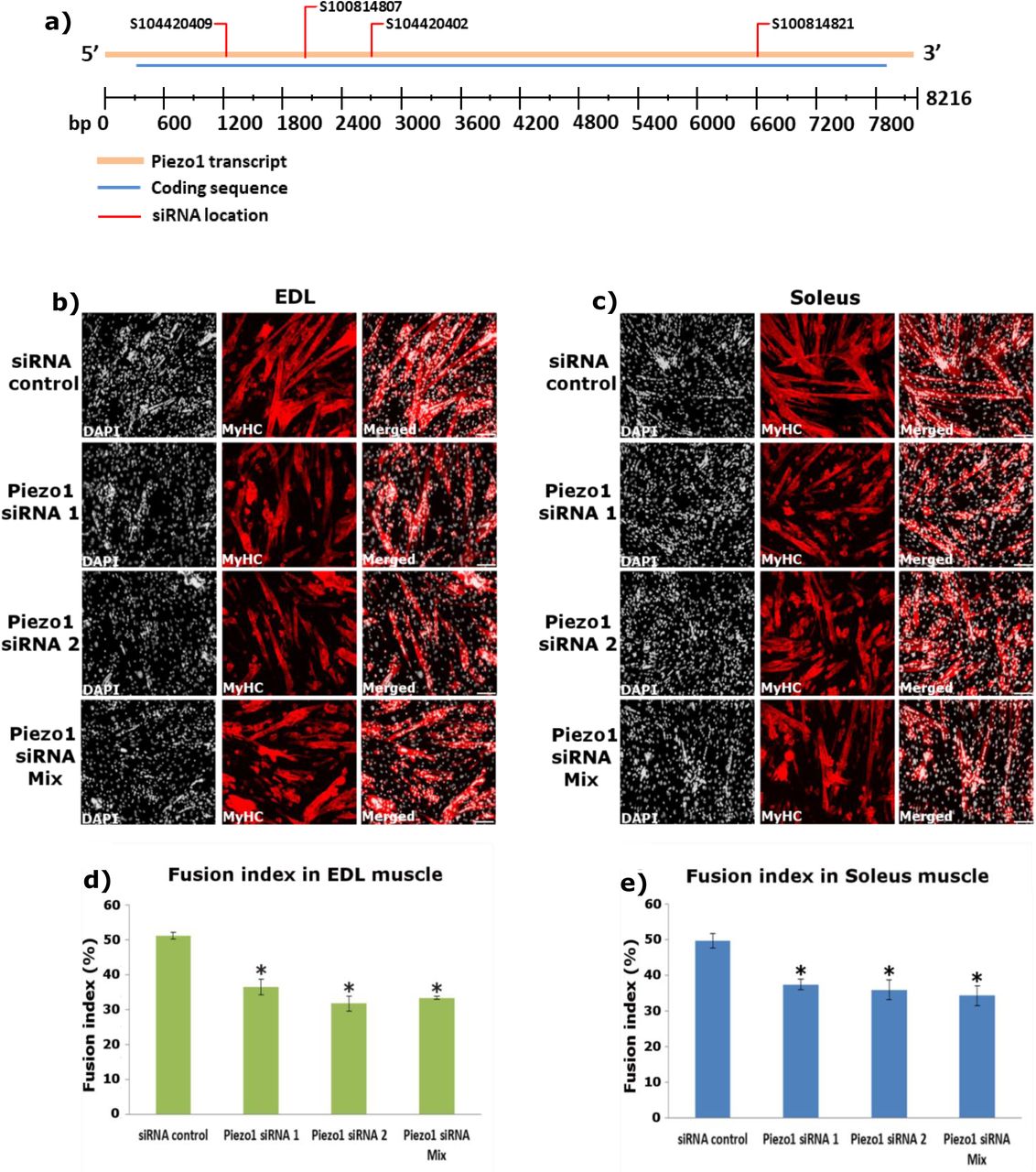
Piezo1 downregulation significantly reduced myocyte fusion during myotube formation and myotube maturation. To the best of our knowledge there is only one other paper published that examined Piezo1 in skeletal muscle by Tsuchiya et al. [17]. Interestingly, the findings from this group showed that Piezo1 inhibition resulted in a sheet-like syncytium of MyHC coupled with increased fusion. Although these findings show contrasting results to the ones presented in this study, we must take into consideration potential factors which may explain why this may be the case. One such factor is the method of Piezo1 inhibition used by Tsuchiya et al, [17]. They carried out many of their experiments using knockout lines of Piezo1 through the gene editing tool CRISPR/Cas9. The fact that these cell lines did not express Piezo1 to begin with (unlike the cells we used) may yield completely different phenotypes, compared to the transient inhibition achieved by siRNA mediated transfection. Therefore, complete lack of Piezo1 expression may favour the activation of a secondary, yet unknown, alternative Piezo1-affected pathway(s) to fusion. Regarding the Piezo1 siRNA transfection experiments, although more than 60% reduction in gene expression is ideal; we nevertheless found that our level of Piezo1 knockdown produced very interesting effects on myogenic regulation. Moreover, Miyamoto et al, [16] also obtained slightly below or just about 60% Piezo1 reduction, yet reported intriguing Piezo1-associated events in urothelial cells. Perhaps Tsuchiya et al, obtained even greater knockdown of Piezo1 in their siRNA-mediated analyses [17], further suggesting that the timing and level of Piezo1 expression may yield varying phenotypes. There was also the likelihood that siRNA used in this study may potentially have off-target effects on other genes which could influence myotube formation by employing other mechanistic pathways. However this is excluded by the fact that we have now tested four different Piezo1-specific siRNAs and all show a marked reduction in the fusion of myoblasts, compared to siRNA controls (Figure S4). Therefore, we conclude that impaired Piezo1 signalling inhibits myotube fusion. Further support comes from our Piezo1 activation experiments. Congruently, we found that selective activation of Piezo1 by the agonist Yoda1 significantly increased the fusion index in a concentration- and time-dependent fashion. These results do not disprove the investigation by Tsuchiya et al, but rather highlight the complexity underlying Piezo1 dynamics. It would be interesting to see the effects of constitutive overexpression of Piezo1 by retroviral transduction or conditional Piezo1 transgenic mice. Furthermore, obtaining the cells used by Tsuchiya et al [17] and conducting myocyte fusion analysis using our setup would undoubtedly prove vital at discerning potential reasons underlying some of the seemingly opposing results.

a) Location of siRNAs shown in Table 1 on Piezo1 mRNA. Diagram shows Piezo1 mRNA transcript (orange bar), coding sequence (blue bar) and location of each siRNA (red line). bp: base pair. Diagram is adapted from Qiagen (https://geneglobe.qiagen.com/product-groups/flexitube-sirna). b and c) Representative images of EDL and soleus muscle-derived myotubes, transfected with 10nM of control-siRNA (siScrambled) or different siRNAs specific for Piezo1 (Piezo1-siRNA 1 and Piezo1-siRNA 2). A mixture of four different Piezo1-siRNAs (Piezo1-siRNA Mix) including the one in the main text (Table 2) was also used at 10nM (2.5 nm each). Myoblasts were transfected and incubated overnight; cells were incubated for a further 72 hours. Cells were immunolabelled for Myosin heavy chain (MyHC) (red panels) and counterstained with DAPI (black and white panels). d) and e) The fusion index was calculated by counting the total number of nuclei within each myofibre and representing this as a percentage relative to the total number nuclei in the image taken. Data is mean ± SEM from three experiments (n = 3 mice). *: Significant at p < 0.05 compared to siScrambled conditions using one-way ANOVA followed by the Tukey-Kramer post-hoc. From table 2, Piezo1 siRNA 1 corresponds to Qiagen, S104420402. Piezo1 siRNA 2 corresponds to Qiagen, S100814807.
Additional support for the involvement of Piezo1 in myogenic fusion comes from our results which showed that its downregulation significantly reduced the expression of Myomaker - a muscle specific protein that localises to the plasma membrane and is crucial for vertebrate myocyte fusion [23,35,36]. Whether this is a direct response for the downregulation of Piezo1 or an indirect event remains to be determined. In addition, the expression level of Myomixer, another essential molecule for cell fusion, showed a tendency to reduce upon Piezo1 knockdown further suggesting that Piezo1 contributes in the regulation of the cell fusion machinery. Removal of either Myomaker or Myomixer results in decreased myoblast fusion, whereas overexpression results in enhanced fusion [36, 37].
A feature that became apparent in our study was the seemingly “thinner” myotubes in agonist treated samples which showed increased fusion. Indeed, compared to DMSO controls, the average myotube width was smaller in Yoda1 treated cohorts (Figure 7 and Figure S5). Thus, we wonder whether the enhanced fusion, measured by the number of myonuclei within MyHC positive myotubes, comes at the expense of myotube syncytial maturation. In other words, Piezo1 activation may have hastened myonuclear fusion leading to premature myotube formation. Perhaps increased PIEZO1 expression (Figure 9f) contributes to the inadequate regenerative state of FSHD stricken muscle.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Myotube width was divided into incremental bins of 5 μm and represented as percentages relative to the total number of myotubes counted. The overall proportion of myotube width distribution in DMSO controls and Yoda1-treated samples is summarised as line graphs in a) EDL and b) soleus-derived myotubes. c-l) Bar graphs comparing DMSO controls and samples which showed increased fusion post Yoda1 treatment in EDL (left) and soleus (right)-derived myotubes Data is mean ± SEM from three experiments (n = 3 mice). *: Significant at p < 0.05 compared to DMSO controls conditions using one-way ANOVA followed by the Tukey-Kramer post-hoc. Please refer to Figure 6 a) and b) for representative images.
DMSO controls at the 4 hours mark showed a trending decrease in fusion in soleus-derived myotubes, compared to DMSO counterparts from other timepoints. EDL-derived DMSO-treated myotubes at the 4-hour incubation timepoint, showed a statistically significant decline in fusion compared to DMSO counterparts. Although these differences may be negligible at lower timepoints, we should warrant concern over the potential for DMSO-mediated inhibition of primary derived myotubes during longer timeframes. Indeed, the observed myotube inhibition may be slightly overestimated in Yoda1-treated samples at the 4-hour timepoint.
Piezo1 activation and Ca2+ influx
The present study showed that selective downregulation of Piezo1 dramatically suppressed [Ca2+]i, which most likely translates in the depression of the influx of Ca2+ into cultured myotubes exposed to stretch. In contrast we showed that activation of Piezo1 significantly increased [Ca2+]i, which means the enhancement of Ca2+ influx. Our results propose that Piezo1 is a novel intracellular Ca2+ regulatory protein in skeletal muscle function. Ca2+ plays a crucial role in skeletal muscle function, maintenance and plasticity. All muscle fibres use Ca2+ as their main regulatory and signalling molecule [38–40]. Therefore, the contractile functionalities of muscle fibres are dependent on the highly regulated expression of proteins involved in Ca2+ handling and signalling. Our study showed that Piezo1 mediated regulation of Ca2+ influx is a key driving factor in the respective decrease and increase in myotube fusion in response to Piezo1 inhibition and activation. To the best of our knowledge this is the first time this has been demonstrated.
The silicon bio-chamber experiments revealed that at relatively low stretch conditions of 3% (0.3 mm) neither EDL- nor soleus-derived myotubes elicited a significant increase in Ca2+ influx. However at higher stretch distances (6% and 9% stretch) this mechanical barrier was crossed as demonstrated by the net increase in [Ca2+]i, compared to 0% stretch counterparts. In a similar set of experiments (albeit using urothelial cells), Miyamoto et al. also showed a distance dependent increase of Ca2+ influx. Interestingly this response was blunted in Piezo1-siRNA-treated conditions [16]. The researchers also showed that a high enough [Ca2+]i must be attained in order to elicit a response, in their case ATP efflux. The data presented in this study supports the presence of a stretch-dependent increase in Ca2+ influx. Remarkably we found that activation of Piezo1 resulted in increased [Ca2+]i at 3% stretch conditions, suggesting that the activation threshold of Piezo1 was lowered. Furthermore the data showed that reduction of Piezo1 expression significantly blunted any significant increase of [Ca2+]i in response to stretch. These results, for the first time show the need for Piezo1 to respond to stretch and permeate Ca2+ into myotubes. The findings also propose the presence of a physical threshold that must be attained before Piezo1 mediated Ca2+ influx is significantly increased. Like Miyamoto et al [14], we find that a stretch-dependent increase in Ca2+ influx is suppressed when Piezo1 expression is decreased. Conversely, we see an increase of [Ca2+]i when Piezo1 is activated. Whether this leads to altered cellular/myotube viability in the form of ATP release remains a subject for future research.
Conclusion
The data presented in this study showed that the Piezo1 channel is present in primary derived myoblasts and myotubes but expressed at a higher proportion in the latter. Downregulation of Piezo1 significantly lowered the fusion capacity during myotube formation and maturation. In contrast, Piezo1 activation increased fusion. Future research examining changes in myotube function (integrity, Ca2+ influx, cytoskeletal organisation and fusion) that are directly the result of mechanical stress should consider analysis of Piezo1. In the context of therapeutic strategies against muscular dystrophies such as FSHD, not only must we unravel the spatiotemporal regulation of Piezo1 expression, but we must be aware of this channel’s ability to alter it Ca2+ influx threshold by adapting or inactivating its gating capacity in response to repetitive stimuli. Pharmaceutically, small activating molecules such as Yoda1 (and others like it) may prove beneficial. However careful attention must be given to the half-life and pharmacokinetics of these agonists in vivo before even considering them as viable drugs for human consumption. Piezo1’s importance in skeletal muscle maintenance and function will undoubtedly grow as new research aims to explore the mechanisms and signalling pathways this remarkable mechanosensor employs.
Funding
This work was supported by KAKENHI (JP17K01762, K.G.; JP18H03160, K.G.; JP19K22825, K.G.; 19KK0254. K.G.) from the Japan Society for the Promotion of Science, the Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan, and Graduate School of Health Sciences, Toyohashi SOZO University (K.G.).
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The authors thank Ms. Yumiko Asakura and from Department of Physiology, Graduate School of Health Sciences, Toyohashi SOZO University for her assistance.
References




