

Abstract
Trajectory inference methods are used to infer the developmental dynamics of a continuous biological process such as stem cell differentiation and cancer cell development. Most of the current trajectory inference methods infer cell developmental trajectories based on the transcriptome similarity between cells, using single cell RNA-Sequencing (scRNA-Seq) data. These methods are often restricted to certain trajectory structures like trees or cycles, and the directions of the trajectory can only be partly inferred when the root cell is provided. We present CellPaths, a single cell trajectory inference method that infers developmental trajectories by integrating RNA velocity information. CellPaths is able to find multiple high-resolution trajectories instead of one single trajectory from traditional trajectory inference methods, and the trajectory structure is no longer constrained to be of any specific topology. The direction information provided by RNA-velocity also allows CellPaths to automatically detect root cell and differentiation direction. We evaluate CellPaths on both real and synthetic datasets. The result shows that CellPaths finds more accurate and detailed trajectories compared to current state-of-the-art trajectory inference methods.
1 Introduction
The availability of large scale single cell RNA-Sequencing (scRNA-Seq) data allows researchers to study the mechanisms of how cells change during a dynamic process, such as stem cell differentiation and cancer cell development. Trajectory inference methods are often used to infer the trajectory of this dynamic, which can be linear, tree, cycle or other complex graph structure [Street et al., 2018, Saelens et al., 2019], and then assign pseudo-time for each cell along the trajectory.
Although scRNA-Seq data provides genome-wide transcriptome profile of a cell, it captures only a snapshot of this profile and we can not profile the same cell more than once. However, directional information of the dynamic process is intrinsically required for trajectory inference. Most traditional trajectory inference methods ameliorate the loss of directional information in scRNA-Seq dataset by assuming developmental root cell is known as a prior, and cells with similar gene-expression profiles should be sorted next to each other on the trajectory, which may not be true in real world scenario. Other methods uses time-series data [Schiebinger et al., 2019, Fischer et al., 2019] to infer the differentiation direction. The recent RNA velocity methods [La Manno et al., 2018, Bergen et al., 2019] can predict the cell gene-expression profile at the next time point by using the abundance of both nascent mRNA and mature mRNA. This information can potentially reveal “flows” of cell dynamics, which provides an alternative for resolving the loss of direction information in scRNA-Seq data for trajectory inference.
Moreover, most traditional trajectory inference methods tend to have a strong assumption on the structure of the lineages, e.g. linear [Campbell et al., 2015], bifurcating [Haghverdi et al., 2016], tree-like structure [Street et al., 2018], which confines traditional trajectory inference methods to be only applicable for a small subset of real world dataset, where there exist one backbone trajectory and one starting point. In reality, cells in most datasets have multiple lineage backbone structure with multiple root cells, even for cells within the same lineage backbone, small cell sub-flows [Qiu et al., 2020] may also exist, which create multiple heterogeneous sub-trajectories.
We hereby present CellPaths, a computational method that can pick multiple root cells and infer multiple trajectories automatically in a complex single-cell dataset. These trajectories connect cells or small clusters of cells based on the predicted cell states calculated with RNA velocity methods.
CellPaths takes in the nascent and mature mRNA count matrix, and calculate RNA velocity using the dynamical model of scVelo [Bergen et al., 2019], an RNA velocity calculation package that is recently proposed. Single-cell RNA-seq data is extremely noisy, and RNA velocity estimated based on the noisy count matrix is even noisier, which can be problematic for downstream trajectory inference methods. CellPaths ameliorates the noise in the data by constructing meta-cells and perform regression model to smooth the calculated velocity. The use of meta-cells can also reduce the downstream calculation complexity. A modified version of shortest-path algorithm is performed on the meta-cells to construct a pool of all possible trajectories within the dataset. A greedy selection strategy is used to pick out a small number of feasible trajectories within the pool, which correspond to the meta-cell level trajectories. Further, we developed a efficient method for cell-level pseudotime assignment. Current cell-level pseudotime assignment can usually be classified into two sub-types: Slingshot [Street et al., 2018] constructs principal curve and projects cells onto the curve, and Diffusion pseudotime [Haghverdi et al., 2016] utilizes diffusion distance to measure the time difference of cells from root cell. Two methods already provide highly accurate cell-level pseudotime assignment, but hard to be integrated into our methods as they cannot efficiently use the inferred upstream meta-cell paths. We propose first-order pseudotime reconstruction method to assign order of true cells in each meta-cell separately and merge the order together according to the meta-cell path order. The whole schematics of CellPaths is shown in Fig.1. CellPaths is implemented as an open-source Python package (https://github.com/PeterZZQ/CellPaths).
2 Results and discussion
We test our method on both real and synthetic datasets. We select dentate-gyrus dataset [Hochgerner et al., 2018], Pancreatic Endocrinogenesis dataset [Bastidas-Ponce et al., 2019] and human forebrain dataset [La Manno et al., 2018] to analyse the performance of CellPaths. We also test CellPaths on synthetic dataset along with other state-of the art trajectory inference methods to further demonstrate CellPaths ability in detecting high-resolution branching structure and robustness to various branching structure in a quantitative manner. We generate synthetic datasets using dyngen [Cannoodt et al., 2020] and VeloSim (Methods) simulators. The synthetic datasets includes four different branching structures: simple trifurcating structure, multiple-batches bifurcating structure, cycle-tree structure and multiple-cycles structure. The testing results on those datasets shows that CellPaths can produce accurate trajectory inference even in highly complex branching structure compared to traditional trajectory inference methods.
2.1 Results on Real data
2.1.1 CellPaths captures both lineages and small differentiation pathways in dentate gyrus dataset
To test the ability of CellPaths in detecting complex lineage structure, we perform CellPaths on a mouse dentate gyrus dataset [Hochgerner et al., 2018]. The original paper studied the dentate-gyrus neurogenesis process in developing and mature mice datate gyrus region. The dataset has 2930 cells, and a UMAP [McInnes et al., 2018] visualization with cell types annotated is shown in Fig.2a. This dataset has multiple lineages and cannot be summarized using a tree-like differentiation structure, which is beyond the assumption of most traditional trajectory inference methods. CellPaths, on the contrary, shows promising results on dentate-gyrus dataset and detects most highly time-coupled sub cell pathways in addition to all the mainstream lineages in the dataset.

Schematics of CellPaths. CellPaths first constructs meta-cell with corresponding RNA velocity, then constructs directed neighborhood graph, path-finding algorithm is utilized to find potential trajectories and greedy path-selection scheme is used to further refine the possible trajectories set. Finally first-order pseudo-time approximation algorithm is designed to assign the cell-level pseudo-time. The Color bar denotes the change of pseudo-time.

a). Dentate-gyrus dataset with cell type annotated. (b). CellPaths meta-cell level result on dentate-gyrus dataset. (c). First order pseudo-time inferred from CellPaths, time annotated by color. Only path 0 and path 6 is shown. (d). Gene ontology analysis on path 0 and path 6.
In Fig.2b, the first 14 paths inferred by CellPaths are drawn at meta-cell level. The algorithm infers multiple highly time-coupled trajectories that follows the mainstream granule cells lineage, i.e. the differentiation path from neuronal intermediate progenitor cells, to original neuralblast cell, immature granule cell and mature granule cell(Path 0, 3 and 4). In addition, algorithm also detects paths correspond to other small lineages: Radial Glia to Astrocytes lineage(Path 1, 2, 5, 7 and 8), Oligodendrocyte Precursor Cells to Myelinating Oligodendrocytes lineage(Path 11). Apart from the high-level lineages that correspond to distinct cell differentiation, CellPaths also captures multiple small sub-flows of cells with the same cell-types, e.g. inferred trajectories the small cell flows within the mature Granule cell (Path 6) and Endothelial(Path 13).
Cell-level pseudo-time is inferred using first-order approximation pseudo-time assignment, and several selected results are shown in Fig.2c. Differentially expressed (DE) genes and gene ontology (GO) analysis are conducted to analyze the functionality of the inferred trajectories. With each inferred trajectory, DE genes analysis is conducted by fitting a Generalized Additive Model (GAM) as a function of pseudotime and corresponding p-value is calculated using likelihood ratio test. After selecting DE genes using p-value, gene ontology analysis is conducted to summarize the function of genes within each trajectory, with results shown in Fig. 2d. The result of the analysis further certifies the correctness of the inferred trajectories.
The 1st reconstructed trajectory in Fig. 2c corresponds to the main Granule generation process. The genes that change most abruptly with time are found to correspond well to the neuron morphogenesis, long-term synaptic potentiation(LTP) and neuron development, which shows an significant sign of neuron developmental process.
The 7th reconstructed trajectory in Fig. 2c shows a small sub-flow within the mature Granule cell cluster, with gene ontology analysis, genes correspond to neurogenesis process can still be found, which shows that even within the mature granule cell cluster, cell development and maturation is still happening.
2.1.2 CellPaths captures cell-cycle and branching process in Pancreatic Endocrinogenesis dataset
We further test CellPaths on mouse Pancreatic Endocrinogenesis dataset [Bastidas-Ponce et al., 2019]. The dataset profiles 3696 cells and includes endocrine cell differentiation process from Ductal cells to four different endocrine cell sub-types, α, β, δ and E endocrine cell, through Ngn3low endocrine progenitor and Ngn3high endocrine progenitor cell. The UMAP visualization of the dataset and corresponding cell-type annotation is shown in Fig.3a. CellPaths discovers multiple distinct lineages that correspond to α, β, δ endocrine cell genesis, with the meta-cell-level paths shown in Fig. 3b. Interestingly, we found a path where the E cells turn into α cells (Path 7, Fig. 3b).

(a).Pancreatic Endocrinogenesis dataset with cell type annotated using different color. (b). CellPaths meta-cell level result on Pancreatic Endocrinogenesis dataset, a. (c).First order pseudo-time inferred from CellPaths, time annotated by color. Path 0, 1 and 6 is shown. (d). Gene ontology analysis on path 0, 1 and 4.
DE gene analysis discovered multiple featured genes for different endocrine cell sub-types generation process, Within the insulin-producing β-cells generation trajectory, i.e. trajectory 1 in Fig. 3b, DE analysis discovers Pcsk2, Ero1lb, Cpe genes that function in insulin process. The strong time-correlationship of Pax4 is discovered in the somatostatin-producing δ-cells trajectory, trajectory 2 in Fig. 3b, which is known to have control over the endocrine cell type specification along with Arx and abundant in δ-cell lineage [Collombat et al., 2003]. And within glucagon-producing α-cells generation path, trajectory 5, strong time-correlationship of Arx gene is discovered, which also correspond well to previous study [Collombat et al., 2003]. Along with Arx and Pax4, a bunch of other highly time-coupled cell sub-type specific genes are discovered through CellPaths, which allows for further study of cell sub-type specification mechanism. On the other hand, CellPaths also discover a clear cell-cycle pattern on the left side of Fig. 3b, and multiple cell-cycle related genes are found through time-resolved DE analysis, such as Kif 23, Clspn, Aurkb, Spc24, which further shows the high cell-level resolution of CellPaths inferred trajectories.
2.1.3 CellPaths finds multiple cell-flows in forebrain linear dataset
We further test CellPaths on human forebrain glutamatergic neuron genesis dataset. The dataset profiles 1720 cells during the glutamatergic neuron differentiation process. Fig. 4 shows a linear trajectory from Radial glia progenitor to fully differentiated neuron. CellPaths is able to find multiple differentiation paths which are in line with the overall linear trajectory structure. All those paths correspond well to the glutamatergic neuron differentiation process while each path has their own developmental differences.

(a). Human forebrain glutamatergic neuronal lineage dataset with cell type annotated using different colors. The dataset shows a linear trajectory from radial glia to fully differentiated glutamatergic neuron. (b). Multiple meta-cell paths within the linear trajectory are discovered using CellPaths. (c). Cell-level pseudo-time of each meta-cell path is inferred using CellPaths. The gradual change of cell states correspond well to the differentiation process of human forebrain glutamatergic neuron.
2.2 Results on Simulated Data
2.2.1 Experiment design
To further evaluate CellPaths performance compared to other state-of-the-art trajectories inference methods, we create four synthetic datasets using different simulators. We generate first two datasets using dyngen, one with a simple trifurcating structure, another with data from two batches, each with a tree structure. To reduce the bias from one simulator, we incorporate two additional synthetic datasets generated using VeloSim (Methods). One with cycle and tree structure merged together, another with multiple-cycle structure.
We compare our result with Slingshot, a scRNA-seq-based trajectory inference algorithm. Slingshot shows the state-of-art performance among all the other trajectory inference algorithms that utilize only scRNA-seq data, according to the recent benchmark conducted by dynverse [Saelens et al., 2019] and SymSim [Zhang et al., 2019]. We provide root cell in Slingshot since it can not detect the root cell. In addition, we also compare CellPaths with trajectory inference method that incorporate RNA-velocity information. Velocity diffusion pseudo-time (Vdpt) is a trajectory inference method in scVelo package that is developed based on diffusion pseudo-time [Haghverdi et al., 2016] and utilize RNA-velocity for transition matrix construction and root-cell finding. The Comparison shows that CellPaths has the advantage of high-resolution cell sub-flow detection like cell cycle within the branching trajectory, the ability of performing trajectories searching with non-tree-like lineage, and the scalability for large datasets.
2.2.2 CellPaths provides high-resolution branching point detection for trifurcating dataset
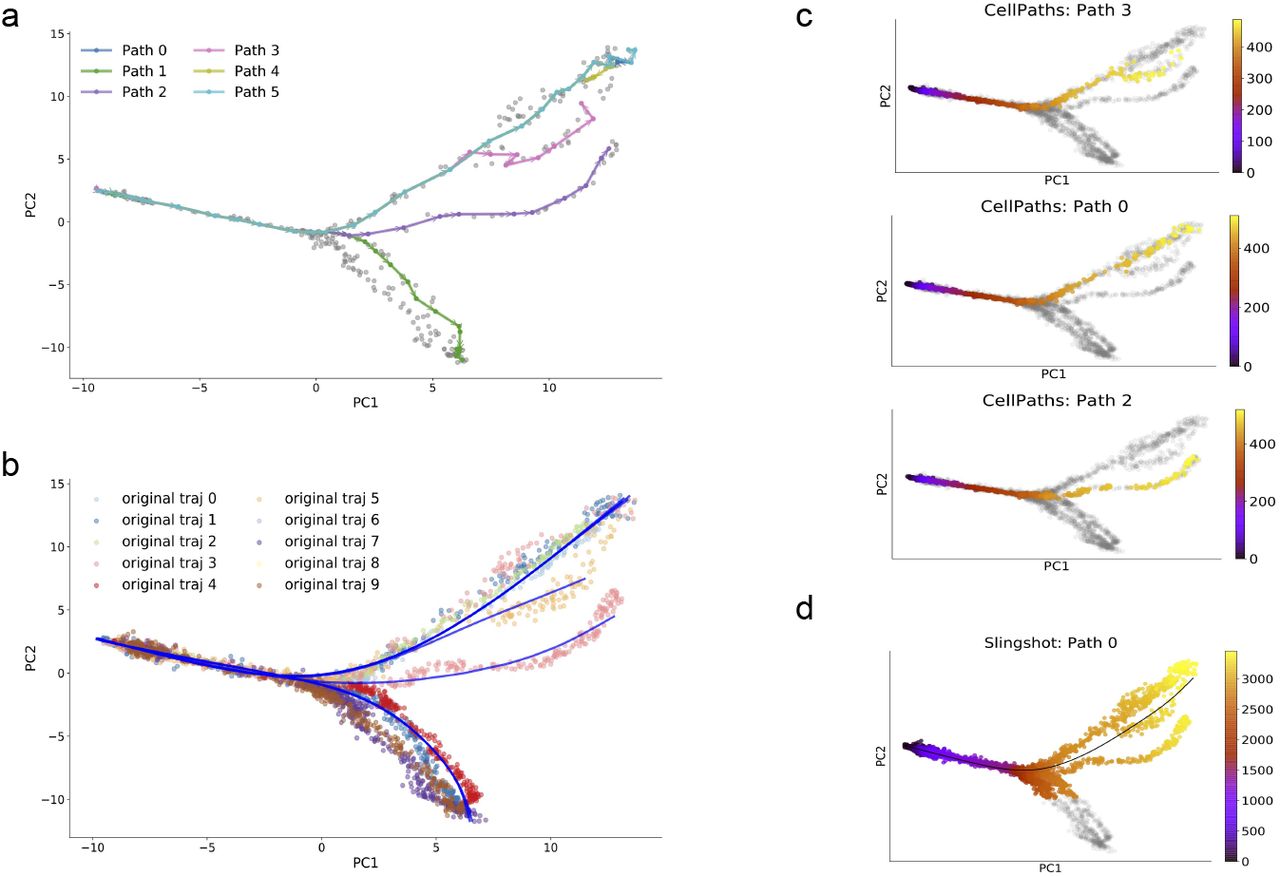
We first compare our results with Slingshot [Street et al., 2018] on a simple trifurcating dataset. With simple datasets, Slingshot has a comparative performance with CellPaths. However, CellPaths detects the branching point more accurately. In Fig. 5, Slingshot misclassifies cells around the branching area into wrong branches, while in Fig. 5, CellPaths correctly classifies cells into the branches that they belong to.

(a). mate-cell level paths detected using CellPaths, First six trajectories are shown. With each gray dot correspond to the location of each meta-cell in the space constructed with first two principal components. (b). Blue curve visualize the true-cell level backbone of the trajectories detected using CellPaths, principal curve is utilized to perform smoothing of the paths on true-cell level. Scattered dots correspond to the true cell in the PCA space, with different color correspond to the cells that belong to different original ground truth trajectories. (c).The pseudo-time ordering on true-cell level with CellPaths, the pseudo-time of each cell is constructed using first order approximation pseudo-time assignment (Methods). The gray cells correspond to the cells that do not belong to current trajectory, the color of all the other colored cells (from yellow to purple) correspond to the ordering of cell in current trajectory.(d).The reconstructed trajectory backbones and pseudo-time recovery using Slingshot method in trifurcating dataset
CellPaths can detect trajectories with high resolution mainly due to the incorporation of RNA velocity information. RNA velocity predicts a cell’s potential differentiation direction, and cells with similar expression data can possibly have distinct differentiation direction. The incorporation of RNA velocity serves as additional information to separate cells in the branching area with a high resolution.
2.2.3 CellPaths detects more correct branches in complex branching dataset
We have designed the second simulated dataset to have more complex structure. We run the process of generating the tri-furcating structure twice, and obtain two highly similar and overlaid datasets. The two datasets are close to each other in the gene-expression space but have a minor difference branching with regarding the branching position. We then simply merge these two datasets which creates a setting that is particularly difficult for the detection of the branching point.
We perform both CellPaths and Slingshot on this dataset. In Fig.6, CellPaths detects all four sub-branches and accurate corresponding branching points, while as in Fig.6, Slingshot merges two adjacency branches together and only detects two branches. With dimension reduction methods that cannot separate the trajectories into distinct spatial position, high-level clustering algorithms tend to cluster cells from different but closely located trajectories together, which results in fewer branch detection. Current cluster-level trajectory inference methods produce a trajectory inference result more robust to the noise within the scRNA-seq data compared to cell-level trajectory inference methods, but its performance are severely affected by the upstream dimension reduction and clustering method. And Slingshot, being one of those methods, tends to predict fewer branches and ignore detailed cell differentiation information. CellPaths, on the other hand, utilizes meta-cell methodology, where clusters of moderate sizes are constructed. This strategy increases the robustness towards the expression measurement and RNA velocity calculation noise while also reduce the possibility of misclassification.

(a). mate-cell level paths detected using CellPaths, first six trajectories are shown. With each gray dot corresponds to the location of each meta-cell in the space constructed with first two principal components. (b). The true-cell level backbone of the trajectories detected using CellPaths, principal curve is utilized to perform smoothing of the paths on true-cell level. Scattered dots correspond to the true cell in the PCA space, with different color correspond to the cells that belong to different original ground truth trajectories. (c).The pseudo-time ordering on true-cell level with CellPaths, the pseudo-time of each cell is constructed using first order approximation pseudo-time assignment (Methods). The gray cells correspond to the cells that do not belong to current trajectory, the color of all the other colored cells (from yellow to purple) correspond to the ordering of cell in current trajectory. (d).The reconstructed trajectory backbones and pseudo-time recovery using Slingshot method in double batches dataset
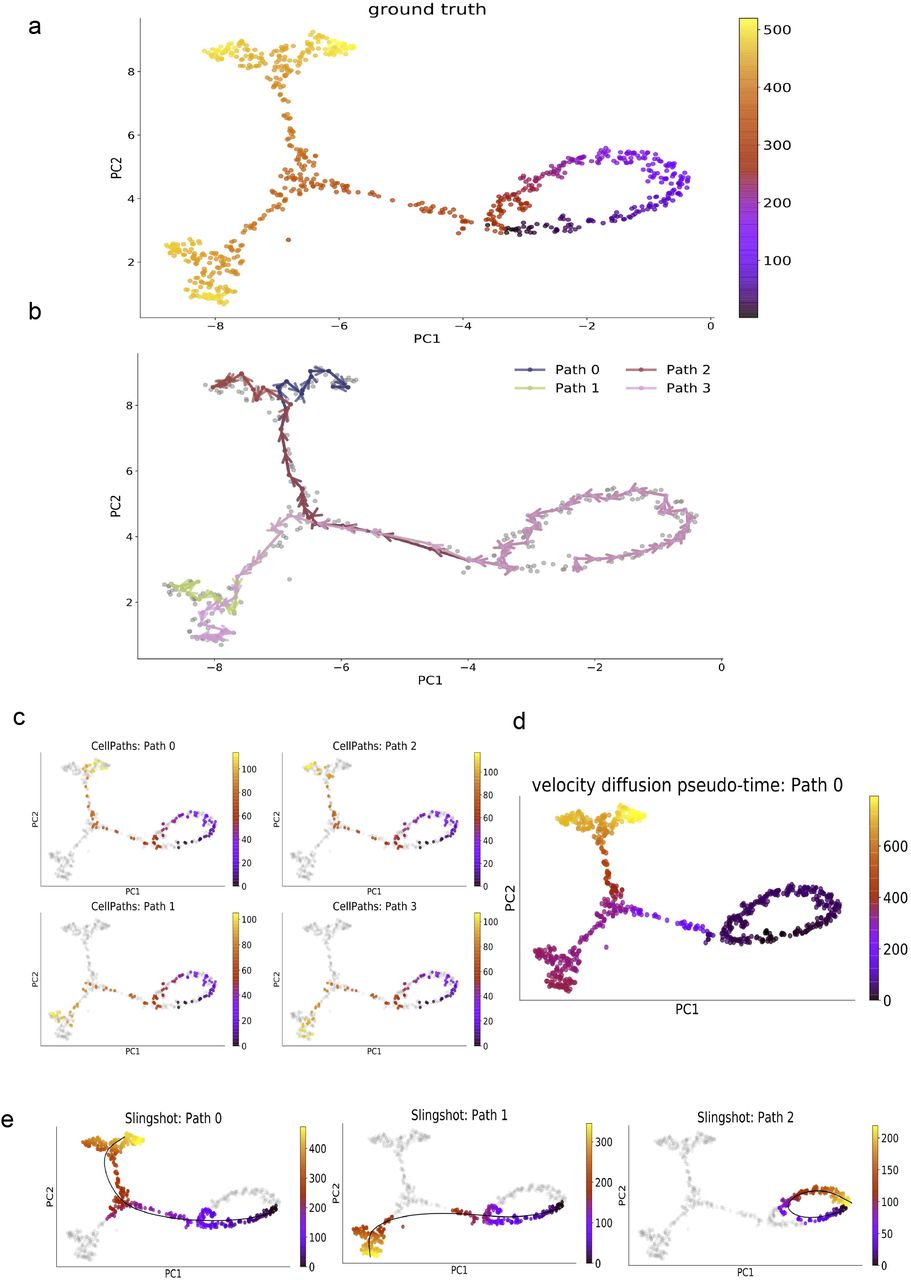
2.2.4 CellPaths infers all branches in complex cycle-tree trajectory structure
Many real world dataset has both tree-like branching structure and small cell-cycles within the branching trajectory. To test CellPaths ability in detecting high-level differentiation lineages while preserving subtle cell-cycle structure. We generate a cycle-tree trajectory structure, shown in Fig. 7a. CellPaths successfully find all four branches and cell-cycle process. Vdpt successfully infers the branching trajectory but smooth out the cell-cycle process. Slingshot, again, fails to infer the trajectory with only root cell provided.

a. Umap visualization of cycle-tree dataset, with cell color annotated using ground truth pseudo-time. b. meta-cell-level path inferred by CellPaths. CellPaths finds all four branches and cell cycle structure. c. Cell-level pseudo-time result, inferred four branches are shown, with cell color annotating the inferred pseudo-time. d. Vdpt result with cell color annotated using inferred pseudo-time. e. Slingshot result, Slingshot infers two branches and one cell-cycle structure. The inferred principal curve and corresponding pseudo-time of each branch is shown.
2.2.5 CellPaths accurately infers multiple cycles trajectory
We perform CellPaths, Slingshot and Vdpt on a synthetic dataset with multiple cycles, with the result shown in Fig. 8. CellPaths can accurately find multiple-cycle structure. On the other hand, Slingshot wrongly infer the differentiation process into a bifurcating structure without the directional information. Vdpt can accurately infer the differentiation direction, but it mixes cells from different cycles together and finds only one cycle. With a careful selection of meta-cell size, CellPaths is able to find a good balance between finding high-resolution trajectory and being robust to the noise within the dataset.

a. PCA visualization of multiple-cycles dataset, with cell color annotated using ground truth pseudo-time. b. Vdpt result with cell color annotated using inferred pseudo-time. c. CellPaths result. Cell color and arrows linked in between annotating the gradual change of inferred pseudo-time. d. Slingshot result, Slingshot infers two branches. The inferred principal curve and corresponding pseudo-time of each branch is shown.
2.2.6 Pseudotime reconstruction Accuracy
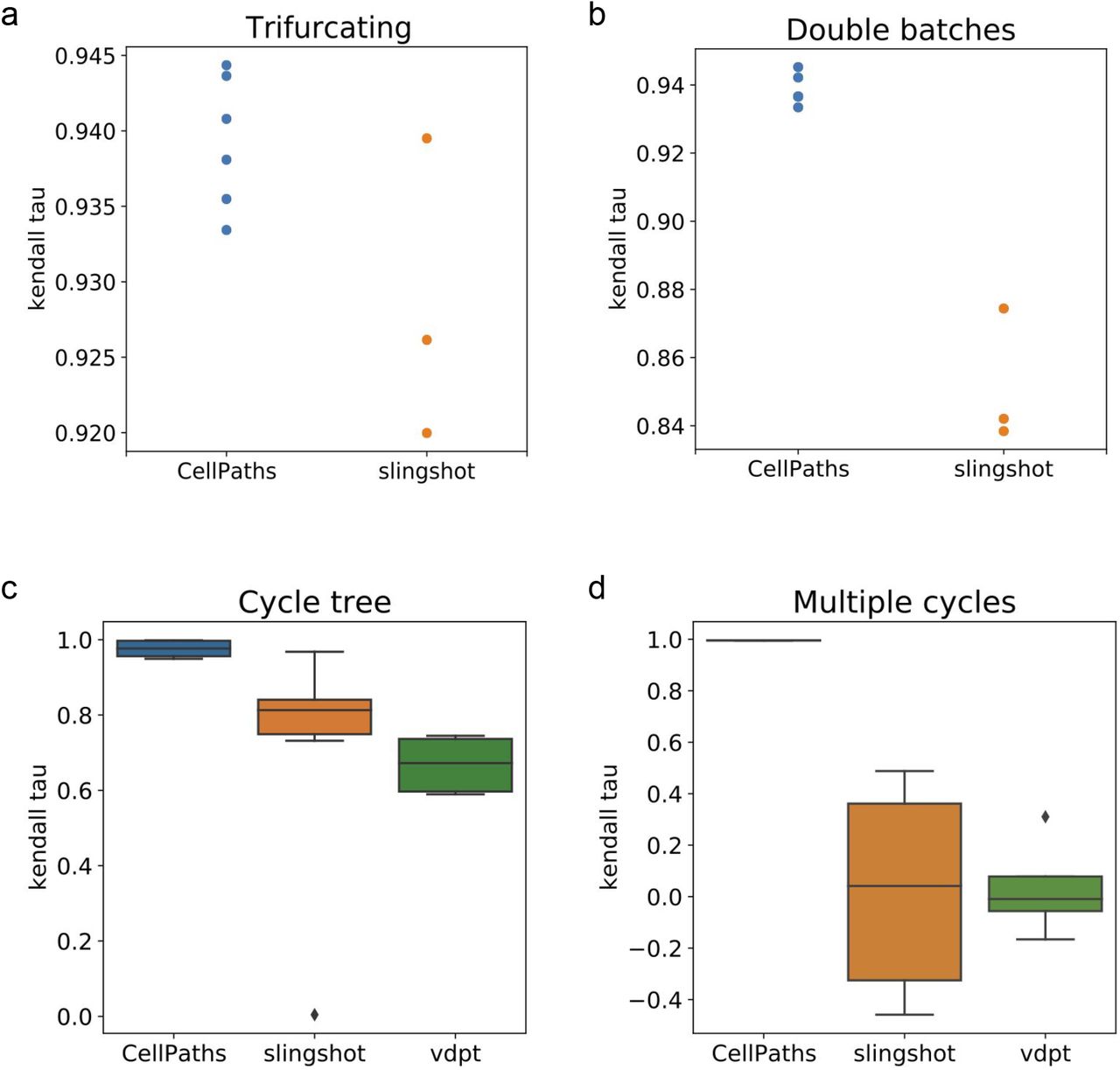
We further quantify the reconstruction accuracy of CellPaths and other trajectory inference methods using kendall tau correlation coefficient between the ground truth pseudotime and the predicted pseudotime by each respective method. The performance are measured in all four types of structures and the result is summarized in Fig. 9. In the trifurcating dataset and two batches branching tree dataset, the coefficient is measured on each individual branches inferred by Slingshot and CellPaths. The result shows that CellPaths provides better reconstruction accuracy, especially in more complex structure. In cycle tree and multiple cycles structures, multiple simulated datasets of each structure are generated using different random seeds, the algorithms are tests on each dataset to provide a statistically significant evaluation result. The running result of different algorithms are summarized using boxplots. The preformance of CellPaths is again better than Slingshot and velocity diffusion pseudo-time. Especially in the multiple-cycle dataset, without the direction annotation, Slingshot tend to infer the cycle structure as bifurcating structure. As a result, Slingshot provides results with both positive and negative correlations. Vdpt, on the other hand, incorporate velocity information, but still provide almost random results as it mix the cells in two cycles together. CellPaths, on the contrary, still provide accurate inference results as it successfully differentiates cells in two difference cycles and correctly detects the differentiation starting point.

The reconstruction accuracy in synthetic dataset, measured using kendall tau correlation. a. Kendall tau correlation coefficient of each individual trajectory inferred using CellPaths and Slingshot. The accuracy of CellPaths is in general a little bit higher than Slingshot in simple trifurcating structure. b. Kendall tau correlation coefficient of CellPaths and Slingshot in double batches dataset. CellPaths provides results with significantly better accuracy in complex dataset. c. Kendall tau correlation of CellPaths, Slingshot and Vdpt in Cycle-tree dataset. Multiple datasets with the same structure are generated, and the results of multiple runs are visualized using boxplot. CellPaths provides better accuracy result. d. Kendall tau correlation of CellPaths, Slingshot and Vdpt in Multiple-cycles dataset. The results of multiple runs are summarized in the boxplot. CellPaths provides significantly better result as it fully utilizes RNA-velocity information.
3 Conclusion
We have presented CellPaths, a method to detect multiple high-resolution trajectories in scRNA-Seq datasets. Noise in the single-cell RNA-seq data has always been one major problem for trajectory inference methods. Cell-level approaches [Trapnell et al., 2014, Haghverdi et al., 2016, Setty et al., 2016], can detect branching point in a high resolution, but is extremely sensitive to measurement noise, while cluster-level methods [Street et al., 2018, Wolf et al., 2019, Shin et al., 2015, Ji and Ji, 2016] find more comprehensive lineage structures that robust to noise at the expense of the loss accuracy in branching point detection. The construction of meta-cell is a method that lies in between, by creating meta-cells, we denoise the original expression and velocity matrix while still preserve the detailed structure of the dataset.
The shortest-path algorithm and greedy selection strategy allow for the discovery of trajectories in a fully automatic way. Since shortest-path algorithm finds almost all possible trajectories, the method does not have any assumption on the underlying backbone structure. Greedy selection strategy assumes that the true trajectories structure of the dataset should have low average weights and cover the cells in the dataset as possible. The assumption fit in the real circumstance and synthetic dataset extremely well, which also proves the correctness of the assumption.
4 Methods
In our result the RNA velocity is calculated using the dynamical model of scVelo [Bergen et al., 2019], but CellPaths is intrinsically flexible with the upstream RNA velocity calculation methods. Different upstream RNA velocity estimation methods, such as scVelo [Bergen et al., 2019] and velocyto [La Manno et al., 2018], can be chosen as an upstream velocity inference method for CellPaths.
4.1 VeloSim: simulation of scRNA-Seq data with RNA velocity
VeloSim is a novel procedure to simulate scRNA-Seq data including the amount of nascent RNAs and the true velocity. The main advantage of VeloSim compared to dyngen is that VeloSim allows users to input a specific trajectory structure.
Given a trajectory structure, VeloSim simulates time-series data of the expression levels of both the nascent and mature mRNAs. VeloSim follows the kinetic model [Munsky et al., 2012] and considers that a gene is either in an on state or in an off state. At every time step:
determine if the current state of the gene is on or off, depending on its state at the previous time point and the transition probabilities between the two states. The transition probabilities are calculated from kinetic parameters kon and koff, which respectively represents the frequency of a gene being in an on and off state.
if the gene is in an on state, more nascent RNAs are generated according to synthesis rate s; and the nascent RNAs are transformed into mature mRNAs according to rate β; the mature mRNAs degrade with rate d.
if the gene is in an off state, no new nascent RNAs are generated. If the current amount of nascent RNAs is not zero, it will continue converting into mature mRNAs and the mRNAs continue to degrade.
4.2 Meta-cell construction
RNA-velocity estimation at single-cell level can be extremely noisy and even possibly erroneous, given the noisy measurement of count matrix especially the nascent mRNA count matrix and the stringent assumption on RNA-velocity estimation. Even though current RNA-velocity estimation methods propose multiple ways to ameliorate the inaccurate estimation problem, e.g. velocyto [La Manno et al., 2018] and scVelo [Bergen et al., 2019] use k-nearest neighbor (kNN) graphs to denoise the measurement, scVelo [Bergen et al., 2019] relaxes the steady-state assumption of velocyto [La Manno et al., 2018] to dynamical model. Using RNA velocity for trajectory inference still suffers from the inaccuracy of upstream RNA velocity calculation. Here we propose meta-cell construction as an additional denoising step prior to the trajectory inference algorithm.
We assume the cell expression data that share strong similarities in the expression space are the noisy realization of the underlying meta-cell expression data [Baran et al., 2019]. Meta-cell is constructed from clustering algorithms. Both k-means and Leiden clustering are implemented in CellPaths for more flexibility. CellPaths also allows the inclusion of nascent mRNA counts to further improve the accuracy of clustering results. For each meta-cell, a denoised expression vector is estimated efficiently by calculating the average of the expression data within the group. The smoothed RNA velocity measurement is estimated using Gaussian radial basis function (RBF) kernel regression method based on the data within the cluster.
The additional regression step is used to reconstruct a smooth function
 given the input single-cell expression samples
given the input single-cell expression samples  and output velocity samples
and output velocity samples  . And kernel regression considers the function lies within the reproducing kernel hilbert space
. And kernel regression considers the function lies within the reproducing kernel hilbert space  with projection
with projection  . And the kernel function can be calculated using the projection:
. And the kernel function can be calculated using the projection:

Gaussian kernel is one of the most widely used smooth kernel for the regression model, which takes the form

The final function is the linear combination of kernel functions
 with coefficient α calculated as
with coefficient α calculated as
 Kernel matrix K can be easily calculated from kernel function and
Kernel matrix K can be easily calculated from kernel function and

Compared to other regression model, Gaussian kernel regression model is more expressive and fully utilizes smoothness assumption of the function, and thus suitable for reconstructing noiseless RNA-velocity value from noisy single cell expression and velocity samples.
Given the relatively small amount of cells within a cluster, a regularization term is needed for the meta-cell RNA velocity estimation process. Here we use the L2-regularization.
4.3 Neighborhood graph construction
The cell differentiation mechanism can be modeled mathematically as a low dimensional manifold within a continuous high dimensional expression space [Morris et al., 2014, Tritschler et al., 2019], which provide a strong theoretical support of manifold learning method in single-cell data analysis. Currently, manifold-learning-based methods [Moon et al., 2019, Haghverdi et al., 2016, Weinreb et al., 2018], for single-cell dataset construct neighborhood graph with different kinds of kernels to approximate the underlying manifold, which achieves promising results in single-cell dataset.
Existing neighborhood graph construction methods usually use similarity measurement between cells and construct an weighted undirected graph. With the incorporation of RNA-velocity measurement, we construct a weighted directed graph that penalize both the direction difference and distance. The graph construction process can be separated by two steps: K-nearest neighbor graph construction with selected K, and weight assignment.
The weight function can be expressed as
 With direction penalty
With direction penalty
 And distance penalty
And distance penalty
 α and λ hyper-parameters.
α and λ hyper-parameters.
The graph construction pseudo-code:

Build Graph
4.4 Detection of trajectories
To reconstruct the trajectories on meta-cell level we conduct two steps: first, we find possible paths on the neighborhood graph, then we select paths using a greedy strategy as our reconstructed trajectories.
Shortest-paths between vertices can approximate the distance within the manifold and is intrinsically suitable for the incorporation of direction notation. However, shortest path suffers from the noisy measurement, and the classical Floyd-Warshall algorithm which finds all-pairs shortest paths for a graph have O(N3) time complexity. These problems are ameliorated through the following: 1) the incorporation of meta-cell in the first step can increase robustness to noise; 2) we use Dijkstra algorithm where the starting nodes are selected using in-degree with O(N2) time complexity, and this algorithm achieves comparable final results to those of the Floyd-Warshall algorithm.
With all the suitable paths found, possibly as large as O(N2), further path selection strategy is necessary to eliminate erroneous paths and pick up the correct path, which should only cover a small subset of all the paths.
We construct a greedily path selection strategy. Firstly, unsuitable paths are filtered out from the pool of paths. The paths that cover too less cells, which have a higher probability to be erroneous, were eliminated via a thresholding process. In addition, paths are filtered based on the average edge weight within the path. Shortest-path algorithm finds path between two nodes as long as those two nodes are connected. As a result, some directed shortest paths that connect two nodes but has large average edge weight usually have low time-coupling between neighboring nodes within the path and do not convey true biological causality relationship.
Then the paths are picked in an iterative way, with each iteration finds the path that has the highest greedy score, which consists of two parts, the length of path itself ℓ and the number of uncovered cells ℓu, and the score calculation follows:

The pseudo-code for greedy path selection algorithm:

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Greedy Paths Selection
With the greedy selection strategy, most meta-cell would be covered within first several paths.
4.5 First order approximation pseudo-time assignment
After finding all the high quality meta-cell paths, pseudo-time assignment for the true cells within each meta-cell path is necessary. The meta-cell paths can be considered as a linear trajectory structure for true cells that covered by the meta-cell. Current best practise of pseudo-time assignment for linear trajectory can basically be separated into two sub-types: principal-curve-based pseudo-time assignment [Campbell et al., 2015, Street et al., 2018] and random-walk-based pseudo-time assignment [Haghverdi et al., 2016, Weinreb et al., 2018, Farrell et al., 2018]. Two methods perform almost equally for simple linear trajectory pseudo-time assignment, but both methods cannot fully utilized the order provided by meta-cell paths, as root cell is the only information that is needed to be provided. Here we propose first order approximation pseudo-time assignment, which is a highly efficient method with linear complexity.
After obtaining meta-cell paths, the relative order of cells in different meta-cell is known, and all we need to do is assign cells within each meta-cell an order, and simply merge the inner meta-cell order with inter meta-cell order.
The inner meta-cell order is obtained by projecting cells onto the direction of meta-cell velocity and order according to the projection position. The backbone trajectory can be considered as a smooth curve that pass through each cluster, we can safely consider that the backbone trajectory pass through the cluster center, which should be the denoised version of all the cells within the cluster.
As a result, the backbone can be considered as a smooth function f(t): ℝ → ℝd, t is the pseudo-time and d is the number of genes. And f(t) should pass through all the cluster center, or the meta-cell xi, i.e. there exist f(ti) = xi, and the derivative of f(·) at ti is the RNA velocity vi of the meta-cell xi.
Using the first order taylor expansion, f(t) can be approximated by

When  . Here
. Here  is one dot(cell) on the underlying continuous true (noiseless) trajectory.
is one dot(cell) on the underlying continuous true (noiseless) trajectory.
As a result, for all the cells xj, j ∈ Ni within the cluster (with center xi), we can find the corresponding true xby projecting xj onto f(t),

We get the pseudo-time of the cells xj, j ∈ Ni within the cluster by simply complare their corresponding projected pseudo-time  on f(t). With simple mathematics, we can prove that
on f(t). With simple mathematics, we can prove that

As a result, within each cluster, we can calculate  , where xi is the meta-cell expression, vi is the velocity of the meta-cell, xj is the true cells within the cluster, and then sort the result.
, where xi is the meta-cell expression, vi is the velocity of the meta-cell, xj is the true cells within the cluster, and then sort the result.
In addition, principal curve and random walk based methods are also implemented in the package. We use mean first passage time as the pseudo-time the random walk based method.
4.6 Differentially Expressed Gene analysis
We consider the gene expression value during a differentiation process to be a function of inferred pseudo-time, and build a generalized additive model for the gene expression data. We assume the gene expression data follows a negative binomial distribution, and using spline function as the building block for the regression model.

We test the significance of a specific gene using likelihood ratio test, and select DE genes according to the p-value.
4.7 Evaluation method
The accuracy of inferred pseudo-time in each trajectory is measured using Kendall rank correlation coefficient.
4.8 Real datasets
We demonstrate the performance of CellPaths using two previously published single-cell RNA-seq datasets, with RNA velocity calculated using the dynamical model in scVelo [Bergen et al., 2019].
Dentate Gyrus dataset
The original paper collected multiple dentate gyrus samples at different time points during mouse development. The scRNA-seq process is performed using droplet-based approach and 10x Genomics Chromium Single Cell Kit V1. The dentate gyrus dataset is created from P12 and P35 time point, which correspond to the dentate gyrus structure in developing and mature mice. Totally 2930 cells are incorporated that covers the full developmental process of granule cells from neuronal intermediate progenitor cells (nIPCs). The dataset can be accessed through GSE95753.
Pancreatic Endocrinogenesis dataset
The dataset is sampled from E15.5 of the original Pancreatic Endocrinogenesis dataset, totally 3696 cells are incorporated in the dataset, which covers the whole lineage from Ductal cell through Endocrine progenitor cells and pre-endocrine cells to four different endocrine cell sub-types. The dataset is obtained through droplet-based approach and 10x Genomics Chromium. The dataset can be accessed through GSE132188.
Human forebrain dataset
The dataset profiles 1720 using droplet-based scRNA-seq method, which incorporates cells spans from radial glia to mature glutamatergic neuron within glutamatergic neuronal lineage in developing human forebrain.
Footnotes
ziqi.zhang{at}gatech.edu, xiuwei.zhang{at}gatech.edu
References
- [Baran et al., 2019].↵
- [Bastidas-Ponce et al., 2019].↵
- [Bergen et al., 2019].↵
- [Campbell et al., 2015].↵
- [Cannoodt et al., 2020].↵
- [Collombat et al., 2003].↵
- [Farrell et al., 2018].↵
- [Fischer et al., 2019].↵
- [Haghverdi et al., 2016].↵
- [Hochgerner et al., 2018].↵
- [Ji and Ji, 2016].↵
- [La Manno et al., 2018].↵
- [McInnes et al., 2018].↵
- [Moon et al., 2019].↵
- [Morris et al., 2014].↵
- [Munsky et al., 2012].↵
- [Qiu et al., 2020].↵
- [Saelens et al., 2019].↵
- [Schiebinger et al., 2019].↵
- [Setty et al., 2016].↵
- [Shin et al., 2015].↵
- [Street et al., 2018].↵
- [Trapnell et al., 2014].↵
- [Tritschler et al., 2019].↵
- [Weinreb et al., 2018].↵
- [Wolf et al., 2019].↵
- [Zhang et al., 2019].↵




